
Abstract
Water oxidation by photosystem II (PSII) sustains most life on Earth, but the molecular mechanism of this unique process remains controversial. The ongoing identification of the binding sites and modes of the two water-derived substrate oxygens (‘substrate waters’) in the various intermediates (Si states, i = 0, 1, 2, 3, 4) that the water-splitting tetra-manganese calcium penta-oxygen (Mn4CaO5) cluster attains during the reaction cycle provides central information towards resolving the unique chemistry of biological water oxidation. Mass spectrometric measurements of single- and double-labeled dioxygen species after various incubation times of PSII with H218O provide insight into the substrate binding modes and sites via determination of exchange rates. Such experiments have revealed that the two substrate waters exchange with different rates that vary independently with the Si state and are hence referred to as the fast (Wf) and the slow (WS) substrate waters. New insight for the molecular interpretation of these rates arises from our recent finding that in the S2 state, under special experimental conditions, two different rates of WS exchange are observed that appear to correlate with the high spin and low spin conformations of the Mn4CaO5 cluster. Here, we reexamine and unite various proposed methods for extracting and assigning rate constants from this recent data set. The analysis results in a molecular model for substrate-water binding and exchange that reconciles the expected non-exchangeability of the central oxo bridge O5 when located between two Mn(IV) ions with the experimental and theoretical assignment of O5 as WS in all S states. The analysis also excludes other published proposals for explaining the water exchange kinetics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Photosystem II (PSII) catalyzes the oxidation of water into molecular oxygen and protons. The reaction happens at the Mn4CaO5 cluster (Fig. 1A) in the oxygen-evolving complex (OEC) of PSII, which is oxidized stepwise within a catalytic cycle that is driven by light-induced charge separations in the reaction center of PSII. Thus, the OEC is going through five intermediate states, S0 through S4, where the subscript indicates the number of stored oxidizing equivalents (Fig. 1B) (Kok et al. 1970). Molecular oxygen is released in the transition of the highly reactive S4 state to S0 in which also one substrate water binds, while the second substrate water is inserted into the cluster during the S2 → S3 transition (Dau et al. 2010; Pantazis 2018; Kern et al. 2018; Lubitz et al. 2019; Junge 2019; Suga et al. 2019; Ibrahim et al. 2020; Yamaguchi et al. 2022b; Shevela et al. 2023). High-resolution structures have been reported first for the dark-stable S1 state (Umena et al. 2011; Suga et al. 2015; Tanaka et al. 2017; Young et al. 2016), and recently also for the S2, S3, and S0 states (Kern et al. 2018; Suga et al. 2017) and several time points during the S3 → S4 → S0 transition (Bhowmick et al. 2023).

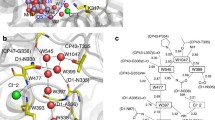
The oxygen-evolving complex of PSII, its reaction cycle, and substrate-water exchange experiments. A Structure of the Mn4CaO5 cluster and its surrounding (PDB: 7RF1). Manganese is shown in magenta, oxygen in red, calcium in green, water molecules around the active site in blue. Water-filled channels are represented with red (O1 channel), blue (O4 channel), and green (Cl1 channel) areas. B S state cycle of the Mn cluster. The formal oxidation states of the four Mn atoms in the four quasi-stable states are given in the boxes next to the state. C, D TR-MIMS measurements of the water exchange in the S2 state of Ca-PSII showing the single 18O-labeled (C) and double-labeled (D) O2 yield at pH 6. E double-labeled O2 yield of Sr-PSII. Points in C–E show individual experimental data points, and lines show exponential fits; redrawn from Ref. de Lichtenberg and Messinger (2020)
Despite the great advancement in the structural resolution of PSII in its reaction cycle, the mechanism of the O–O bond formation remains controversial (Vinyard et al. 2013, 2017; Li and Siegbahn 2015; Lubitz et al. 2019; Yamaguchi et al. 2022a; Greife et al. 2023; Bhowmick et al. 2023; Shevela et al. 2023). An important step towards resolving some of the open questions would be to identify the two substrate waters in all the S states. Presently, the only technique that can provide a unique signature for the substrate water molecules is time-resolved membrane inlet mass spectrometry (TR-MIMS) in combination with H216O/H218O exchange (Messinger et al. 1995; Messinger 2004; Hillier and Wydrzynski 2008; Cox and Messinger 2013). In this method, H218O is rapidly injected into a PSII sample and the isotopic composition of the O2 produced by flash illumination after various incubation times is measured in order to determine the exchange rates of the two substrate waters.
The TR-MIMS measurements reveal a fast-exchanging (Wf) and a slowly exchanging (WS) substrate water. The exchange of WS can be resolved in all quasi-stable S states and usually happens on time scales from tens of milliseconds (S0 state) through seconds (S2 and S3) to tens of seconds (S1). The half-time of Wf exchange in the S2 and S3 states is also resolvable in the TR-MIMS measurements, at around 7 to 25 ms, while in the S0 and S1 states Wf exchanges faster than it can be resolved with this method (Hillier et al. 1998; Hillier and Wydrzynski 2000; Nilsson et al. 2014a; Cox and Messinger 2013). The biphasic behavior of the single 18O-labeled O2 yield (denoted 34Y) reflects the distinct exchange kinetics of the two substrate waters (Fig. 1C), while the yield of the double-labeled O2 (36Y) rises usually monophasic and is mostly determined by the exchange of Ws (Fig. 1D). Therefore, the TR-MIMS data can be analyzed by fitting a single-exponential curve to the 36Y-vs-time plot, while 34Y requires two exponential components (Messinger et al. 1995; Hillier and Wydrzynski 2000):
These equations describe a system with pseudo-first-order kinetics, where exchange happens at two independent sites with apparent rate constants kf and ks, under the condition that kf ≫ ks. The parameter a describes the exponential component proportions of the fast and slow exchange in the 34Y data, and it can be shown (Messinger et al. 1995) that a should be a function of the isotope contents of the bulk water in the sample:
where αin and αf are the initial and final H218O enrichment correspondingly; a is approximately equal to 0.5/(1 − αf) if the initial enrichment (estimated to be around 0.7% in these experiments) is ignored.
The S2 state can exist in at least two different conformations, one having an EPR signal centered at g = 2.0 (low spin, LS) and the other at 4.1 (high spin, HS) (Dismukes and Siderer 1981; Zimmermann and Rutherford 1984; Kim et al. 1992). Only the g = 2.0 conformation (Fig. 1A) is observed in untreated cyanobacterial PSII in serial crystallography experiments at room temperature (Kern et al. 2018; Li et al. 2021), but EPR experiments reveal that by Ca/Sr exchange and/or high pH a HS conformation can be induced that occurs at g = 4.8–4.9, indicating that it may differ in structure compared to S2HS of plant PSII (Boussac et al. 2018). At least three different conformations have been suggested for the Mn4CaO5/6 cluster in the S2HS state (Fig. 2), which all were shown by DFT calculations to give the S = 5/2 spin state (Pantazis et al. 2012; Corry and O’Malley 2019; Pushkar et al. 2019), while models B and C were also favorably compared to x-ray spectroscopy results of S2HS (Pushkar et al. 2019; Chatterjee et al. 2019). It is noted that the three proposals are not mutually exclusive and that the ability of the Mn4CaO5 cluster to transiently attain additional conformations in the S2 state forms the basis of the proposal for exchanging the central O5 bridge (Siegbahn 2013).

