
Abstract
Here we review the emerging neurobiological understanding of the role of the brain’s reward system in the regulation of body weight in health and in disease. Common obesity is characterized by the over-consumption of palatable/rewarding foods, reflecting an imbalance in the relative importance of hedonic versus homeostatic signals. The popular ‘incentive salience theory’ of food reward recognises not only a hedonic/pleasure component (‘liking’) but also an incentive motivation component (‘wanting’ or ‘reward-seeking’). Central to the neurobiology of the reward mechanism is the mesoaccumbal dopamine system that confers incentive motivation not only for natural rewards such as food but also by artificial rewards (eg. addictive drugs). Indeed, this mesoaccumbal dopamine system receives and integrates information about the incentive (rewarding) value of foods with information about metabolic status. Problematic over-eating likely reflects a changing balance in the control exerted by hypothalamic versus reward circuits and/or it could reflect an allostatic shift in the hedonic set point for food reward. Certainly, for obesity to prevail, metabolic satiety signals such as leptin and insulin fail to regain control of appetitive brain networks, including those involved in food reward. On the other hand, metabolic control could reflect increased signalling by the stomach-derived orexigenic hormone, ghrelin. We have shown that ghrelin activates the mesoaccumbal dopamine system and that central ghrelin signalling is required for reward from both chemical drugs (eg alcohol) and also from palatable food. Future therapies for problematic over-eating and obesity may include drugs that interfere with incentive motivation, such as ghrelin antagonists.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction: Food reward and obesity
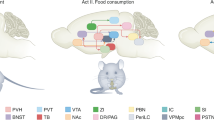
To eat is pleasurable and rewarding. It is not surprising therefore that brain centres involved in pleasure and reward are activated when we eat. Fundamental neurobiological mechanisms involved in food reward are of considerable importance for understanding how body weight is regulated, both in health and in disease. A great deal of obesity research over the past two decades has identified genes and mechanisms that are important for maintaining energy balance. Although body weight is strongly influenced by our genes, it is also influenced by lifestyle and social habits, reflecting a powerful interaction between genes and environment [1]. Food intake, however, is motivated not only by the need to restore energy homeostasis; palatable, rewarding high fat and/or sugar foods such as chocolate can motivate intake despite a state of satiety. Obesity reflects an energy imbalance in which genetically susceptible individuals become increasingly vulnerable to an obesogenic environment. Thus, both the palatability and availability of foods in the Western diet play a major role for the development of this disease [2]. An emerging hypothesis concerns the role of the brain’s reward system, that responds to the stimulus provided by rewarding and palatable ‘obesogenic foods’ and appears to override the homeostatic signals for body weight control [3]. Indeed, mismatch between the hedonic/rewarding value attributed to food and energy needs is characteristic of eating disorders, including those that lead to obesity (Fig. 1).

Schematic illustration of the interactions of between homeostatic/metabolic and hedonic control of food intake in normal weight and obese individuals. Palatable foods reinforce their consumption by increasing both the motivational and hedonic components of the reward process. Whereas homeostatic signals are able to put a brake on food reinforcement in normal weight individuals, this does not appear to be the case for the obese. Moreover, increased food intake in obesity may reflect an allostatic shift in the set-point for food reward, characterized by either an increased hedonic requirement (the reward hyperfunction theory) or an increased motivation to compensate for a hedonic deficit (the reward hypofunction theory). By analogy with chemical drug addiction, problematic over-eating may commence with an increased hedonic requirement but with increased exposure, the hedonic/rewarding value of the food decreases, resulting in an increased motivation for food (ie eating in the absence of pleasure in the ‘food addicted’ state). Unfortunately obesity- associated leptin and insulin resistance likely play an important role to desynchronize these appetitive brain mechanisms. Recent studies have identified the central ghrelin signalling system as having an important role for increasing food reward. Given that obese individuals appear to remain ghrelin sensitive, future therapies for problematic overeating could include ghrelin antagonists
From an evolutionary perspective, it is easy to understand why eating involves hedonic processes. A positive hedonic experience in association with the consumption of food helps ensure an adequate supply of nutritionally diverse foods from our environment. Whereas ‘man the hunter’ would have benefited from the hedonic experience of eating, in our modern obesogenic environment, it may be more advantageous for health and survival to suppress it. Indeed, this concept has inspired research and development of anti-obesity drugs that target the reward mechanism. Such agents would be expected to reduce food intake through the suppression of food reward, involving direct or indirect interruption of food-sensitive reward pathways. Here we review some of the mechanisms and candidate systems, focusing especially on recent advances in the field.
2 Neurobiological mechanisms of food reward
The popular ‘incentive salience theory’ of reward recognises three distinct neurobiological components: ‘liking’, ‘wanting’ and ‘learning’, phenomena that can be applied as readily to natural reinforcers such as food as to artificial reinforcers such as chemical drugs of addiction [4]. ‘Liking’ is the hedonic component that reflects the immediate experience or anticipation of pleasure, for example, from the orosensory stimulation of eating a pleasurable food [5]. ‘Wanting’ is the reward-seeking (or incentive motivation) component that results in increased appetite, food cravings and other behaviours associated with increased motivation to obtain food [1, 5–7]. While ‘liking’ and ‘wanting’ are closely associated, it is clear from individuals with chemical drug addiction that motivated reward-seeking activity can occur in the absence of pleasure. Although it remains much debated as to whether there is a sufficient evidence base for food addiction, there are indications that the ‘wanting’ and ‘liking’ of obesogenic foods can also be dissociated, for example, in individuals with binge eating disorders [8, 9].
