
Abstract
Application of nano-scale supported Au in catalytic hydrogenation delivers high chemoselectivity but low activity using (pressurized) H2 far in excess of stoichiometric requirements. We have tackled the issues of low reaction rate and inefficient hydrogen utilization through a series of approaches taking Au/CeO2 (mean Au size = 2.8 nm) as a test catalyst. Increased spillover hydrogen (using physical mixtures of Au/CeO2 with CeO2 and SiO2) and the promotional effect of water (via catalytic dissociation on surface oxygen vacancies) resulted in a fourfold increase in the selective rate of furfural hydrogenation to furfuryl alcohol. In contrast, Pd/CeO2 and Ni/CeO2 promoted decarbonylation (to furan), hydrogenolysis (to 2-methylfuran) and ring reduction (to tetrahydrofurfuryl alcohol). Coupling (2-butanol → 2-butanone) dehydrogenation over Cu/SiO2 (mean Cu size = 7.8 nm) with furfural hydrogenation over Au/CeO2 further increased rate with full utilization of the hydrogen generated in dehydrogenation. The coupling strategy allows “hydrogen free” hydrogenation that circumvents the limitation of Au in standard catalytic hydrogenation. This can open new avenues to exploit the ultra-selectivity of Au for continuous production of high value commodity products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The application of supported Au catalysts has focused on selective oxidation and environmental remediation in the conversion of volatile organics [1]. By comparison, use in hydrogenation, a key process across the chemical sector [2] is more limited [3]. Interest in Au catalysts for hydrogenation has been driven by the high chemoselectivity achieved in the reduction of polyfunctional molecules [4] but low reaction rate is a decided drawback [5]. Commercial hydrogenation processes are typically operated in excess (pressurized) H2 in order to maximize product yield. Hydrogen is not a naturally occurring feedstock and production is fossil fuel based (by methane steam reforming and coal gasification [6]) where storage and transport represent serious constraints. A move away from reaction using compressed H2 is a critical driver to achieve safer operations for large scale production. The use of a hydrogen carrier in transfer hydrogenation is a promising alternative that has been explored in organic synthesis using homogeneous [7] and heterogeneous catalysis [8, 9] in batch mode but it introduces additional catalyst/product separation and purification steps. Catalytic dehydrogenation of alcohols offers a possible in situ source of reactive hydrogen [10] as demonstrated by Nagaraja et al. [11] in coupling cyclohexanol dehydrogenation with furfural hydrogenation on Cr2O3 promoted Cu–MgO. However, the similarity (10 K difference) in the boiling points of the products necessitated difficult separation/purification while hydrogen utilization was poor (< 10%). 2-Butanone (used as solvent and in the production of plastics, coatings and films [12]) can be obtained from 2-butanol dehydrogenation with hydrogen release.
With the overarching goal of process sustainability, we have examined the hydrogenation of furfural as a biomass (corncob and sugar cane bagasse) derived heterocyclic aldehyde [13]. Selective hydrogenation generates furfuryl alcohol, a high value chemical (130,000 tons per annum global production) used in the manufacture of resins/rubbers/adhesives and as a chemical building block for drug synthesis [14]. Gold on ceria has been shown to promote selective –C=O hydrogenation [15] and is used as a model catalyst in this work. Supported Pd and Ni with higher H2 dissociative chemisorption capacity than Au [16] and enhanced activity in the hydrogenation of carbonyl compounds [17] were also tested to assess the performance of Au/CeO2. Taking full H2 utilization and increased selective hydrogenation rate as our ultimate objectives, we evaluate four approaches to increase the available surface reactive hydrogen: (i) modification of external H2 partial pressure; (ii) enhanced spillover hydrogen associated with the support; (iii) hydrogen donation via catalytic water dissociation; (iv) coupling dehydrogenation with hydrogenation.