Proposals for high spin (g ≥ 4.1) conformation of the OEC in S2 state. A Closed cubane model in which the ‘dangling’ Mn4 is a pentacoordinate Mn(III) ion (Pantazis et al. 2012; Isobe et al. 2012; Bovi et al. 2013); B early water binding between Mn1 and Ca (Pushkar et al. 2019; de Lichtenberg and Messinger 2020) in which Mn2 or Mn3 maybe the only Mn(III) (Pushkar et al. 2019); C proton isomer of the open cubane conformation in which, compared to S2LS, a proton is moved from W1 to O4 (Corry and O’Malley 2019). Manganese atoms are shown in magenta, calcium in green, and oxygen in red with arabic numbers used as indices, whereas the roman numbers are showing the oxidation states of Mn ions
The HS and LS conformations may exhibit different substrate-water exchange kinetics, and if a sample contains significant (> 10%) fractions of both conformations, then Eq. (1) may not be a suitable description of the system. In that case, an additional kinetic component may be observed in the 34Y and 36Y signals, and has been fitted by the following expressions (de Lichtenberg and Messinger 2020):
The kf rate constant, as before, reflects the fast-exchanging water in both conformations; since the data did not reveal two distinct phases for the fast water exchange, Wf was modeled as a single exchange component. For WS, however, two components were resolved, of which the faster, ki, is assigned to the exchange of Ws in HS conformation of the S2 state (ks2 in Scheme 1). For the slower component, ks, of Ws exchange two possible explanations were given: it could correspond to the WS exchange in the LS conformation exhibiting the g = 2.0 EPR multiline signal (ks1 in Scheme 1), or to the rate constant of conformational change, kc2 (Scheme 1) of the LS state (ELS) to the HS state (EHS). The coefficient b depends on the equilibrium ratio of the two conformations.

Substrate-water exchange reactions in a double-conformation model of PSII. Modified after (Huang and Brudvig 2021). EHS and ELS signify the conformations of the HS and LS states of the Mn4CaO5 cluster in PSII, while the superscripts (EWs,Wf) indicate the oxygen isotope bound in the binding sites of the slowly (WS) and fast (Wf) exchanging substrate waters. The exchange rates of Wf and WS in ELS, EHS are denoted kf1, kf2 and ks1, ks2, respectively, while the rates for the EHS/ELS interconversion are noted as kc1 and kc2
Explicit kinetic modeling of the water exchange has also been done. A numerical solution of the differential equations describing a model with two binding sites and two conformations (Scheme 1) has been applied previously and resulted in exchange rates consistent with those derived by Eq. (3) (de Lichtenberg and Messinger 2020). Recently, Huang and Brudvig (2021) developed the analytical solution for this model, potentially allowing for a more accurate description of the water exchange measurements. However, due to the many terms in the complete analytical solution it is not possible to see the physical meaning and derive unique solutions. Thus, Huang and Brudvig examined some approximations to simplify the expressions. Consistent with our previous work, they conclude that the expressions can be reduced to the form shown in Eq. (3) under the following approximations: (a) the rate of conformational change is much slower than the rates of water exchange; (b) the rate of exchange of Ws is significantly slower than that of Wf, so that Wf exchange is essentially complete before WS exchange commences; (c) the fast-exchange rate constants in the two conformations are equal. Due to the first approximation, the rates of conformational change are not present in the exponential factors of the simplified equations, and thus the authors concluded, in contrast to de Lichtenberg and Messinger (2020), that the parameter ks2 in Eq. (3) cannot correspond to the rate of conformational change between the LS and HS isomers, but only to the rate constant of Ws exchange in the LS conformation. However, this specific approximation is not always satisfied, as seen even in some of the simulations presented in the paper of Huang and Brudvig (2021).
In the following, we revisit the analytic description developed by Huang and Brudvig (2021) and examine other possible simplifications and corrections.
Results and discussion
Non-zero initial H2 18O enrichment
The initial conditions (concentrations of each state at time zero) of the system of differential equations are not discussed by Huang and Brudvig (2021). While the coefficients inside of the exponential functions do not depend on the initial conditions, the coefficients in front of the exponentials do. Examining the coefficients given in Ref. Huang and Brudvig (2021), it can be deduced that the authors assumed that at time zero all centers have H216O, and that the equilibrium between the two conformations (dictated by the values of the conformation change rate constants) has already been established at time zero. The first assumption, about all centers holding 16O, is not exactly correct due to the natural abundance of 18O in normal water and due to leakage from the syringe used for injection of labeled water; it has been previously estimated that around 0.7% of the water at time zero has 18O (de Lichtenberg and Messinger 2020). This value is relatively small and does not affect the kinetics of the exchange; however, it may affect the simulations and fitting of the exchange data at early times. Including this value in the model is straightforward, and the equations describing the yield of single- and double-labeled oxygen for the model with two exchange sites and two conformations are now given by:
The equations are identical with the ones in Ref. Huang and Brudvig (2021) (also shown in the Supporting Information), with the addition of the term q that describes the dependence on the initial H218O enrichment:
Here, r is the equilibrium ratio \(\frac{\left[{{\text{H}}}_{2}^{16}{\text{O}}\right]}{\left[{{\text{H}}}_{2}^{18}{\text{O}}\right]}\) after the H218O injection, and r0 is this ratio before the injection. The pre-exponential coefficients c and the exponential coefficients λ have the same expressions as given in Huang and Brudvig (2021) and in the Supporting Information. The inclusion of this correction to the simulations results in a change that is similar to a shift of the simulated curves to earlier times (see SI Fig. S1a).
HS/LS equilibrium during the substrate exchange
The second assumption that the conformation equilibrium between S2LS and S2HS is already reached at time zero of H218O incubation is presumably correct for short incubation times when the H218O injection comes long (10–20 s) after the first flash that advances S1->S2, allowing enough time for the conformation equilibration to occur before the injection of labeled water. By contrast, for long incubation times, the injection happens soon after the first flash that generates S2. Here, the situation is less clear, since low temperature illumination (200 K) of PSII samples in the S1 state leads predominantly to the formation of S2LS, which only upon short warming transforms under suitable conditions into S2HS (Boussac et al. 2018). The rate of this process is presently not well characterized. By contrast, others have concluded on the basis of DFT calculations that the ratio of S2HS to S2LS depends on the distribution of two conformations of the S1 state (Drosou et al. 2021). Even if that were the case, the equilibrium concentration between the respective conformations would be likely S state dependent and require a presently unknown time to establish. Due to these uncertainties, and the complexity of incorporating this additional equilibration that occurs over variable times into an already highly sophisticated model, we accept the assumption and make no attempt to include it here.
Non-instant injection
In reality, the injection of H218O into the PSII suspension and the subsequent mixing is not instant. The injection and mixing can be observed using a fluorescent dye (PSII or fluorescein) (Messinger et al. 1995; Nilsson et al. 2014a), and is approximately linear, taking place within around 6 ms. To evaluate the effect of the mixing on the exchange curves, we consider the exchange at a single site. The relative fraction of sites with labeled water, E18, can be described by:
where k is the apparent rate constant of the exchange, and α is the enrichment of labeled water in the bulk. If α is a constant equal to the final enrichment αf, this has the solution:
This would describe the situation where the injection of labeled water is instantaneous and happens at t = 0.