2.1 The role of dopamine in food reward
A great deal is known about the circuits and mechanisms underpinning both hedonic and motivational components of reward, including mechanisms that are common to natural and artificial rewards. As these circuits and mechanisms are covered extensively in other reviews [8, 10–12], only the key elements are described here. The neurotransmitter ‘dopamine’ is of primary importance for incentive motivation [13], involving especially a key ‘mesoaccumbal’ projection from the ventral tegmental area (VTA) of the midbrain to the nucleus accumbens (NAcc) [14], but also via a neuronal network that includes VTA projections to the prefrontal cortex, amygdala and hypothalamus. Studies measuring accumbal dopamine release in rodents have revealed that the mesoaccumbal dopamine pathway is activated in response to sweet tastants [15] by binging on sugar [16] and by corn oil [17]. Consistent with these effects on the dopamine system, such foods also stimulate motivated behaviour for a food reward [18–21]. In these studies motivated behaviour for a food reward was demonstrated using either operant conditioning experiments in which the animals have to work increasingly hard (eg by pressing a lever) in order to obtain a food reward or by studies incorporating the ‘incentive runway’ paradigm, in which motivated behaviour is reflected in the time taken to reach a goal box. Operant responding for food can be increased by drugs that increase accumbal dopamine signalling (eg amphetamine)[22] and decreased in models of suppressed dopamine signalling [23]. As reviewed elsewhere [12, 13], the mesolimbic dopamine system does not appear to be directly involved in the ‘liking’ of sweet tastants, although it is involved in the reinforcement of their consumption; thus, animals will repeat behaviours that increase accumbal dopamine levels, such as the consumption of food rewards. Dopamine lesions (using 6-OHDA) that deplete forebrain dopamine do not alter food intake per se but do alter facial ‘liking’ reactions (facial affective expressions of taste pleasure) to sweet tastants [24]. In a model of enhanced dopamine signalling in mice (by genetic knockdown of a dopamine transporter gene), ‘liking’ reactions to sucrose did not increase, even although such mice did show increased motivated behaviour for sweet rewards [25, 26]. Indeed, it has been suggested that taste may not be required for food reward. Mice that are unable to process sweet tastes (trpm5 knockout mice) appear to experience reward from sucrose reflected by an increased sucrose preference and by the ability of sucrose to activate the mesoaccumbal system in these mice [27].
2.2 The role of mu-opioid signalling in food reward
The neural networks involved in the ‘liking’ hedonic component of reward include pathways involved in taste processing in the brainstem, pons, nucleus accumbens, ventral pallidum, amygdala and prefrontal cortex [28, 29]. Within these circuits, the mu-opioid system emerges as a key target for the hedonic experience of feeding [26, 30]. Indeed, mu opioid receptor stimulation of the NAcc has been shown to increases the intake of (and preference for) sweet and high fat foods [31, 32]. Indeed, within the extensive opioid-responsive feeding circuits the NAcc has been identified as a primary target for food intake that is associated with “liking” orofacial responses [33]. It has recently been questioned whether the NAcc mu-opioid system may not also be of importance for ‘wanting’ [34]. These authors found that suppression of endogenous mu-opioid in the nucleus accumbens shell using selective antagonists decreased both ‘liking’ of sucrose (reflected by fewer positive hedonic orofacial responses) and the incentive value (‘wanting’) of a food reward, assessed in the incentive runway paradigm. Consistent with this, it was earlier reported that a mu-opioid receptor agonist increases motivated behaviour for a food reward, reflected by the elevated break point for progressive ratio lever pressing [35].
2.3 The role of cannabinoids and orexin in food reward
Suppression of the endogenous cannabinoid (endocannabinoid) system resulted in a successful anti-obesity therapy, rimonabant that unfortunately was withdrawn due to adverse psychiatric effects. The cannabinoid receptor 1 (CB1) is widely expressed in the CNS, including areas associated with food intake, food reward and appetitive behaviour. Various parenchymal targets appear to be important for the orexigenic effects of the endocannabinoids, including several nuclei in the hypothalamus and hindbrain [36–38] as well as the NAcc (shell) [39]. CB1 signalling also appears to be important for food reward, both hedonic and incentive motivation components [40] involving interactions with both dopaminergic and opioid mechanisms [41, 42]. Interestingly, endocannabinoid signalling in the parabrachial nucleus appears to be especially important for the intake of palatable foods [38].
The central orexin signalling system also figures rather prominently in motivated behaviour for artificial and natural rewards (including food), involving a key projection from the lateral hypothalamus to the VTA, where orexin appears to directly target the dopamine cells that project to the NAcc shell [43, 44]. Within the appetitive/reward circuits CB1 and orexin A receptors colocalize and appear to interact, reflected by the effects of a subeffective dose of rimonabant to suppress the effects of centrally administered orexin [45].
3 Palatability and food reward
From a neurobiological perspective, it remains difficult to identify what makes ‘Luxury Creamy Dark Chocolate Fudge Cake’ more rewarding to eat than a bowl of piping hot porridge, so much so that we are prepared to consume a large portion or two at the end of a satiating meal. How do rewarding foods trigger neural responses that reinforce their consumption? Is it the ingredients or even the combination of ingredients that elicit special visual, olfactory and/or oro-sensory experiences that make them more palatable and heighten their rewarding value? Rats, like humans, have a “sweet tooth” and show preference for sweet and/or fatty foods. Indeed, a well-recognised and highly reproducible finding is that animals show dietary hyperphagia (an increased kcal consumed per day) when switched from normal chow to an obesogenic diet [46]. The most likely explanation for this diet-induced hyperphagia is that the obesogenic diet is more rewarding and invites increased consumption. Conceivably, increased exposure to such foods could even increase the ‘hedonic set point’ leading to problematic over-consumption. Indeed, sweet taste alone appears to be sufficient to activate the reward system, reflected by the effects of high sucrose solutions to increase motivated behaviour for food in the operant conditioning model [19, 47]. Moreover there are indications that the calorie content of sucrose, not the taste, that is rewarding [27]. Sugar and fat are especially effective for inducing motivated behaviour for food in rats, especially in combination [18–21]. On the other hand, properties of the food such as palatability (i.e. the hedonic evaluation of a flavour stimulus) enhance pleasure and motivational drives that induce further consumption, providing positive reinforcement (Fig. 1). In the context of chemical drug reward, reinforcement forms part of the addiction mechanism. For survival, evolutionary pressures have clearly promoted reinforcement of the oro-sensory pleasurable experience of eating in order to help maintain energy balance. The problem emerges of how to put on the break during times of food excess.