Experimental
Catalyst preparation
Ceria (Sigma-Aldrich, 99%) supported Au (0.7 mol%), Pd (0.8 mol%), Ni (0.5 mol%) and Cu/SiO2 (15.2 mol%) were prepared by deposition–precipitation. In the synthesis of Au/CeO2 an aqueous solution of urea (100-fold excess, Riedel-de Haën, 99%) with HAuCl4 (2 × 10−3 mol dm−3, 400 cm3, Sigma-Aldrich, 99%) was mixed with the ceria support (10 g). The suspension was stirred (600 rpm) and heated (2 K min−1) to 353 K in air where the pH progressively increased (to ca. 7) after 3 h as a result of urea decomposition. The solid was separated by filtration and washed with distilled water until Cl free (based on AgNO3 test). In the synthesis of Pd/CeO2 and Ni/CeO2, an aqueous solution (2 × 10−3 mol dm−3, 300 cm3) of the metal precursor (Pd(NO3)2 or Ni(NO3)2, Sigma-Aldrich, 99%) was added to CeO2 (10 g), Na2CO3 (2 mol dm−3, Riedel-de Haën, 99%) slowly added to the resultant suspension until pH > 10, heating (2 K min−1) to 353 K for 4 h resulting in deposition of Pd(OH)2 and NiCO3 [18]. The filtered solid was washed with distilled water and dried in a vacuum oven at 333 K for 12 h. Silica supported Cu was prepared with NaOH (Riedel-de Haën, 99%) as precipitation agent where 20 g (fumed) silica powder (Sigma-Aldrich, 99%) were dispersed in a solution of Cu(NO3)2 (25 × 10−2 mol dm−3, 200 cm3, Sigma-Aldrich, 99%). The suspension was stirred (600 rpm) at room temperature for 1 h with the addition of aqueous NaOH (2 M) until pH > 10 and subsequent heating to 353 K maintained for 4 h to ensure homogeneous Cu(OH)2 deposition [19]. The solid was separated by filtration, washed with distilled water until pH 7 and dried at 393 K overnight. The dried sample was calcined (in air) at 10 K min−1 to 723 K for 4 h to generate copper oxide (CuO) [19]. All the samples were sieved (ATM fine test sieves) to mean particle diameter = 75 μm. The samples were activated in 60 cm3 min−1 H2 to 523–723 K (at 2–10 K min−1) with a final isothermal hold for 1–2 h and passivated at ambient temperature in 1% v/v O2/He for ex situ characterization.
Catalyst characterization
Metal content was measured by atomic absorption spectroscopy using a Shimadzu AA-6650 spectrometer with an air-acetylene flame from the diluted extract in aqua regia (25% v/v HNO3/HCl). Temperature programmed reduction (TPR), H2 chemisorption (at 498 K) and subsequent temperature programmed desorption (TPD) were conducted on the CHEM-BET 3000 (Quantachrome) unit equipped with a thermal conductivity detector (TCD) for continuous monitoring of gas composition and the TPR Win™ software for data acquisition/manipulation. Samples were loaded into a U-shaped Pyrex glass cell (3.76 mm i.d.) and reduced in 17 cm3 min−1 5% v/v H2/N2 to 523–723 K (at 2–10 K min−1) where the effluent gas passed through a liquid N2 trap. The activated samples were swept with 65 cm3 min−1 N2 for 1.5 h, cooled to 498 K and subjected to H2 chemisorption (BOC, 99.99%) using pulse (10–50 µl) titration. The hydrogen pulses were repeated until the signal area was constant, indicating surface saturation. In blank tests, there was no measurable H2 uptake on the supports. Samples were thoroughly flushed in N2 (65 cm3 min−1) to remove weakly bound hydrogen and subjected to TPD (at 50 K min−1) to 973 K with a final isothermal hold until the signal returned to baseline. Scanning transmission electron microscopy (STEM) was performed using a JEOL 2200FS field emission gun-equipped transmission electron microscope unit, employing Gatan Digital Micrograph 1.82 for data acquisition/manipulation. Samples for analysis were prepared by dispersion in acetone and deposited on a holey carbon/Cu grid (300 Mesh). Surface area weighted mean metal particle size (d) was determined from a count of up to 600 particles according to:
Here ni is the number of particles of diameter di.