Due to the time course for the injection and mixing, the enrichment is initially not constant but increases linearly, starting at t = 0 at αin until a time t = tm (which is approximately 6 ms for our experiments), when it reaches αf:
In the two regions, the solution of (6) is now:
Since all points of the water exchange data are measured at times t ≥ tm = 6 ms, only the second time region in Eq. (9) is relevant. In this region, the expression for non-instantaneous injection (9) differs from the one for instantaneous injection (7) only by the factor tk, in a way that one is a time-shifted version of the other, with a time shift given by tk:
The coefficient tk depends on the apparent rate constant of water exchange k:
The value of tk approaches tm/2 = 3 ms for small k, staying close to that value for most of the relevant values of k, only increasing above 3.3 ms for k > 200 s−1 (see SI Fig. S2 for a plot of tk as a function of k). At higher values of k, the exchange happens faster than the time resolution of the TR-MIMS experiment, with or without the linear mixing correction. Therefore, we simply use a value of tk = 3 ms and expression (10) to approximate the effect of non-instant injection and mixing. The inclusion of this correction to the simulations results in a shift of the simulated curves to later times (see SI Fig. S1b), which is in opposite direction of the correction for non-zero initial H218O enrichment.
Water exchange in the S3 state affecting the S2 state exchange measurements
In the TR-MIMS experiments, when water exchange in the S2 state is measured, the PSII sample is first brought into the S2 state by one excitation light flash, then labeled water is injected into the sample, and after a varying incubation time two more light flashes are given to drive the O2 evolution reaction. The first of the two extra flashes brings the sample to the S3 state, where it spends a very short time, usually 10 ms, before the next flash that drives the S3 → S0 + O2 step. The time is short, in order to minimize the effect on the observed isotope ratios due to exchange in the S3 state, but must remain long enough to allow re-opening of the acceptor side and, thereby, advancement of a significant fraction to S0 coupled to O2 production. We note that for short H218O incubation times in the S2 state, and for relatively fast S3 exchange kinetics, the effect might be significant and we thus recently started to account for it [see SI of de Lichtenberg et al. (2021)]. Knowing the water exchange rates in the S3 state, it is possible to account for the 10 ms S3 state exchange on the simulated S2 state exchange kinetics by doing an extra simulation for each time point, where one takes the final concentrations of the components from the S2 exchange simulation and uses them as initial concentrations for a 10 ms exchange using the S3 exchange rate constants [see SI of de Lichtenberg et al. (2021)]. This assumes that the fast- and slow-exchanging sites observed in S2 correspond to the fast- and slow-exchanging sites in S3. This correction results in the apparent shift of the simulated curves to earlier times (see SI Fig. S1c), opposite to the effect of the correction for non-instant mixing. This extra exchange in the S3 state is ignored in the model of Huang and Brudvig (2021).
Applying all three corrections to the simulation has their effects mostly canceling each other (see SI Fig. S1d), and thus not considering them should not affect the interpretation of the data in a major way, at least in most cases when the exchange in S3 is relatively slow. Nevertheless, in the rest of this work, all simulations are performed applying all three corrections.
Interpretation of the water exchange data with the analytical solution of the two-site double-conformation model
The analytical solution of the two-site double-conformation model (Scheme 1) is a linear combination of a total of 8 exponential terms (Eq. 4) (Huang and Brudvig 2021). The arguments of the exponential functions are the H218O incubation time multiplied by the eigenvalues of the rate constants matrix (one of them is zero, yielding a constant term). To make physical sense of the results, Huang and Brudvig applied two approximations that simplify the expressions: firstly, the rates of conversion between the two conformations (with rate constants \({k}_{{\text{c}}1}\) and \({k}_{{\text{c}}2}\)) are both significantly slower than all other rates, and secondly, the fast exchange (with apparent rate constants \({k}_{{\text{f}}1}^{*}\) and \({k}_{{\text{f}}2}^{*}\)) is significantly faster than the slow exchange (\({k}_{{\text{s}}1}^{*}\) and \({k}_{{\text{s}}2}^{*}\)). The authors then arrive at simple approximate expressions that show that the yield of double-labeled O2 (36Y) is a sum of two exponentials with rate constants equal to the two slow exchange constants, \({k}_{{\text{s}}1}^{*}\) and \({k}_{{\text{s}}2}^{*}\). Both the constants come from the third eigenvalue pair, \({\lambda }_{3}^{+/-}\) in Eq. (4) [Eq. 30 in Huang and Brudvig (2021)]; due to the assumption that \({k}_{f}^{*}\gg {k}_{s}^{*}\), the terms containing \({\lambda }_{1}^{+/-}\) and \({\lambda }_{2}^{+/-}\) cancel each other. The yield of single-labeled O2 (34Y) adds two more exponentials with constants equal to the fast-exchange constants. The rate constants of interconversion between the two conformations appear only in the pre-exponential coefficients, but not as parameters of the exponential functions in this simplification. However, while the approximation that \({k}_{f}^{*}\gg {k}_{s}^{*}\) is presumably valid under most circumstances, the assumption that \({k}_{s}^{*}{\gg k}_{c}\) may not hold in all cases; for example, in the simulations of the Sr2+-PSII data at pH 8.3 presented in Table 2 of Ref. Huang and Brudvig (2021) the conformation change rate constants \({k}_{{\text{c}}}\) are significantly larger than one of the rate constant of the slow exchange \({k}_{{\text{s}}}^{*}\).
A different simplification of the equations and a different interpretation of the experimental data is possible, which is not discussed by Huang and Brudvig (2021). In this approximation, only one of the slow exchange rates (\({k}_{{\text{s}}1}^{*}\) or \({k}_{{\text{s}}2}^{*}\)) needs to be significantly faster than only one of the rate constants of conformation change (\({k}_{{\text{c}}1}\) or \({k}_{{\text{c}}2}\)). This would lead to the other conformation change rate appearing in the approximate expressions for 34Y and 36Y. We first note that the (exact) expression for the third pair of eigenvalues can be rewritten as:
With the relaxed approximation requirements, we can neglect only the term \(4{k}_{{\text{c}}1}{k}_{{\text{c}}2}\) in the above expression, which allows us to cancel the square and square root, yielding the following two approximate expressions for the eigenvalues:
If both \({k}_{{\text{s}}1}^{*}\) and \({k}_{{\text{s}}2}^{*}\) are significantly larger than \({k}_{{\text{c}}1}\) and \({k}_{{\text{c}}2}\) we still get, as in Huang and Brudvig (2021):
However, in the case when e.g., \({k}_{{\text{s}}1}^{*}\) ≪ \({k}_{{\text{c}}2}\), we get instead:
This would mean that the two phases observed in the yield of double-labeled O2 (36Y) would not reflect the exchange of WS in the two conformations, but instead the rate of exchange in EHS and the conversion of ELS to EHS (Scheme 1). This corresponds to the second interpretation obtained by de Lichtenberg and Messinger (2020) employing Eq. (3).
For example, using the values from the first simulation for the Sr-PSII sample at pH 8.3 presented in Ref. Huang and Brudvig (2021) and shown in Table 1, the exact values of the two eigenvalues (Eq. 12, not approximated) are \({\lambda }_{3}^{+}\) = − 11.96 s−1 and \({\lambda }_{3}^{-}\) = − 54.64 s−1. It is only these two kinetic phases that can be seen in the 36Y trace, although they are not clearly visibly separated, as the two values are not sufficiently different (Fig. 3C, blue trace). The \({\lambda }_{3}^{-}\) = − 54.64 s−1 component is associated with the slow water exchange in EHS, \({k}_{{\text{s}}2}^{*}\) = 50 s−1. The \({\lambda }_{3}^{+}\) = − 11.96 s−1 component, however, matches the approximations given (Eq. 15) very well (\({\lambda }_{3}^{+}\) = − 11.96 s−1 ≈ − \({k}_{{\text{c}}2}\) = − 12 s−1) and does not match the approximations (Eq. 14) given in Ref. Huang and Brudvig (2021) (− 11.96 s−1 ≠ − \({k}_{s1}^{*}\) = − 1 s−1), and thus reflects the conversion of ELS to EHS and not the exchange in ELS.