4 Obesity: an altered hedonic “set point”?
The rewarding value attributed to a given food can be rather subjective, influenced not only by food palatability and availability but also by individual genetic/trait/psychosocial differences. In a recent report, it has even been suggested that for some individuals, exercise increases the rewarding value of food and hence, diminishes the impact of exercise on fat loss [48]. Obese individuals may have allostatic changes in the hedonic set point for food and hence, attribute inappropriate rewarding values to foods. For example, according to the reward hyperfunction model of obesity, over-consumption could reflect a heightened responsiveness of the reward circuits to rewarding foods [49]. This would be rather analogous to the enhanced responsiveness of the reward circuits to addictive drugs in susceptible individuals. Supportively, obese individuals self-report increased pleasure and displayed stronger reinforcement from eating high-fat and high-sugar foods than lean individuals [50, 51]. Alternatively (or perhaps additionally), it has been suggested that over-eating, like gambling or substance use disorder, could reflect a reward deficiency syndrome, the consequences of which would be to increase motivation to obtain the reward, in this case for food [52]. Consistent with this, animals fed rewarding diets (eg high fat feeding or saccharine-enhanced chow) appear to have reduced sensitivity to psychostimulant rewards, reflected by an impaired acquisition of an operant response task reinforced by cocaine [53, 54].
It seems rather likely, as is the case for chemical drug addiction, that individuals predisposed to obesity may exhibit increased hedonic drives during early stages of the disease but, with increased exposure to rewarding foods, their hedonic value wanes while the motivational drives to obtain reward increase (ie increased eating to compensate for an increasing hedonic reward deficit). Supportively, studies in rodents, including obesity-prone rats, reveal addiction-like neuro-adaptive responses of the reward circuits to obesogenic diets, in terms of operant conditioning for rewarding foods, craving behaviour following cessation of the diet and self-administered reward (evoked by electrical stimulation of the lateral hypothalamus) [55–57]. Indeed, rats self-restrict their ad libitum intake of standard chow during “abstinence” from palatable/rewarding food [56, 57]. These adaptations, appear to be associated with suppressed dopamine release/turnover and dopamine receptor signalling/expression [58–60].
More direct evidence for an altered reward mechanism in obese individuals is provided from studies examining the brain response to food intake [61] or visual food cues [62]. Those pathways responding to food intake are likely involved in the oro-sensory pleasure experience of eating, whereas those responding to visual food cues are likely important for incentive motivation. It has been shown that individuals that have a tendency to over-eat, show an increased activity in brain areas associated with reward (ventral striatum, amygdala, anterior cingulate and premotor cortex) when shown appetizing compared with bland foods [63]. Obese individuals have a greater striatal response to visual food cues relative to lean individuals [64–66]. By contrast, the striatal response to actual food intake was actually reduced in obese individuals [67]. Collectively, these data suggest that obese individuals differ from lean individuals in reward processing; conceivably, the reduced activity of pathways involved in ‘liking’ is compensated for by a heightened activity of pathways involved in incentive motivation (‘wanting’). Consistent with this hypothesis, obese individuals have been shown to have reduced dopamine receptor D2 availability in the striatum relative to lean individuals, assessed by positron emission tomography [68, 69]. A decrease in D2 receptor availability, in other contexts, such as chemical drug addiction, has been thought to reflect increased dopamine release (ie increased activity of pathways involved in incentive motivation) [68]. Indeed, according to this model problematic over-eating would be driven by a hypofunction of the reward mechanism that compensates for a hedonic reward deficit.
Human genetic association studies also implicate dopamine signalling in obesity; for example associations were found between body weight and genetic variants in a dopamine transporter [70], catechol-o-methyl transferase (COMT) [71], and D2 receptor [72]. Indeed, functional studies have associated genetic variants of the D2 gene with increased activity in the dorsal striatum in response to palatable food [67].
The question remains as to whether long term exposure to obesogenic foods reprograms our hedonic set point for food reward. In rodents, diet-induced obesity causes temporal changes in the mesoaccumbal dopamine system, reflected by a suppression of basal accumbal dopamine levels [59], a reduced accumbal dopamine turnover [60] as well as a reduction in D2 receptors in the striatum [73]. Diet-induced obese animals display a suppressed motivated behaviour (operant responding) for palatable/rewarding foods [55] that appears to be independent of the obesity [60]. Conceivably, such animals receive sufficient reward from their obesogenic diet and therefore do not need to display motivated behaviour to increase their food reward. In obesity-prone animals, increased motivated behaviour for a palatable food reward only became apparent when the obesogenic diet has been terminated, when animals typically express craving behaviour [56].
There are indications that the successful outcome of Roux-en-Y gastric bypass surgery (RYGB), in terms of weight loss, may reflect allostatic changes in the hedonic set point for food. Patients that have undergone this bariatric surgery not only make healthier food choices but also have an altered attitude to (and desire for) food [74]. Moreover, recent studies in RYGP rats reveal an altered food reward phenotype [75, 76]. After surgery, these rats show a suppressed preference/acceptance of high fat food together with a restoration of normal “wanting” and “liking” responses, effects that likely reflect an altered gut-reward signalling mechanism.
5 Metabolic regulators of food reward
There is much evidence from rodent studies to suggest that the rewarding effect (and hedonic experience) of a given food is powerfully modulated by nutritional state and by metabolic regulators of hunger and satiety [77–81]. Thus, hungry animals show increased operant responding [80] and hedonic reactivity [82] to palatable foods. Conversely, caloric satiety has been shown to reduce positive hedonic reactions to sweet tastants [82]. Studies in human subjects have found that a food stimulus that induces reward when hungry can cause aversion when satiated, involving changes in neural activity in several brain regions that include the amygdala and orbitofrontal cortex [83]. Interestingly, nutritional status also impacts upon the rewarding value of reinforcers other than food, including addictive drugs [84]. Importantly, while the rewarding value attributed to a given food appears to reflect internal nutritional state, it also seems clear that hedonic drives are able to promote intake independently of nutritional need (i.e. over-eating palatable/rewarding foods when satiated). Thus, exposure to highly appetizing food cues can override satiety signals and promote overeating [85].