Catalytic procedure
Reactions (independent hydrogenation of furfural (in H2) and coupled dehydrogenation/hydrogenation of 2-butanol/furfural) were carried out at atmospheric pressure and 498 K in situ after activation in a continuous flow fixed-bed tubular reactor (i.d. = 15 mm). Reaction conditions ensured negligible internal/external mass and heat transfer limitations. A layer of borosilicate glass beads served as preheating zone where the reactant was vaporized and reached reaction temperature before contacting the catalyst bed. Isothermal conditions (±1 K) were maintained by diluting the catalyst with ground glass (75 µm); reaction temperature was continuously monitored by a thermocouple inserted in a thermowell within the catalyst bed. The reactant(s) was(were) delivered to the reactor via a glass/Teflon air-tight syringe and Teflon line using a microprocessor controlled infusion pump (Model 100 kd Scientific). The independent hydrogenation of furfural was tested in a co-current flow of H2 with furfural (GHSV = 5 × 103 h−1 and molar metal to furfural feed rate (n/F) = 5 × 10−3 h). Coupled dehydrogenation/hydrogenation was conducted in N2 (GHSV = 3 × 103 h−1, nAu/F = 5 × 10−3–11 × 10−3 h, molar Cu/Au = 65–150). The reactor effluent was analyzed by capillary GC (Perkin-Elmer Auto System XL gas chromatograph equipped with a programmed split/splitless injector and a flame ionization detector, employing a DB-1 (50 m × 0.33 mm i.d., 0.20 μm film thickness) capillary column (J&W Scientific)). In control experiments, passage of each reactant in a stream of H2 or N2 through the empty reactor or over the support alone did not result in any detectable conversion. Reactant (i) fractional conversion (X) is defined by:
The selectivity (S) to product (j) is given by:
Here the subscripts “in” and “out” refer to the inlet and outlet gas streams, respectively. Repeated reactions using different samples from the same batch of catalyst delivered raw data reproducibility and mass balance within ± 5%. Catalytic activity is quantified in terms of initial conversion/hydrogenation rate obtained from time on-stream measurements [20].
Results and discussion
The TPR of Au/CeO2 (Fig. 1aI) exhibits a single positive peak (H2 consumption, Tmax = 418 K), which can be ascribed to Au3+→ Au0 reduction and is in line with published data (Tmax = 375–500 K) for ceria supported Au involving a range of syntheses [21]. Hydrogen consumption during TPR \(\left( { 1 2\;{\text{mol}}_{{{\text{H}}_{ 2} }} \;{\text{mol}}_{\text{Au}}^{ - 1} } \right)\) was eight times greater than that required for reduction to metallic gold, indicative of partial support reduction with the formation of surface oxygen vacancies [22]. STEM analysis (Fig. 1bI) revealed pseudo-spherical metal particles at the nano-scale (1–5 nm, Fig. 1cI) with an associated surface area weighted mean of 2.8 nm. It has been noted elsewhere [23] that Au particle size < 10 nm is critical for significant hydrogenation activity. Hydrogen chemisorption on Au/CeO2 was appreciably lower than that measured under reaction conditions for the benchmark Pd/CeO2 and Ni/CeO2 (Table 1). This is due to the filled 3d band in Au that results in a high energy barrier for dissociative adsorption and low H–Au binding strength [24]. Given that H2 activation is a limiting factor, we explored the effect of increasing inlet H2/furfural on reaction rate (Fig. 2). In every case, reaction over Au/CeO2 was fully selective to the target furfuryl alcohol, an important result in the light of reported batch liquid phase furfural hydrogenation over Au/SiO2 where conversion was negligible even at high H2 pressure (1.0 MPa) [25]. We observed no detectable catalytic activity at the reaction stoichiometry (H2/furfural = 1) but rate increased to reach an upper value (26 molfurfural mol −1Au h−1) at H2/furfural > 60 (Fig. 2). Hydrogen utilization efficiency (represented by inlet H2 to outlet furfuryl alcohol ratio) was enhanced with increasing inlet H2/furfural (to 45, Fig. 2). The restricted capacity of Au for H2 adsorption/dissociation resulted in an upper limit in terms of rate with measurably lower efficiencies at higher H2/furfural (> 45). Hydrogen utilization is a critical consideration in terms of process sustainability where the best result (H2/furfuryl alcohol = 420) is far removed from the target stoichiometry (= 1). We evaluated Pd/CeO2 and Ni/CeO2 to assess the catalytic performance of Au/CeO2 and the results are presented in Table 1. There was no detectable furfuryl alcohol formation over either catalyst. Pd/CeO2 with the greatest H2 uptake capacity (Table 1) delivered the highest rate but promoted secondary decarbonylation to furan, hydrogenolysis of furfuryl alcohol to 2-methylfuran and ring reduction to tetrahydrofurfuryl alcohol (Fig. 3). Furan was the main product formed over Ni/CeO2. Sitthisa and Resasco [26] recorded decarbonylation (to furan) over Pd/SiO2 and ring opening (to butanal, butanol and butane) over Ni/SiO2 in the gas phase hydrogenation of furfural. Our results demonstrate a dependence of product distribution on the catalytic metal where Au facilitates exclusive reduction of the carbonyl group to the target alcohol.