Substrate-water exchange data in two-conformation systems in the S2 state taken from Ref. de Lichtenberg and Messinger (2020) and simulated using our extended form of the double-conformation model (Scheme 1) presented in Huang and Brudvig (2021). The single-labeled O2 yield (34Y) is shown on the left, and the double-labeled O2 yield (36Y) is shown on the right. Black dots show individual experimental data points. The black and the blue curves (mostly overlapping) show simulations using rate constants given in Table 1, for the cases when the slowest kinetic component corresponds either to the slow water exchange in ELS (\({k}_{{\text{s}}1}^{*}\), black) or to the rate constant for the conversion of ELS to EHS (\({k}_{{\text{c}}2}\), blue). The intermediate kinetic component is explained in all simulations by the slow water exchange in EHS. Corrections for initial enrichment (\({\alpha }_{{\text{in}}}\) = 0.7%), non-instant injection (tk = 3 ms), and exchange in S3 (with additional 10 ms exchange using rate constants \({k}_{{\text{f}}}^{*}=19.5,{k}_{{\text{s}}}^{*}=0.25\) s−1) are applied for all simulations. A Ca2+-PSII, pH 8.6; B Sr2+-PSII, pH 6.0; C Sr2+-PSII, pH 8.3
In other simulations, the kinetic components observed in 36Y do correspond to the slow water exchange in the two conformations, \({k}_{{\text{s}}1}^{*}\) and \({k}_{{\text{s}}2}^{*}\), see the Ca2+-PSII, pH 8.6 and Sr2+-PSII, pH 6.0 simulations taken from Ref. Huang and Brudvig (2021) in Table 1 and Fig. 3A and B, black traces (mostly covered). However, these are not the only possible simulations that give the same fit quality. Depending on the values at which we choose to fix \({k}_{{\text{s}}1}^{*}\), we can obtain simulations in which the simulated 34Y and 36Y curves are practically identical, but for which the slowest observed kinetic component (with a rate of around 1 s−1 for both samples) corresponds not to \({k}_{{\text{s}}1}^{*}\) but to \({k}_{{\text{c}}2}\) (blue traces in Fig. 3A and B). The two kinetic components in 36Y are especially well separated in the Sr2+-PSII, pH 6.0 simulations (Figs. 1B, S3), and correspond to the eigenvalues \({\lambda }_{3}^{+}\) = − 1.31 s−1 ≈ − \({k}_{{\text{c}}2}\) and \({\lambda }_{3}^{-}\) = − 27 s−1 ≈ − \({k}_{{\text{s}}2}^{*}\) for the blue trace in Fig. 3B. Two interpretations of the exchange data in the Sr2+-PSII sample at pH 8.3 are also possible, see for example the last simulation in Table 1 and the black traces in Fig. 3C, where the two kinetic components in 36Y correspond to \({k}_{{\text{s}}1}^{*}\) and \({k}_{{\text{s}}2}^{*}\). We, therefore, conclude, in line with our previous work (de Lichtenberg and Messinger 2020), that it is not possible to tell from the simulations if the slowest observed kinetic component in the TR-MIMS traces corresponds to the slow water exchange in the LS state or to the rate of conversion of ELS to EHS.
Discussion
The present re-evaluation of the analytical solution obtained by Huang and Brudvig (2021) for water exchange in the high spin (EHS) and low spin (ELS) conformations of the S2 state of photosystem II shows that the simplifications required for interpreting the results lead to the same two possibilities as proposed previously by de Lichtenberg and Messinger (2020): firstly (solution 1), the two kinetic phases in the 36Y data correspond to the slow exchange constants (ks1 and ks2) in the two conformations; this requires that the conformational equilibrium is much slower than the slow exchange rates in the two conformations. Secondly (solution 2), the faster of the slow exchange rates corresponds to the slow exchange in the S2HS state (EHS) that promotes Ws exchange, while the slower 36Y rise reflects the rate of conversion of ELS to EHS that promotes Ws exchange [see also de Lichtenberg and Messinger (2020)]. A molecular interpretation of this important result with regard to the binding site of Ws in the Mn4CaO5 cluster is complicated by the fact that the structure of EHS remains controversial due to the absence of reliable x-ray diffraction or cryoEM data of PSII in the S2HS state(s) (Fig. 2A–C).
Previous substrate-water exchange measurements, advanced EPR experiments with 17O labeling, and computational studies have indicated O5 as the slow-exchanging substrate (Messinger 2004; Siegbahn 2009, 2013; Rapatskiy et al. 2012; Cox and Messinger 2013). In the well-established low spin (multiline) conformation of the S2 state (ELS in Scheme 1), O5 is bound as a µ3-oxo bridge between Mn4, Mn3, and Ca, with both Mn ions being in the formal oxidation state IV (Fig. 1). Thus, O5 is expected to be rather exchange inert in the ELS conformation of the S2 state (Hillier and Wydrzynski 2001, 2008; Tagore et al. 2006, 2007), consistent with solution 2 in which one of the states is exchange inert and the slower phase of the 36Y rise is reflecting the rate of ELS to EHS conversion. Siegbahn calculated an exchange mechanism for O5 in the S2LS state (ELS in Scheme 1) that involves water binding to Mn1 and proton transfer to O5 coupled with stepwise electron transfer from Mn1 via Mn3 to Mn4 (Siegbahn 2013). In this sequence, O5 finally ends up being fully protonated and ligated as terminal ligand at Mn4(III), where it then can exchange with water surrounding the cluster (‘cavity’ water). The calculated rate-limiting barrier for this transition is surprisingly close in energy to the experimentally determined value (Hillier and Wydrzynski 2000; Siegbahn 2013). However, it may also be possible that a modified solution 1 is at play in which the two rate constants \({k}_{{\text{s}}1}^{*}\) and \({k}_{{\text{s}}2}^{*}\) reflect the exchange in ELS and EHS, respectively, and where the exchange rates are limited by the different activation barriers for water binding to Mn1, thus a structural change not explicitly included in Scheme 1 [see also discussion in de Lichtenberg and Messinger (2020)].
The second conformation (EHS in Scheme 1), in which O5 exchanges faster, was associated with the high spin state on the basis of its prominence at high pH and its sensitivity to ammonia addition as well as Ca/Sr exchange (de Lichtenberg and Messinger 2020), properties of the high spin state established earlier by Boussac and coworkers (Boussac et al. 2018). Nevertheless, while the correlation appears convincing, it cannot be fully excluded that the state promoting Ws exchange is not identical to the high spin state.
Of the three dominant proposals for the high spin S2 state (S2HS), one involves a closed cubane conformation Fig. 2A) (Pantazis et al. 2012; Isobe et al. 2012; Bovi et al. 2013). In this conformation, O5 is bound as µ3-oxo bridge between Mn1, Mn3, and Ca, and both ligating Mn ions are in oxidation state IV. Thus, it is not obvious as to why O5 would exchange more rapidly in this conformation. By contrast, such a structure would be more likely to promote a faster W2 or O4 exchange, which are alternative assignments for Ws in the literature [for discussion, see de Lichtenberg and Messinger (2020), Huang and Brudvig (2021)]. However, while several theoretical studies consistently identify the high spin state with a closed cubane structure (Pantazis et al. 2012; Isobe et al. 2012; Bovi et al. 2013), several experimental approaches targeted to elucidating the high spin structure found no evidence for this conformation (Chatterjee et al. 2019; Pushkar et al. 2019).