5.1 Food reward: regulation by leptin
The discovery that 1 week of treatment of the adipose-derived hormone, leptin to leptin-deficient obese individuals is able to alter the response of their reward system (especially striatum) to visual food cues [86], confronts us with the realization that metabolic hormones are able to exert a powerful influence on the way we process visual information about food from our environment (eg by advertising). Metabolic status is signalled to the brain, not only by circulating nutrients but also via a number of circulating hormones that include those produced by adipose tissue (eg leptin, adiponectin), by the gut (eg. ghrelin, PYY(3–36), oxyntomodulin, cholecystokinin) and also by hormones regulating glucose homeostasis (eg. insulin, glucagon-like peptide-1). As discussed in a number of recent reviews [87–89] these circulating signals inform diverse neurobiological circuits, including especially those involved in energy homeostasis in the hypothalamus and brainstem. For many of these hormones, there is increasing evidence that they also target diverse brain areas involved in reward, emotion and cognitive function, including those depicted in Fig. 1. Thus, for example, it is clear that leptin regulates both hedonic and motivational components of reward [80, 81, 90, 91]. Central leptin treatment to rodents suppresses the ability of sucrose and high fat food to condition a place preference [92, 93] and also suppresses operant responding for sucrose [79]. There is considerable evidence that the mesoaccumbal dopamine system appears to be a key target for leptin. Leptin receptors are present in the VTA, including on dopaminergic cells in this region and, moreover, direct injection of leptin into the VTA suppresses food intake, and leptin has been shown to suppress both accumbal dopamine release (basal and food-induced) and the electrical activity of VTA dopamine neurones [94–96]. Interestingly a sub-population of leptin-responsive VTA neurones have been shown to project to the central nucleus of the amygdala [97], an area strongly implicated in the addiction process. Additional reward targets for leptin include the lateral hypothalamus; leptin receptors have recently been shown to be present on discrete populations of cells in this region, including cells that project both to the VTA and to orexin-cells in the lateral hypothalamus [98].
An important question to address is whether the reward system remains responsive to leptin in obesity, given the key role attributed to leptin resistance in the development of the disease. Rats fed a higher fat diet for 5 weeks were resistant to the effects of centrally-administered leptin on operant responding for a sucrose reward [79]. Interestingly, low serum leptin levels have not been associated with binging episodes (ie a behaviour connected to food reward) in obese patients diagnosed with binge eating disorder [99], arguing against leptin as an acute regulator of food reward in obese patients. In such patients, chronic leptin resistance may be a more important factor, regulating the sensitivity of the brain’s feeding networks to rewarding foods in the long term and thereby increasing the likelihood of binge-eating, rather than having a direct role in individual binge episodes.
5.2 Food reward: regulation by insulin and PYY (3-36)
Of the other circulating anti-obesity satiety/anorexigenic hormones, several are rather well-studied in the context of food reward. The work of Diane Figlewicz, amongst others, has highlighted the importance of insulin in food reward, acting via similar mechanisms to leptin at the level of the VTA [81]. This could indicate that leptin and insulin potentiate each other’s effects at the level of the VTA as shown previously for the arcuate nucleus [100]. Unfortunately, obesity-associated insulin resistance in rats has also been shown to impact on the ability of the reward circuits to respond to insulin [79] (Fig. 1). PYY(3-36), a lower gut hormone, is released in association with food intake [101], has also been shown to impact upon key reward circuits in the VTA and ventral striatum [102]. Interestingly, these fMRI studies have shown that PYY(3-36) may play an important role in shifting feeding control from hypothalamic homeostatic circuits when hungry to the reward circuits when satiated.
For obesity to prevail, it seems clear that metabolic satiety signals are failing to regain control of appetitive brain networks, including those involved in food reward. There remains good reason for hope, however. Altered gut-brain signalling for appetite control remains a major topic of investigation, not least because the successful outcome of gastric bypass surgery (a bariatric weight loss procedure) appears to include not only a reduction in the amount of food eaten but also an altered attitude to, and preference for, healthier food [74]. Indeed, many metabolic/endocrine signalling systems, especially gut hormones, have been implicated in the successful outcome.
5.3 Food reward: role of ghrelin
In our research group, we have been especially interested recently in the possibility that future therapies for obesity may include a suppression of the central ghrelin signalling system. Ghrelin is the first identified circulating hormone to be attributed an orexigenic role. Ghrelin levels increase preprandially in association with meal initiation [103, 104] and studies in rodents have shown orexigenic effects after acute central or peripheral administration [105, 106]. It seems clear that ghrelin and synthetic ghrelin mimetics target cells in the hypothalamic arcuate nucleus [107, 108], notably the orexigenic neuropeptide Y cells in this region [109] that are likely involved in ghrelin’s orexigenic effects. The ghrelin receptor, GHS-R1A, is also expressed in tegmental and mesolimbic areas involved in reward, such as the VTA and laterodorsal tegmental areas (LDTg) [110, 111]. Recently, we provided the first evidence that ghrelin targets a key reward circuit, the so-called “cholinergic-dopaminergic reward link”. This link includes a cholinergic afferent projection from the LDTg onto the VTA dopamine cells. We found that central, intra-VTA or intra-LDTg administration of ghrelin increases accumbal dopamine release and locomotor activity, effects abolished by nicotinic cholinergic receptor blockade [112, 113]. Consistent with this, GHS-R1A has been shown to be co-localised both in dopamine (tyrosine-hydroxylase)-containing cells in the VTA [114] and with cholinergic (choline acetyl transferase)-containing cells in the LDTg [115]. In addition to these cholinergic afferents, we also recently found that pharmacological suppression of glutamatergic signalling suppresses ghrelin’s effects on the mesoaccumbal dopamine system [116]. These findings emerged as having direct relevance for reward from addictive drugs [117, 118] as well as from palatable food [119, 120]. Intracerebroventricular injection of ghrelin has been shown to stimulate food intake [121], especially the intake of palatable food [119]. Ghrelin signalling at the level of the VTA appears to be important for these feeding effects as intra-VTA injection of ghrelin increases the intake of palatable food [119]. Moreover, the effects of peripheral ghrelin on food intake were blunted by intra-VTA administration of a GHS-R1A antagonist [114]. More specifically, the cholinergic-dopaminergic reward link is implicated in ghrelin-induced feeding; nicotinic blockade suppressed ghrelin-induced and fasting-induced feeding and also suppresses the ability of food to condition a place preference [115]. Consistent with this, peripheral treatment with a GHS-R1A antagonist decreased preference for palatable food, suppressed the ability of sweet treats to condition a place preference [119] and suppressed motivated behaviour for rewarding foods, both sweet [122] (Fig. 2) and high fat [120] foods. Collectively these data support the idea that the physiological role of ghrelin is to increase the incentive motivation for natural rewards such as food.