a TPR profile, b representative STEM image (arrows identify Au nanoparticles) with c associated metal particle size distribution histogram for I Au/CeO2 and II Cu/SiO2

Furfural hydrogenation rate (R) and H2 utilization (H2/furfuryl alcohol) as a function of inlet H2/furfural over Au/CeO2. Reaction conditions T = 498 K, P = 1 atm, nAu/F = 5 × 10−3 h

Reaction pathways for the hydrogenation of furfural over CeO2 supported Au (solid arrow) and Pd and Ni (open arrows)
Hydrogen dissociated on metal sites “spills” across the metal/support interface onto the support surface and even across solid/solid grain boundaries (e.g. catalyst + support physical mixtures) [27]. This spillover hydrogen contributes to hydrogenation activity [28]. Spillover has been reported for supported Au catalysts following hydrogen treatment at 423–673 K and can be quantified by TPD [29]. The TPD profile for Au/CeO2 (Fig. 4) is characterized by H2 release (180 μmol g−1, Tmax = 788 K) that far exceeded H2 chemisorption (Table 1) and can be attributed to spillover desorption [28]. The incorporation of CeO2 or SiO2 in a physical mixture with Au/CeO2 resulted in measurably greater H2 desorption (Fig. 4). A consequent increase in furfural hydrogenation rate (from 26 to 31 molfurfural mol −1Au h−1) was obtained while retaining full selectivity to furfuryl alcohol and confirms contribution from spillover hydrogen to increase selective hydrogenation rate.

Furfural hydrogenation rate (R) and H2 TPD profiles (as insets) for Au/CeO2 and physical mixtures of Au/CeO2 with CeO2 and SiO2. Reaction conditions T = 498 K, P = 1 atm, nAu/F = 5 × 10−3 h, H2/furfural = 66
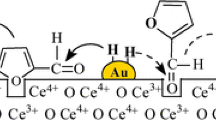
Water is an environmentally benign solvent that can act as hydrogen donor in water gas shift and steam reforming over (CeO2 and Fe2O3) supported Au [30, 31]. The effect of incorporating water in the furfural feed was tested for reaction over Au/CeO2 (Fig. 5) where a threefold higher rate was achieved at H2O/furfural = 0.2–1. We envision a surface mechanism (schematic in Fig. 5) where spillover hydrogen results in the formation of oxygen vacancies (steps I–II). Dissociative interaction of H2O generates OH that is consumed in a re-oxidation of the support (step III) with the generation of reactive protons (step IV) that attack the furfural carbonyl group to produce the alcohol. At H2O/furfural = 0.2, a switch from H2 to N2 in the feed did not result in any detectable conversion (Fig. 5). This suggests an activated process that involves continuous creation, consumption and regeneration of oxygen vacancies, requiring a co-current H2/H2O inlet with hydroxyl consumption and release of reactive hydrogen for –C=O reduction.