In the second proposed structure of the S2HS conformation (Fig. 2B), early water binding to the Mn1 site is suggested, mostly for alkaline conditions (Pushkar et al. 2019; de Lichtenberg and Messinger 2020). Such a state is similar in structure to one of the intermediates of Siegbahn’s proposed O5 exchange pathway (Siegbahn 2013; de Lichtenberg and Messinger 2020). Thus, it may be plausible that in this state O5 would exchange more rapidly, assuming that reaching the water(hydroxide)-bound state would be rate limiting in ELS at neutral pH [see discussion in de Lichtenberg and Messinger (2020)].
In the third suggested S2HS state structure (Fig. 2C), mostly proposed for neutral or slightly acidic conditions, a proton from W1 is shifted to the O4-bidge (Corry and O’Malley 2019). This proton shift would likely promote O5 exchange by either (i) making deprotonation of water to hydroxide easier during binding to Mn1 via providing a suitable base (W1 = OH−) and/or (ii) enabling O5 protonation via the trans effect of OH− in the W1 position. Thus, also this structural proposal is consistent with the S2HS (EHS) state being faster exchanging than the S2LS (ELS) state.
Vinyard et al. (2015) and Vinyard and Brudvig (2017) proposed an exchange model for substrate water in which they aimed to reconcile their expectation that O5 is not exchangeable in the S2 and S3 states with experimental evidence that O5 can exchange in the S1 state with rates consistent with those of Ws (Rapatskiy et al. 2012). In their proposal, W2 bound to Mn4(III) is, in the S0 and S1 states, in exchange equilibrium with both the bulk water and O5; thus, it exchanges at an unresolved fast rate (together with W3) with bulk water, and additionally with a slow rate with O5. In the S2 and S3 states, the slow exchange of W2 with O5 is proposed to be blocked because O5 then binds between two Mn(IV) ions. Instead, it is proposed that the accumulation of a positive charge during the S1 → S2 transition dramatically slows the exchange of the Ca-bound W3. In this explanation, Ws binding would be consistent with the acceleration of its exchange upon Ca/Sr substitution, but in conflict with known exchange rates for Ca-bound water (Helm and Merbach 2005; Hillier and Wydrzynski 2008). On that basis, Vinyard and Brudvig propose that O–O bond formation occurs between W2 and W3 via nucleophilic attack. While interesting, this proposal is inconsistent with available substrate-water exchange data and kinetic considerations: since the two proposed substrates W2 and W3 exchange rapidly with the essentially endless pool of bulk water in the S1 and S2 states, and W2 equilibration with O5 is more than 100-fold slower than the unresolved rapid exchange with bulk water, no slow phase but instead an essentially instantaneous complete exchange of ‘Ws’ (W2) would be observable in both the 34Y and 36Y data for the S0 and S1 states, in stark contrast to experimental observations that clearly reveal the slow exchange phase.
Huang and Brudvig (2021) concluded, on the basis of their analysis and by favoring the closed cube model for S2HS (EHS), that W1 and/or W2 may be substrate waters. In contrast the team’s earlier publications (Vinyard et al. 2015; Vinyard and Brudvig 2017), they excluded that Ca-bound W3 and W4 can be a substrate due to their weak association with Ca. However, no specific suggestion regarding a mechanism was made.
In 2013, Vinyard and Dismukes published a molecular explanation for the substrate exchange rates (Vinyard et al. 2013). While several features are similar to our earlier proposal (Messinger 2004), such as employing a closed cube conformation and assigning Wf to W2 and Ws to O5, this interpretation is based on the low-oxidation state paradigm in which the S1 state has the oxidation states Mn4(III,III,III,III). Additionally, special emphasis was given to explain the Si state dependence of the exchange rates by changes in the Jahn–Teller axes of the Mn(III) ions connected to O5. The main argument against this mechanism is the convincing evidence for the high-oxidation state paradigm (Yachandra et al. 1996; Haumann et al. 2005; Kulik et al. 2007; Siegbahn 2009; Cox et al. 2014; Krewald et al. 2015; Cheah et al. 2020) and the experimental verification that the S3 state contains an extra oxygen bridge (Suga et al. 2017; Kern et al. 2018), see however Wang et al. (2021).
The analysis obtained here is fully consistent with the picture derived by us over the years by combining substrate-water exchange results with structural information from advanced EPR and snapshot crystallography at XFELs (Messinger 2004; Rapatskiy et al. 2012; Cox and Messinger 2013; Navarro et al. 2013; Nilsson et al. 2014b; Kern et al. 2018; de Lichtenberg and Messinger 2020; Ibrahim et al. 2020; de Lichtenberg et al. 2021; Hussein et al. 2021; Bhowmick et al. 2023). In this model, for substrate-water binding and O–O bond formation (Fig. 4), O5 is the slowly exchanging substrate in all S states, while W3 bound as terminal ligand to Ca may be identified as the fast-exchanging substrate in the S0, S1, and S2 states [although in rapid exchange equilibrium with all other water molecules in the inner cavity (de Lichtenberg et al. 2021)]. In these early S states, Wf exchange is limited by isotopic equilibration through barriers in the channels connecting the bulk and the Mn4CaO5 cluster (Vassiliev et al. 2012; de Lichtenberg et al. 2021), while during the S2 → S3 transition Wf moves into the OX/O6 position that bridges Ca and Mn1. The binding of Wf to Mn in the S3 state is consistent with its slower exchange in S3 compared to the earlier S states. Since in the major S3 state conformation all Mn ions of the cluster are in the Mn(IV) state (Haumann et al. 2005; Cox et al. 2014; Kern et al. 2018; Schuth et al. 2018) and thus are exchange inert, its rate of exchange may be limited by the back donation of one electron from YZ (Siegbahn 2013; Nilsson et al. 2014b; de Lichtenberg et al. 2021), i.e., by the formation of the S2′YZox state. Nevertheless, also S3 minority species are proposed in which Mn(III) ions may be formed by partial oxygen ligand oxidation, such as oxyl radical or peroxide formation (Isobe et al. 2019; Corry and O’Malley 2021). While peroxide formation between the substrates is expected to fully block exchange, oxyl radical formation may initiate water exchange under certain conditions.

A schematic presentation of the OEC conformations of the S states during the Kok cycle, based on previous spectroscopic, structural, and DFT results, see Text. The oxygens proposed to be the fast and slowly exchanging substrate waters are denoted as Wf and Ws, respectively, for states their exchange rates have been measured. N1 signifies a new water molecule that replaces W3 at Ca during the S2 → S3 transition. A second water binding event during the reconstruction of the cluster after O2 release in the S4 → S0 transition, as well as proton and O2 release are not indicated for simplicity of presentation. Manganese atoms are shown in magenta, calcium in green, and oxygen in red with Arabic numbers. Roman numbers indicate the oxidation states of manganese ions
In principle, also two kinetic phases for Wf exchange may be expected and are indeed included in Scheme 1. However, the apparent rates, \({k}_{{\text{f}}1}^{*}\) and \({k}_{{\text{f}}2}^{*}\), are within a factor of 2 (Table 1), consistent with the experimental observation that under most circumstances only one fast phase can be observed. The inability to resolve two fast rates is consistent with the above picture that Wf exchange in the S2 state is limited by equilibration of all ‘cavity’ waters around the Mn4CaO5 cluster with bulk water (de Lichtenberg et al. 2021), which effectively prevents the observation of possible chemical differences in Wf binding between ELS and EHS.
In our model, exchange of Ws (O5) in S3 state is dependent on Mn(III) formation, e.g., via YZ back donation, but slower than Wf exchange because additional significant barriers are involved for its exchange.