Central ghrelin signalling is required for the incentive motivation (“wanting”) for sweet rewards in rats. Operant responding (lever-pressing) for a sucrose reward is a increased by peripheral administration of ghrelin to satiated rats and b decreased by peripheral administration of a GHS-R1A (ghrelin receptor) antagonist to hungry rats. Reproduced with permission from Addiction Biology (122)
There are indications that these studies in rodents are relevant in man as systemic ghrelin administration has been shown to alter the brain response to visual food cues in relevant reward targets area, including the striatum [123]. Although common obesity is associated with a reduction in circulating ghrelin levels [124], food intake appears to be less effective in suppressing ghrelin levels in obese subjects [125]. The relevance of the peripheral ghrelin signal in common obesity could also be questioned as brain ghrelin production may be increased in obese subjects [126] and, moreover, the ghrelin receptor may not require ghrelin for activity as it is possesses a high level of constitutive activity [127]. It remains to be determined whether the central ghrelin signalling system has a role in the pathophysiology of obesity and whether ghrelin antagonists or inverse agonists will provide an effective future therapy, either alone or in combination with drugs/hormones that interfere with overlapping signalling mechanisms.
6 Conclusions
From a neurobiological perspective, it is clear that the decision to eat is very complex, involving genetic, environmental, psychosocial and physiological processes. To understand these processes is to regain control of the obesity epidemic and develop a better relationship with food in our modern obesogenic lifestyle. The reward system remains a key target for the development of future therapies, especially those that alter the rewarding value of food. In this context, the central ghrelin signalling system emerges as a novel and interesting therapeutic target as studies in rodents have shown that ghrelin antagonists suppress the mesocummbal dopamine system, suppress the intake of (and preference for) palatable food, suppress the ability of rewarding foods to condition a place preference and decrease operant responding for rewarding foods. Indeed, such compounds may form part of a future combination therapy that attempts to mimic the altered gut-brain signalling observed in patients that have undergone gastric bypass surgery.
References
Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu Rev Psychol. 2008;59:55–92.
Yeomans MR, Blundell JE, Leshem M. Palatability: response to nutritional need or need-free stimulation of appetite? Br J Nutr. 2004;92:S3–S14.
Zheng HY, Berthoud HR. Neural systems controlling the drive to eat: mind versus metabolism. Physiology. 2008;23(2):75–83.
Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Rev. 1998;28(3):309–69.
Mela DJ. Eating for pleasure or just wanting to eat? Reconsidering sensory hedonic responses as a driver of obesity. Appetite. 2006;47(1):10–7.
Berridge KC. Food reward: brain substrates of wanting and liking. Neurosci Biobehav Rev. 1996;20(1):1–25.
Finlayson G, King N, Blundell JE. Is it possible to dissociate ‘liking’ and ‘wanting’ for foods in humans? A novel experimental procedure. Physiol Behav. 2007;90(1):36–42.
Avena NM, Rada P, Hoebel BG. Evidence for sugar addiction: behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev. 2008;32(1):20–39.
Benton D. The plausibility of sugar addiction and its role in obesity and eating disorders. Clin Nutr. 2010;29(3):288–303.
Berthoud HR. Interactions between the “cognitive” and “metabolic” brain in the control of food intake. Physiol Behav. 2007;91(5):486–98.
Stice E, Spoor S, Ng J, Zald DH. Relation of obesity to consummatory and anticipatory food reward. Physiol Behav. 2009;97(5):551–60.
Vucetic Z, Reyes TM. Central dopaminergic circuitry controlling food intake and reward: implications for the regulation of obesity. Wiley Interdiscip Rev Syst Biol Med. 2010;2:577–93.
Berridge KC. The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology. 2007;191(3):391–431.
Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94(4):469–92.
Hajnal A, Smith GP, Norgren R. Oral sucrose stimulation increases accumbens dopamine in the rat. Am J Physiol Regul Integr Comp Physiol. 2004;286(1):R31–7.
Rada P, Avena NM, Hoebel BG. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience. 2005;134(3):737–44.
Liang NC, Hajnal A, Norgren R. Sham feeding corn oil increases accumbens dopamine in the rat. Am J Physiol Regul Integr Comp Physiol. 2006;291(5):R1236–9.
Reilly S. Reinforcement value of gustatory stimuli determined by progressive ratio performance. Pharmacol Biochem Behav. 1999;63(2):301–11.
Sclafani A, Ackroff K. Reinforcement value of sucrose measured by progressive ratio operant licking in the rat. Physiol Behav. 2003;79(4–5):663–70.
Wojnicki FHE, Babbs RK, Corwin RLW. Reinforcing efficacy of fat, as assessed by progressive ratio responding, depends upon availability not amount consumed. Physiol Behav. 2010;100(4):316–21.
Yoneda T, Taka Y, Okamura M, Mizushige T, Matsumura S, Manabe Y, et al. Reinforcing effect for corn oil stimulus was concentration dependent in an operant task in mice. Life Sci. 2007;81(23–24):1585–92.
Baldo BA, Kelley AE. Discrete neurochemical coding of distinguishable motivational processes: insights from nucleus accumbens control of feeding. Psychopharmacology. 2007;191(3):439–59.
Salamone JD, Cousins MS, Snyder BJ. Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev. 1997;21(3):341–59.
Berridge KC, Venier IL, Robinson TE. Taste reactivity analysis of 6-hydroxydopamine-induced aphagia—implications for arousal and anhedonia hypotheses of dopamine function. Behav Neurosci. 1989;103(1):36–45.