Furfural hydrogenation rate (R) as a function of inlet H2O/furfural molar ratio with proposed reaction mechanism (steps (I)–(IV)) illustrating the effect of water addition on furfural hydrogenation (to furfuryl alcohol). Reaction conditions T = 498 K, P = 1 atm, nAu/F = 5 × 10−3 h, H2/furfural = 66
Viable commercial application requires more efficient hydrogen utilization. Coupling 2-butanol dehydrogenation with furfural hydrogenation generates two high value products (2-butanone and furfuryl alcohol) that can be readily separated (100 K difference in boiling points at 1 atm) by standard distillation [32]. Au/CeO2 exhibited negligible activity in the dehydrogenation of 2-butanol and there is a requirement for an alternative (dehydrogenation) catalytic metal. Copper is effective in dehydrogenation [33] and Cu/SiO2 was chosen as a suitable candidate to work in tandem with Au/CeO2. TPR of Cu/SiO2 (Fig. 1aII) resulted in H2 consumption \(\left( { 1\;{\text{mol}}_{{{\text{H}}_{ 2} }} \;{\text{mol}}_{\text{Cu}}^{ - 1} } \right)\) that matched the requirement for the Cu2+ → Cu0 reduction step. This is consistent with the work of Smith et al. who demonstrated formation of zero valent Cu at T > 523 K [34]. The activated catalyst exhibited Cu particles in the 1–15 nm range with a surface area weighted mean of 7.8 nm (Fig. 1bII, cII). Dehydrogenation of 2-butanol over Cu/SiO2 generated 2-butanone as sole product at a rate (42 molfurfural mol −1Cu h−1) that was greater than hydrogenation over Au/CeO2, guaranteeing sufficient hydrogen in the coupled reaction (Fig. 6a). Under the same reaction conditions, Cu/SiO2 delivered negligible activity (< 2 molfurfural mol −1Cu h−1) in furfural conversion and consequently does not contribute significantly to hydrogenation in the coupled reaction (Fig. 6a). We evaluated catalytic coupling (in N2) over a physical mixture of Au/CeO2 + Cu/SiO2 with varying Cu/Au ratio and the results are presented in Fig. 6b. In every case, 2-butanone and furfuryl alcohol were the only products obtained. Rate increased (from 26 to 85 molfurfural mol −1Au h−1) with decreasing Cu/Au (from 150 to 65). Our results demonstrate successful utilization by Au/CeO2 of the hydrogen generated in situ by Cu/SiO2. Dehydrogenation proceeds via a two-step H abstraction mechanism [35]. This supply of reactive hydrogen circumvents the limitations associated with hydrogen activation/dissociation by Au and delivers an improved furfural hydrogenation rate. Activity in the coupled system was not affected by water addition unlike the stand alone furfural hydrogenation. This can be attributed to insufficient hydrogen availability for ceria reduction to form the oxygen vacancies necessary for water dissociation and release of reactive hydrogen (see Fig. 5).

a Reaction rate in stand-alone furfural hydrogenation (R, filled square) and dehydrogenation of 2-butanol (R, bars) over Au/CeO2 and Cu/SiO2; b furfural hydrogenation rate (R) as a function of Cu/Au molar ratio in coupled 2-butanol dehydrogenation with furfural hydrogenation. Inset schematic representation of mechanism involved in coupling of 2-butanol dehydrogenation over Cu with furfural hydrogenation over Au. Reaction conditions T = 498 K, P = 1 atm, 2-BuOH/furfural = 15
The different approaches we have considered in this work to enhance hydrogen utilization and selective furfural hydrogenation rate over supported Au are summarized in Fig. 7. The use of a conventional compressed H2 source suffers from low utilization efficiency (H2/furfuryl alcohol = 420) and the reaction is conducted in excess H2. Incorporation of oxides (CeO2 or SiO2) in a physical mixture with Au/CeO2 and inclusion of water in the feed generate reactive hydrogen via spillover and water dissociation, respectively. This resulted in a fourfold increase in rate but hydrogen utilization was still far removed from stoichiometric requirements. The dehydrogenation/hydrogenation coupling strategy enabled full utilization of (in situ generated) hydrogen via dehydrogenation where H2/furfuryl alcohol converged at reaction stoichiometry. This approach delivers improved process efficiency and offers clear advantages over standard hydrogenation applications with new opportunities for exploitation of the chemoselectivity exhibited by supported Au catalysts.

Comparison of catalytic response for different hydrogen supply strategies: furfural hydrogenation rate (R; filled square) and H2 utilization (H2/furfuryl alcohol, bars) over Au/CeO2 as a result of variation in inlet H2/furfural, increased spillover H2, promotion by water dissociation and coupling with dehydrogenation. Note horizontal line represents full H2 utilization under stoichiometric conditions. Reaction conditions T = 498 K, P = 1 atm
Conclusions
We have demonstrated that Au/CeO2 (Au mean size = 2.8 nm) promotes continuous gas phase hydrogenation of furfural to furfuryl alcohol. Under the same reaction conditions, Pd/CeO2 and Ni/CeO2 promoted decarbonylation, hydrogenolysis and furan ring reduction. An increase in available surface hydrogen on Au/CeO2 via (i) increased H2 content in the feed, (ii) spillover hydrogen and (iii) catalytic water dissociation resulted in an increase in selective hydrogenation rate. Full hydrogen utilization and higher alcohol production rate was achieved by coupling catalytic dehydrogenation (2-butanol → 2-butanone) over Cu/SiO2 (mean Cu size = 7.8 nm) with furfural hydrogenation over Au/CeO2. This circumvents use of external pressurized H2 with the production of two high value products that are readily separated by distillation. Our results demonstrate a feasible solution to the limitations of Au in reductive processes and can open new directions in “hydrogen free” hydrogenation.