Conclusion
The present extension of the analytical analysis of the two-state two-conformation model for the S2 state (Scheme 1) and its interpretation in the context of literature data reconciles the expected non-exchangeability of O5 when bound between Mn(IV) ions (Tagore et al. 2006, 2007) with the experimental and theoretical assignment of O5 as the slowly exchanging substrate water in all S states (Messinger 2004; Siegbahn 2009; Rapatskiy et al. 2012; Cox and Messinger 2013), and excludes the alternative substrate water assignments in the literature (Vinyard et al. 2013, 2015; Vinyard and Brudvig 2017).
Data availability
Original data will be provided upon request.
Abbreviations
- EPR:
-
Electron paramagnetic resonance
- DFT:
-
Density functional theory
- E:
-
Conformational state of enzyme
- k c :
-
Rate of equilibration between HS and LS state
- k f, k S :
-
Fast and slow rate for substrate-water exchange
- k*:
-
Apparent rate constant
- HS, LS:
-
High spin, low spin
- MIMS:
-
Membrane inlet mass spectrometry
- Mn4CaO5/6 :
-
Inorganic cofactor of the OEC with 5 (S0–S2) or 6 (S3, S4) oxo bridges
- OEC:
-
Oxygen-evolving complex: functional unit of PSII that catalyzes water oxidation
- O5:
-
Central oxygen bridge of the Mn4CaO5/6 cluster
- PSII:
-
Photosystem II
- Si states:
-
Oxidation states of the Mn4CaO5/6 cluster (i = 0, 1, 2, 3, 4)
- Wf, WS :
-
Fast and slowly exchanging substrate water bound to the Mn4CaO5/6 cluster
- WN :
-
New water added to the Mn4CaO5 cluster during the S2 → S3 transition
- W1–W4:
-
Terminal water ligands of the OEC
- 34Y, 36Y:
-
Oxygen yield at mass-to-charge ratio 34 and 36
- XFEL:
-
X-ray free electron laser
- α in, α f :
-
Initial and final 18O isotope enrichment of the buffer
References
Bhowmick A, Hussein R, Bogacz I, Simon PS, Ibrahim M, Chatterjee R, Doyle MD, Cheah MH, Fransson T, Chernev P, Kim I-S, Makita H, Dasgupta M, Kaminsky CJ, Zhang M, Gätcke J, Haupt S, Nangca II, Keable SM, Aydin AO, Tono K, Owada S, Gee LB, Fuller FD, Batyuk A, Alonso-Mori R, Holton JM, Paley DW, Moriarty NW, Mamedov F, Adams PD, Brewster AS, Dobbek H, Sauter NK, Bergmann U, Zouni A, Messinger J, Kern J, Yano J, Yachandra VK (2023) Structural evidence for intermediates during O2 formation in photosystem II. Nature 617:629–636. https://doi.org/10.1038/s41586-023-06038-z
Boussac A, Ugur I, Marion A, Sugiura M, Kaila VRI, Rutherford AW (2018) The low spin–high spin equilibrium in the S2-state of the water oxidizing enzyme. Biochim Biophys Acta 1859(5):342–356. https://doi.org/10.1016/j.bbabio.2018.02.010
Bovi D, Narzi D, Guidoni L (2013) The S2 state of the oxygen-evolving complex of photosystem II explored by QM/MM dynamics: spin surfaces and metastable states suggest a reaction path towards the S3 state. Angew Chem Int Ed 52(45):11744–11749. https://doi.org/10.1002/anie.201306667
Chatterjee R, Lassalle L, Gul S, Fuller FD, Young ID, Ibrahim M, de Lichtenberg C, Cheah MH, Zouni A, Messinger J, Yachandra VK, Kern J, Yano J (2019) Structural isomers of the S2 state in photosystem II: do they exist at room temperature and are they important for function? Physiol Plant 166(1):60–72. https://doi.org/10.1111/ppl.12947
Cheah MH, Zhang M, Shevela D, Mamedov F, Zouni A, Messinger J (2020) Assessment of the manganese cluster’s oxidation state via photoactivation of photosystem II microcrystals. Proc Natl Acad Sci USA 117(1):141–145. https://doi.org/10.1073/pnas.1915879117
Corry TA, O’Malley PJ (2019) Proton isomers rationalize the high- and low-spin forms of the S2 state intermediate in the water-oxidizing reaction of photosystem II. J Phys Chem Lett 10(17):5226–5230. https://doi.org/10.1021/acs.jpclett.9b01372
Corry TA, O’Malley PJ (2021) S3 state models of nature’s water oxidizing complex: analysis of bonding and magnetic exchange pathways, assessment of experimental electron paramagnetic resonance data, and implications for the water oxidation mechanism. J Phys Chem B 125(36):10097–10107. https://doi.org/10.1021/acs.jpcb.1c04459
Cox N, Messinger J (2013) Reflections on substrate water and dioxygen formation. Biochim Biophys Acta 1827:1020–1030. https://doi.org/10.1016/j.bbabio.2013.01.013
Cox N, Retegan M, Neese F, Pantazis DA, Boussac A, Lubitz W (2014) Electronic structure of the oxygen-evolving complex in photosystem II prior to O-O bond formation. Science 345(6198):804–808. https://doi.org/10.1126/science.1254910
Dau H, Limberg C, Reier T, Risch M, Roggan S, Strasser P (2010) The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis. ChemCatChem 2(7):724–761. https://doi.org/10.1002/cctc.201000126
de Lichtenberg C, Messinger J (2020) Substrate water exchange in the S2 state of photosystem II is dependent on the conformation of the Mn4Ca cluster. Phys Chem Chem Phys 22(23):12894–12908. https://doi.org/10.1039/D0CP01380C
de Lichtenberg C, Kim CJ, Chernev P, Debus RJ, Messinger J (2021) The exchange of the fast substrate water in the S2 state of photosystem II is limited by diffusion of bulk water through channels—implications for the water oxidation mechanism. Chem Sci 12(38):12763–12775. https://doi.org/10.1039/d1sc02265b
Dismukes GC, Siderer Y (1981) Intermediates of a polynuclear manganese cluster involved in photosynthetic oxidation of water. Proc Natl Acad Sci USA 78(1):274–278. https://doi.org/10.1073/pnas.78.1.274
Drosou M, Zahariou G, Pantazis DA (2021) Orientational Jahn-Teller isomerism in the dark-stable state of Nature’s water oxidase. Angew Chem Int Ed 60(24):13493–13499. https://doi.org/10.1002/anie.202103425
Greife P, Schönborn M, Capone M, Assunção R, Narzi D, Guidoni L, Dau H (2023) The electron–proton bottleneck of photosynthetic oxygen evolution. Nature 617:623–628. https://doi.org/10.1038/s41586-023-06008-5
Haumann M, Müller C, Liebisch P, Iuzzolino L, Dittmer J, Grabolle M, Neisius T, Meyer-Klaucke W, Dau H (2005) Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3, S4 → S0) characterized by X-ray absorption spectroscopy at 20 K and room temperature. Biochemistry 44(6):1894–1908. https://doi.org/10.1021/bi048697e
Helm L, Merbach AE (2005) Inorganic and bioinorganic solvent exchange mechanisms. Chem Rev 105(6):1923–1959. https://doi.org/10.1021/cr030726o