Cagniard B, Balsam PD, Brunner D, Zhuang XX. Mice with chronically elevated dopamine exhibit enhanced motivation, but not learning, for a food reward. Neuropsychopharmacol. 2006;31(7):1362–70.
Pecina S, Cagniard B, Berridge KC, Aldridge JW, Zhuang XX. Hyperdopaminergic mutant mice have higher “wanting” but not “liking” for sweet rewards. J Neurosci. 2003;23(28):9395–402.
de Araujo IE, Oliveira-Maia AJ, Sotnikova TD, Gainetdinov RR, Caron MG, Nicolelis MAL, et al. Food reward in the absence of taste receptor signaling. Neuron. 2008;57(6):930–41.
Grill HJ, Norgren R. Taste reactivity test.2. Mimetic responses to gustatory stimuli in chronic thalamic and chronic decerebrate rats. Brain Res. 1978;143(2):281–97.
Pecina S, Berridge KC. Opioid site in nucleus accumbens shell mediates eating and hedonic ‘liking’ for food: map based on microinjection Fos plumes. Brain Res. 2000;863(1–2):71–86.
Berridge KC, Robinson TE. Parsing reward. Trends Neurosci. 2003;26(9):507–13.
Zhang M, Kelley AE. Intake of saccharin, salt, and ethanol solutions is increased by infusion of a mu opioid agonist into the nucleus accumbens. Psychopharmacology. 2002;159(4):415–23.
Zhang M, Gosnell BA, Kelley AE. Intake of high-fat food is selectively enhanced by mu opioid receptor stimulation within the nucleus accumbens. J Pharmacol Exp Ther. 1998;285(2):908–14.
Pecina S, Berridge KC. Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J Neurosci. 2005;25(50):11777–86.
Shin AC, Pistell PJ, Phifer CB, Berthoud HR. Reversible suppression of food reward behavior by chronic mu-opioid receptor antagonism in the nucleus accumbens. Neuroscience. 2010;170:580–8.
Zhang M, Balmadrid C, Kelley AE. Nucleus accumbens opioid, GABAergic, and dopaminergic modulation of palatable food motivation: contrasting effects revealed by a progressive ratio study in the rat. Behav Neurosci. 2003;117(2):202–11.
Jamshidi N, Taylor DA. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol. 2001;134(6):1151–4.
Verty ANA, McGregor IS, Mallet PE. Paraventricular hypothalamic CB1 cannabinoid receptors are involved in the feeding stimulatory effects of Delta(9)-tetrahydrocannabinol. Neuropharmacology. 2005;49(8):1101–9.
DiPatrizio NV, Simansky KJ. Activating parabrachial cannabinoid CB1 receptors selectively stimulates feeding of palatable foods in rats. J Neurosci. 2008;28(39):9702–9.
Soria-Gomez E, Matias I, Rueda-Orozco PE, Cisneros M, Petrosino S, Navarro L, et al. Pharmacological enhancement of the endocannabinoid system in the nucleus accumbens shell stimulates food intake and increases c-Fos expression in the hypothalamus. Br J Pharmacol. 2007;151(7):1109–16.
Perio A, Barnouin MC, Poncelet M, Soubrie P. Activity of SR141716 on post-reinforcement pauses in operant responding for sucrose reward in rats. Behav Pharmacol. 2001;12(8):641–5.
Kirkham TC, Williams CM. Synergistic effects of opioid and cannabinoid antagonists on food intake. Psychopharmacology. 2001;153(2):267–70.
Verty ANA, McGregor IS, Mallet PE. The doparnine receptor antagonist SCH 23390 attenuates feeding induced by Delta(9)-tetrahydrocannobinol. Brain Res. 2004;1020(1–2):188–95.
Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437(7058):556–9.
Cason AM, Smith RJ, Tahsili-Fahadan P, Moorman DE, Sartor GC, Aston-Jones G. Role of orexin/hypocretin in reward-seeking and addiction: Implications for obesity. Physiol Behav. 2010;100(5):419–28.
Crespo I, de Heras RG, de Fonseca FR, Navarro M. Pretreatment with subeffective doses of Rimonabant attenuates orexigenic actions of orexin A-hypocretin 1. Neuropharmacology. 2008;54(1):219–25.
Ramirez I, Friedman MI. Dietary hyperphagia in rats—role of fat, carbohydrate, and energy content. Physiol Behav. 1990;47(6):1157–63.
Sclafani A. Oral and postoral determinants of food reward. Physiol Behav. 2004;81(5):773–9.
Finlayson G, Caudwell P, Gibbons C, Hopkins M, King N, Blundell J. Low fat loss response after medium-term supervised exercise in obese is associated with exercise-induced increase in food reward. J Obes. (Epub 2010 Sep 20). 2011.
Davis C, Strachan S, Berkson M. Sensitivity to reward: implications for overeating and overweight. Appetite. 2004;42(2):131–8.
Rissanen A, Hakala P, Lissner L, Mattlar CE, Koskenvuo M, Ronnemaa T. Acquired preference especially for dietary fat and obesity: a study of weight-discordant monozygotic twin pairs. Int J Obes. 2002;26(7):973–7.
Saelens BE, Epstein LH. Reinforcing value of food in obese and non-obese women. Appetite. 1996;27(1):41–50.
Comings DE, Blum K. Reward deficiency syndrome: genetic aspects of behavioral disorders. Prog Brain Res. 2000;126:325–41.
Carroll ME, Lac ST. Dietary additives and the acquisition of cocaine self-administration in rats. Psychopharmacology. 1998;137(1):81–9.
Wellman PJ, Nation JR, Davis KW. Impairment of acquisition of cocaine self-administration in rats maintained on a high-fat diet. Pharmacol Biochem Behav. 2007;88(1):89–93.
la Fleur SE, Vanderschuren LJMJ, Luijendijk MC, Kloeze BM, Tiesjema B, Adan RAH. A reciprocal interaction between food-motivated behavior and diet-induced obesity. Int J Obes. 2007;31(8):1286–94.