References
Scurrell MS (2017) Gold Bull 50:77–84
Irfan M, Glasnov TN, Kappe CO (2011) ChemSusChem 4:300–316
Liu X, He L, Liu YM, Cao Y (2014) Acc Chem Res 47:793–804
Pan M, Gong J, Dong G, Mullins CB (2014) Acc Chem Res 47:750–760
Corma A, Serna P, García H (2007) J Am Chem Soc 129:6358–6359
Kothari R, Buddhi D, Sawhney RL (2008) Renew Sustain Energy Rev 12:553–563
Werkmeister S, Neumann J, Junge K, Beller M (2015) Chem A Eur J 21:12226–12250
Gilkey MJ, Xu B (2016) ACS Catal 6:1420–1436
Wang F, Zhang Z (2017) ACS Sustain Chem Eng 5:942–947
Trincado M, Banerjee D, Grutzmacher H (2014) Energy Environ Sci 7:2464–2503
Nagaraja BM, Padmasri AH, Raju BD, Rao KSR (2011) Int J Hydrog Energy 36:3417–3425
Zhang W, Yu D, Ji X, Huang H (2012) Green Chem 14:3441–3450
Bohre A, Dutta S, Saha B, Abu-Omar MM (2015) ACS Sustain Chem Eng 3:1263–1277
Sharma RV, Das U, Sammynaiken R, Dalai AK (2013) Appl Catal A 454:127–136
Wang M-M, He L, Liu Y-M, Cao Y, He H-Y, Fan K-N (2011) Green Chem 13:602–607
Mäki-Arvela P, Hájek J, Salmi T, Murzin DY (2005) Appl Catal A 292:1–49
Ide MS, Hao B, Neurock M, Davis RJ (2012) ACS Catal 2:671–683
Babu NS, Lingaiah N, Pasha N, Kumar JV, Prasad PSS (2009) Catal Today 141:120–124
Huang Z, Cui F, Xue J, Zuo J, Chen J, Xia C (2012) Catal Today 183:42–51
Li M, Wang X, Hao Y, Cárdenas-Lizana F, Keane MA (2017) Catal Today 279:19–28
Arena F, Famulari P, Interdonato N, Bonura G, Frusteri F, Spadaro L (2006) Catal Today 116:384–390
Andreeva D, Idakiev V, Tabakova T, Ilievaa L, Falaras P, Bourlinos A, Travlos A (2002) Catal Today 72:51–57
Bus E, Miller JT, van Miller JA (2005) J Phys Chem B 109:14581–14587
Pan M, Brush AJ, Pozun ZD, Ham HC, Yu W-Y, Henkelman G, Hwang GS, Mullins CB (2013) Chem Soc Rev 42:5002–5013
Hong Y-C, Sun K-Q, Zhang G-R, Zhong R-Y, Xu B-Q (2011) Chem Commun 47:1300–1302
Sitthisa S, Resasco DE (2011) Catal Lett 141:784–791
Prins R (2012) Chem Rev 112:2714–2738
Amorim C, Keane MA (2012) J Hazard Mater 211–212:208–217
Cárdenas-Lizana F, Gómez-Quero S, Perret N, Keane MA (2011) Catal Sci Technol 1:652–661
Andreeva D (2002) Gold Bull 35:82–88
Palo DR, Dagle RA, Holladay JD (2007) Chem Rev 107:3992–4021
Javaid A, Bildea CS (2014) Chem Eng Technol 37:1515–1524
Torresi PA, Díez VK, Luggren PJ, Cosimo JID (2013) Appl Catal A 458:119–129
Smith ML, Campos A, Spivey JJ (2012) Catal Today 182:60–66
Fridman VZ, Davydov AA, Titievsky K (2004) J Catal 222:545–557
Acknowledgements
The work was supported by the Engineering & Physical Sciences Research Council (Grant EP/M029476/1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Keane, M.A., Li, M., Collado, L. et al. Overcoming the limitations of gold catalysts in hydrogenation: enhanced activity with full hydrogen utilization. Reac Kinet Mech Cat 125, 25–36 (2018). https://doi.org/10.1007/s11144-018-1417-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-018-1417-x