Hillier W, Wydrzynski T (2000) The affinities for the two substrate water binding sites in the O2 evolving complex of photosystem II vary independently during S-state turnover. Biochemistry 39(15):4399–4405. https://doi.org/10.1021/bi992318d
Hillier W, Wydrzynski T (2001) Oxygen ligand exchange at metal sites: implications for the O2 evolving mechanism of photosystem II. Biochim Biophys Acta 1503(1–2):197–209. https://doi.org/10.1016/S0005-2728(00)00225-5
Hillier W, Wydrzynski T (2008) 18O-Water exchange in photosystem II: substrate binding and intermediates of the water splitting cycle. Coord Chem Rev 252:306–317. https://doi.org/10.1016/j.ccr.2007.09.004
Hillier W, Messinger J, Wydrzynski T (1998) Kinetic determination of the fast exchanging substrate water molecule in the S3 state of photosystem II. Biochemistry 37(48):16908–16914. https://doi.org/10.1021/bi980756z
Huang H-L, Brudvig GW (2021) Kinetic modeling of substrate-water exchange in photosystem II. BBA Adv 1:100014. https://doi.org/10.1016/j.bbadva.2021.100014
Hussein R, Ibrahim M, Bhowmick A, Simon PS, Chatterjee R, Lassalle L, Doyle M, Bogacz I, Kim IS, Cheah MH, Gul S, de Lichtenberg C, Chernev P, Pham CC, Young ID, Carbajo S, Fuller FD, Alonso-Mori R, Batyuk A, Sutherlin KD, Brewster AS, Bolotovsky R, Mendez D, Holton JM, Moriarty NW, Adams PD, Bergmann U, Sauter NK, Dobbek H, Messinger J, Zouni A, Kern J, Yachandra VK, Yano J (2021) Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat Commun. https://doi.org/10.1038/s41467-021-26781-z
Ibrahim M, Fransson T, Chatterjee R, Cheah MH, Hussein R, Lassalle L, Sutherlin KD, Young ID, Fuller FD, Gul S, Kim IS, Simon PS, de Lichtenberg C, Chernev P, Bogacz I, Pham CC, Orville AM, Saichek N, Northen T, Batyuk A, Carbajo S, Alonso-Mori R, Tono K, Owada S, Bhowmick A, Bolotovsky R, Mendez D, Moriarty NW, Holton JM, Dobbek H, Brewster AS, Adams PD, Sauter NK, Bergmann U, Zouni A, Messinger J, Kern J, Yachandra VK, Yano J (2020) Untangling the sequence of events during the S2 → S3 transition in photosystem II and implications for the water oxidation mechanism. Proc Natl Acad Sci USA 117(23):12624–12635. https://doi.org/10.1073/pnas.2000529117
Isobe H, Shoji M, Yamanaka S, Umena Y, Kawakami K, Kamiya N, Shen JR, Yamaguchi K (2012) Theoretical illumination of water-inserted structures of the CaMn4O5 cluster in the S2 and S3 states of oxygen-evolving complex of photosystem II: full geometry optimizations by B3LYP hybrid density functional. Dalton Trans 41(44):13727–13740. https://doi.org/10.1039/C2dt31420g
Isobe H, Shoji M, Suzuki T, Shen JR, Yamaguchi K (2019) Spin, valence, and structural isomerism in the S3 state of the oxygen-evolving complex of photosystem II as a manifestation of multimetallic cooperativity. J Chem Theory Comput 15(4):2375–2391. https://doi.org/10.1021/acs.jctc.8b01055
Junge W (2019) Oxygenic photosynthesis: history, status and perspective. Q Rev Biophys. https://doi.org/10.1017/S0033583518000112
Kern J, Chatterjee R, Young ID, Fuller FD, Lassalle L, Ibrahim M, Gul S, Fransson T, Brewster AS, Alonso-Mori R, Hussein R, Zhang M, Douthit L, de Lichtenberg C, Cheah MH, Shevela D, Wersig J, Seuffert I, Sokaras D, Pastor E, Weninger C, Kroll T, Sierra RG, Aller P, Butryn A, Orville AM, Liang MN, Batyuk A, Koglin JE, Carbajo S, Boutet S, Moriarty NW, Holton JM, Dobbek H, Adams PD, Bergmann U, Sauter NK, Zouni A, Messinger J, Yano J, Yachandra VK (2018) Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 563(7731):421–425. https://doi.org/10.1038/s41586-018-0681-2
Kim DH, Britt RD, Klein MP, Sauer K (1992) The manganese site of the photosynthetic oxygen-evolving complex probed by EPR spectroscopy of oriented photosystem II membranes: the g = 4 and g = 2 multiline signals. Biochemistry 31:541–547. https://doi.org/10.1021/bi00117a034
Kok B, Forbush B, McGloin M (1970) Cooperation of charges in photosynthetic O2 evolution. Photochem Photobiol 11:457–476. https://doi.org/10.1111/j.1751-1097.1970.tb06017.x
Krewald V, Retegan M, Cox N, Messinger J, Lubitz W, DeBeer S, Neese F, Pantazis DA (2015) Metal oxidation states in biological water splitting. Chem Sci 6(3):1676–1695. https://doi.org/10.1039/c4sc03720k
Kulik LV, Epel B, Lubitz W, Messinger J (2007) Electronic structure of the Mn4OxCa cluster in the S0 and S2 states of the oxygen-evolving complex of photosystem II based on pulse 55Mn-ENDOR and EPR spectroscopy. J Am Chem Soc 129:13421–13435. https://doi.org/10.1021/ja071487f
Li XC, Siegbahn PEM (2015) Alternative mechanisms for O2 release and O-O bond formation in the oxygen evolving complex of photosystem II. Phys Chem Chem Phys 17(18):12168–12174. https://doi.org/10.1039/c5cp00138b
Li HJ, Nakajima Y, Nomura T, Sugahara M, Yonekura S, Chan SK, Nakane T, Yamane T, Umena Y, Suzuki M, Masuda T, Motomura T, Naitow H, Matsuura Y, Kimura T, Tono K, Owada S, Joti Y, Tanaka R, Nango E, Akita F, Kubo M, Iwata S, Shen JR, Suga M (2021) Capturing structural changes of the S1 to S2 transition of photosystem II using time-resolved serial femtosecond crystallography. IUCrJ 8:431–443. https://doi.org/10.1107/S2052252521002177
Lubitz W, Chrysina M, Cox N (2019) Water oxidation in photosystem II. Photosynth Res 142(1):105–125. https://doi.org/10.1007/s11120-019-00648-3
Messinger J (2004) Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys Chem Chem Phys 6:4764–4771. https://doi.org/10.1039/B406437B
Messinger J, Badger M, Wydrzynski T (1995) Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc Natl Acad Sci USA 92:3209–3213. https://doi.org/10.1073/pnas.92.8.3209
Navarro MP, Ames WM, Nilsson H, Lohmiller T, Pantazis DA, Rapatskiy L, Nowaczyk MM, Neese F, Boussac A, Messinger J, Lubitz W, Cox N (2013) Ammonia binding to the oxygen-evolving complex of photosystem II identifies the solvent-exchangeable oxygen bridge (μ-oxo) of the manganese tetramer. Proc Natl Acad Sci USA 110(39):15561–15566. https://doi.org/10.1073/pnas.1304334110
Nilsson H, Krupnik T, Kargul J, Messinger J (2014a) Substrate water exchange in photosystem II core complexes of the extremophilic red alga Cyanidioschyzon merolae. Biochim Biophys Acta 1837(8):1257–1262. https://doi.org/10.1016/j.bbabio.2014.04.001
Nilsson H, Rappaport F, Boussac A, Messinger J (2014b) Substrate-water exchange in photosystem II is arrested before dioxygen formation. Nat Commun 5:4305. https://doi.org/10.1038/ncomms5305