Pickering C, Alsio J, Hulting AL, Schioth HB. Withdrawal from free-choice high-fat high-sugar diet induces craving only in obesity-prone animals. Psychopharmacology. 2009;204(3):431–43.
Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13(5):635–41.
Alsiö J, Olszewski PK, Norbäck AH, Gunnarsson ZEA, Levine AS, Pickering C, Schiöth HB. Dopamine D1 receptor gene expression decreases in the nucleus accumbens upon long-term exposure to palatable food and differs depending on diet-induced obesity phenotype in rats. Neuroscience. 2010;171(3):779–87.
Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG, Pothos EN. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159(4):1193–9.
Davis JF, Tracy AL, Schurdak JD, Tschop MH, Lipton JW, Clegg DJ, et al. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav Neurosci. 2008;122(6):1257–63.
Liu YJ, Gao JH, Liu HL, Fox PT. The temporal response of the brain after eating revealed by functional MRI. Nature. 2000;405(6790):1058–62.
Morris JS, Dolan RJ. Involvement of human amygdala and orbitofrontal cortex in hunger-enhanced memory for food stimuli. J Neurosci. 2001;21(14):5304–10.
Passamonti L, Rowe JB, Schwarzbauer C, Ewbank MP, von dem Hagen E, Calder AJ. Personality predicts the brain’s response to viewing appetizing foods: the neural basis of a risk factor for overeating. J Neurosci. 2009;29(1):43–51.
Rothemund Y, Preuschhof C, Bohner G, Bauknecht HC, Klingebiel R, Flor H, et al. Differential activation of the dorsal striatum by high-calorie visual food stimuli in obese individuals. Neuroimage. 2007;37(2):410–21.
Stoeckel LE, Weller RE, Cook EW, Twieg DB, Knowlton RC, Cox JE. Widespread reward-system activation in obese women in response to pictures of high-calorie foods. Neuroimage. 2008;41(2):636–47.
Stice E, Yokum S, Bohon C, Marti N, Smolen A. Reward circuitry responsivity to food predicts future increases in body mass: moderating effects of DRD2 and DRD4. Neuroimage. 2010;50(4):1618–25.
Stice E, Spoor S, Bohon C, Small DM. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008;322(5900):449–52.
Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, et al. Brain dopamine and obesity. Lancet. 2001;357(9253):354–7.
Volkow ND, Wang GJ, Telang F, Fowler JS, Thanos PK, Logan J, et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. Neuroimage. 2008;42(4):1537–43.
Epstein LH, Jaroni JL, Paluch RA, Leddy JJ, Vahue HE, Hawk L, et al. Dopamine transporter genotype as a risk factor for obesity in African–American smokers. Obes Res. 2002;10(12):1232–40.
Annerbrink K, Westberg L, Nilsson S, Rosmond R, Holm G, Eriksson E. Catechol O-methyltransferase val158-met polymorphism is associated with abdominal obesity and blood pressure in men. Metabolism. 2008;57(5):708–11.
Comings DE, Flanagan SD, Dietz G, Muhleman D, Knell E, Gysin R. The dopamine-D(2) receptor (Drd2) as a major gene in obesity and height. Biochem Med Metabol Biol. 1993;50(2):176–85.
Hajnal A, Margas WM, Covasa M. Altered dopamine D2 receptor function and binding in obese OLETF rat. Brain Res Bull. 2008;75(1):70–6.
Olbers T, Bjorkman S, Lindroos A, Maleckas A, Lonn L, Sjostrom L, et al. Body composition, dietary intake, and energy expenditure after laparoscopic Roux-en-y gastric bypass and laparoscopic vertical banded gastroplasty—A randomized clinical trial. Ann Surg. 2006;244(5):715–22.
Zheng HY, Shin AC, Townsend RL, Patterson LM, Sigalet DL, Berthoud HR. Changes in fat preference, meal patterns, gastrointestinal transit, energy expenditure, and circulating hormones in a rat model of Roux-en-Y gastric bypass surgery. Obesity. 2009;17:S103–S.
Shin AC, Zheng H, Pistell PJ, Berthoud HR. Roux-en-Y gastric bypass surgery changes food reward in rats. Int J Obes (Lond). 2010. doi:10.1038/ijo.2010.174.
Cabanac M, Duclaux R. Specificity of internal signals in producing satiety for taste stimuli. Nature. 1970;227(5261):966–7.
De Jonghe BC, Hajnal A, Covasa M. Conditioned preference for sweet stimuli in OLETF rat: effects of food deprivation. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1819–27.
Figlewicz DP, Bennett JL, Naleid AM, Davis C, Grimm JW. Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol Behav. 2006;89(4):611–6.
Figlewicz DP, Naleid AM, Sipols AJ. Modulation of food reward by adiposity signals. Physiol Behav. 2007;91(5):473–8.
Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. Am J Physiol Regul Integr Comp Physiol. 2009;296(1):R9–R19.
Berridge KC. Modulation of taste affect by hunger, caloric satiety, and sensory-specific satiety in the rat. Appetite. 1991;16(2):103–20.
Small DM, Zatorre RJ, Dagher A, Evans AC, Jones-Gotman M. Changes in brain activity related to eating chocolate—From pleasure to aversion. Brain. 2001;124:1720–33.
Kelley AE, Berridge KC. The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci. 2002;22(9):3306–11.
Cornell CE, Rodin J, Weingarten H. Stimulus-induced eating when satiated. Physiol Behav. 1989;45(4):695–704.
Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. 2007;317(5843):1355.
Zheng H, Lenard NR, Shin AC, Berthoud HR. Appetite control and energy balance regulation in the modern world: reward-driven brain overrides repletion signals. Int J Obes. 2009;33:S8–S13.
Shin AC, Zheng HY, Berthoud HR. An expanded view of energy homeostasis: neural integration of metabolic, cognitive, and emotional drives to eat. Physiol Behav. 2009;97(5):572–80.
Fulton S. Appetite and reward. Front Neuroendocrinol. 2010;31(1):85–103.
Figlewicz DP. Adiposity signals and food reward: expanding the CNS roles of insulin and leptin. Am J Physiol Regul Integr Comp Physiol. 2003;284(4):R882–92.
Opland DM, Leinninger GM, Myers MG. Modulation of the mesolimbic dopamine system by leptin. Brain Res. 2010;1350:65–70.
Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ. Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav. 2001;73(1–2):229–34.
Figlewicz DP, Bennett J, Evans SB, Kaiyala K, Sipols AJ, Benoit SC. Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behav Neurosci. 2004;118(3):479–87.
Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003;964(1):107–15.
Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51(6):801–10.
Krugel U, Schraft T, Kittner H, Kiess W, Illes P. Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin. Eur J Pharmacol. 2003;482(1–3):185–7.
Leshan RL, Opland DM, Louis GW, Leinninger GM, Patterson CM, Rhodes CJ, et al. Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci. 2010;30(16):5713–23.
Louis GW, Leinninger GM, Rhodes CJ, Myers MG. Direct innervation and modulation of orexin neurons by lateral hypothalamic LepRb neurons. J Neurosci. 2010;30(34):11278–87.
Adami GF, Campostano A, Cella F, Scopinaro N. Serum leptin concentration in obese patients with binge eating disorder. Int J Obes. 2002;26(8):1125–8.
Morton GJ, Gelling RW, Niswender KD, Morrison CD, Kim F, Schwartz MW. Leptin signaling in the hypothalamic arcuate nucleus is a key determinant of glucose tolerance and hepatic insulin action. Obes Res. 2005;13(1):201.
Chelikani PK, Haver AC, Reeve JR, Keire DA, Reidelberger RD. Daily, intermittent intravenous infusion of peptide YY(3-36) reduces daily food intake and adiposity in rats. Am J Physiol Regul Integr Comp Physiol. 2006;290(2):R298–305.
Batterham RL, ffytche DH, Rosenthal JM, Zelaya FO, Barker GJ, Withers DJ, et al. PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature. 2007;450(7166):106–9.
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50(8):1714–9.
Cummings DE, Frayo RS, Marmonier C, Aubert R, Chapelot D. Plasma ghrelin levels and hunger scores in humans initiating meals voluntarily without time- and food-related cues. Am J Physiol Endocrinol Metab. 2004;287(2):E297–304.
Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50(11):2540–7.
Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology. 2000;141(11):4325–8.
Hewson AK, Dickson SL. Systemic administration of ghrelin induces Fos and Egr-1 proteins in the hypothalamic arcuate nucleus of fasted and fed rats. J Neuroendocrinol. 2000;12(11):1047–9.
Dickson SL, Leng G, Robinson ICAF. Systemic administration of growth hormone-releasing peptide activates hypothalamic arcuate neurons. Neuroscience. 1993;53(2):303–6.
Dickson SL, Luckman SM. Induction of c-fos messenger ribonucleic acid in neuropeptide Y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection of the GH secretagogue, GH-releasing peptide-6. Endocrinology. 1997;138(2):771–7.
Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJS, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Mol Brain Res. 1997;48(1):23–9.
Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494(3):528–48.
Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol. 2007;12(1):6–16.
Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA. Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol. 2006;11(1):45–54.
Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116(12):3229–39.
Dickson SL, Hrabovszky E, Hansson C, Jerlhag E, Alvarez-Crespo M, Skibicka KP, et al. Blockade of central nicotine acetylcholine receptor signaling attenuate ghrelin-induced food intake in rodents. Neuroscience. 2010.
Jerlhag E, Egecioglu E, Dickson SL, Engel JA. Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict Biol. 2011;16(1):82–91.
Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechard D, et al. Requirement of central ghrelin signaling for alcohol reward. PNAS. 2009;106(27):11318–23.
Jerlhag E, Egecioglu E, Dickson SL, Engel JA. Ghrelin receptor antagonism attenuates cocaine- and amphetamine-induced locomotor stimulation, accumbal dopamine release and conditioned place preference. Psychopharmacology (Berl). 2010;211(4):415–22.
Egecioglu E, Jerlhag E, Skibicka S, Salomé N, Haage D, Bohlooly-YM, et al. Ghrelin increases intake of palatable food in rodents. Addict Biol. 2010;15(3):304–11.
Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, et al. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry. 2010;67(9):880–6.
Naleid AM, Grace MK, Cummings DE, Levine AS. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides. 2005;26(11):2274–9.
Skibicka KP, Hansson C, Egecioglu E, Dickson SL. Role of ghrelin in food reward: impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict Biol. 2010. doi:10.1111/j.1369-1600.2010.00294.x.
Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008;7(5):400–9.
Tschöp M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating Ghrelin levels are decreased in human obesity. Diabetes. 2001;50(4):707–9.
English PJ, Ghatei MA, Malik IA, Bloom SR, Wilding JPH. Food fails to suppress ghrelin levels in obese humans. J Clin Endocrinol Metab. 2002;87(6):2984–7.
Couce ME, Cottam D, Esplen J, Teijeiro R, Schauer P, Burguera B. Potential role of hypothalamic ghrelin in the pathogenesis of human obesity. J Endocrinol Invest. 2006;29(7):599–605.
Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor—Identification of a potent inverse agonist. Mol Endocrinol. 2003;17(11):2201–10.
Acknowledgements
The Swedish Research Council for Medicine (2009-S266, K2006-21X-04247-33-3 and 2009-2782), European Commission 7th Framework (FP7-HEALTH-2009-241592; FP7-KBBE-2009-3-245009 and FP7-KBBE-2010-4-266408), FOU/ALF Göteborg (ALFGBG-138741, ALFGBG-7341), The Swedish Brain Foundation, Torsten and Ragnar Söderberg foundation and the Swedish Foundation for Strategic Research to Sahlgrenska Center for Cardiovascular and Metabolic Research (A305-188). Dr Johan Alsiö and Prof Roger Adan for helpful discussion regarding section IV.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Egecioglu, E., Skibicka, K.P., Hansson, C. et al. Hedonic and incentive signals for body weight control. Rev Endocr Metab Disord 12, 141–151 (2011). https://doi.org/10.1007/s11154-011-9166-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-011-9166-4