Pantazis DA (2018) Missing pieces in the puzzle of biological water oxidation. ACS Catal 8(10):9477–9507. https://doi.org/10.1021/acscatal.8b01928
Pantazis DA, Ames W, Cox N, Lubitz W, Neese F (2012) Two interconvertible structures that explain the spectroscopic properties of the oxygen-evolving complex of photosystem II in the S2 state. Angew Chem Int Ed 51(39):9935–9940. https://doi.org/10.1002/anie.201204705
Pushkar Y, Ravari AK, Jensen SC, Palenik M (2019) Early binding of substrate oxygen is responsible for a spectroscopically distinct S2 state in photosystem II. J Phys Chem Lett 10(17):5284–5291. https://doi.org/10.1021/acs.jpclett.9b01255
Rapatskiy L, Cox N, Savitsky A, Ames WM, Sander J, Nowaczyk MM, Rögner M, Boussac A, Neese F, Messinger J, Lubitz W (2012) Detection of the water-binding sites of the oxygen-evolving complex of photosystem II using W-band 17O electron-electron double resonance-detected NMR spectroscopy. J Am Chem Soc 134(40):16619–16634. https://doi.org/10.1021/Ja3053267
Schuth N, Zaharieva I, Chernev P, Berggren G, Anderlund M, Styring S, Dau H, Haumann M (2018) K alpha X-ray emission spectroscopy on the photosynthetic oxygen-evolving complex supports manganese oxidation and water binding in the S3 state. Inorg Chem 57(16):10424–10430. https://doi.org/10.1021/acs.inorgchem.8b01674
Shevela D, Kern JF, Govindjee G, Messinger J (2023) Solar energy conversion by photosystem II: principles and structures. Photosynth Res 156(3):279–307. https://doi.org/10.1007/s11120-022-00991-y
Siegbahn PEM (2009) Structures and energetics for O2 formation in photosystem II. Acc Chem Res 42(12):1871–1880. https://doi.org/10.1021/Ar900117k
Siegbahn PEM (2013) Substrate water exchange for the oxygen evolving complex in PSII in the S1, S2, and S3 states. J Am Chem Soc 135(25):9442–9449. https://doi.org/10.1021/ja401517e
Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen JR (2015) Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond x-ray pulses. Nature 517(7532):99–103. https://doi.org/10.1038/nature13991
Suga M, Akita F, Sugahara M, Kubo M, Nakajima Y, Nakane T, Yamashita K, Umena Y, Nakabayashi M, Yamane T, Nakano T, Suzuki M, Masuda T, Inoue S, Kimura T, Nomura T, Yonekura S, Yu LJ, Sakamoto T, Motomura T, Chen JH, Kato Y, Noguchi T, Tono K, Joti Y, Kameshima T, Hatsui T, Nango E, Tanaka R, Naitow H, Matsuura Y, Yamashita A, Yamamoto M, Nureki O, Yabashi M, Ishikawa T, Iwata S, Shen JR (2017) Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543(7643):131–135. https://doi.org/10.1038/nature21400
Suga M, Akita F, Yamashita K, Nakajima Y, Ueno G, Li HJ, Yamane T, Hirata K, Umena Y, Yonekura S, Yu LJ, Murakami H, Nomura T, Kimura T, Kubo M, Baba S, Kumasaka T, Tono K, Yabashi M, Isobe H, Yamaguchi K, Yamamoto M, Ago H, Shen JR (2019) An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 366(6463):334–338. https://doi.org/10.1126/science.aax6998
Tagore R, Chen HY, Crabtree RH, Brudvig GW (2006) Determination of μ-oxo exchange rates in di-μ-oxo dimanganese complexes by electrospray ionization mass spectrometry. J Am Chem Soc 128(29):9457–9465. https://doi.org/10.1021/ja061348i
Tagore R, Crabtree RH, Brudvig GW (2007) Distinct mechanisms of bridging-oxo exchange in di-μ-O dimanganese complexes with and without water-binding sites: implications for water binding in the O2-evolving complex of photosystem II. Inorg Chem 46(6):2193–2203. https://doi.org/10.1021/ic061968k
Tanaka A, Fukushima Y, Kamiya N (2017) Two different structures of the oxygen-evolving complex in the same polypeptide frameworks of photosystem II. J Am Chem Soc 139(5):1718–1721. https://doi.org/10.1021/jacs.6b09666
Umena Y, Kawakami K, Shen JR, Kamiya N (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473(7345):55–61. https://doi.org/10.1038/Nature09913
Vassiliev S, Zaraiskaya T, Bruce D (2012) Exploring the energetics of water permeation in photosystem II by multiple steered molecular dynamics simulations. Biochim Biophys Acta 1817(9):1671–1678. https://doi.org/10.1016/j.bbabio.2012.05.016
Vinyard DJ, Brudvig GW (2017) Progress toward a molecular mechanism of water oxidation in photosystem II. Annu Rev Phys Chem 68(1):101–116. https://doi.org/10.1146/annurev-physchem-052516-044820
Vinyard DJ, Ananyev GM, Dismukes GC (2013) Photosystem II: the reaction center of oxygenic photosynthesis. Annu Rev Biochem 82:577–606. https://doi.org/10.1146/annurev-biochem-070511-100425
Vinyard DJ, Khan S, Brudvig GW (2015) Photosynthetic water oxidation: binding and activation of substrate waters for O-O bond formation. Faraday Discuss 185:37–50. https://doi.org/10.1039/c5fd00087d
Vinyard DJ, Khan S, Askerka M, Batista VS, Brudvig GW (2017) Energetics of the S2 state spin isomers of the oxygen-evolving complex of photosystem II. J Phys Chem B 121(5):1020–1025. https://doi.org/10.1021/acs.jpcb.7b00110
Wang J, Armstrong WH, Batista VS (2021) Do crystallographic XFEL data support binding of a water molecule to the oxygen-evolving complex of photosystem II exposed to two flashes of light? Proc Natl Acad Sci USA 118(24):e2023982118. https://doi.org/10.1073/pnas.2023982118
Yachandra VK, Sauer K, Klein MP (1996) Manganese cluster in photosynthesis: where plants oxidize water to dioxygen. Chem Rev 96:2927–2950. https://doi.org/10.1021/cr950052k
Yamaguchi K, Miyagawa K, Shoji M, Isobe H, Kawakami T (2022a) Elucidation of a multiple S3 intermediates model for water oxidation in the oxygen evolving complex of photosystem II. Calcium-assisted concerted O-O bond formation. Chem Phys Lett. https://doi.org/10.1016/j.cplett.2022.140042
Yamaguchi K, Shoji M, Isobe H, Kawakami T, Miyagawa K, Suga M, Akita F, Shen JR (2022b) Geometric, electronic and spin structures of the CaMn4O5 catalyst for water oxidation in oxygen-evolving photosystem II. Interplay between experiments and theoretical computations. Coord Chem Rev. https://doi.org/10.1016/j.ccr.2022.214742
Young ID, Ibrahim M, Chatterjee R, Gul S, Fuller FD, Koroidov S, Brewster AS, Tran R, Alonso-Mori R, Kroll T, Michels-Clark T, Laksmono H, Sierra RG, Stan CA, Hussein R, Zhang M, Douthit L, Kubin M, de Lichtenberg C, Pham LV, Nilsson H, Cheah MH, Shevela D, Saracini C, Bean MA, Seuffert I, Sokaras D, Weng TC, Pastor E, Weninger C, Fransson T, Lassalle L, Brauer P, Aller P, Docker PT, Andi B, Orville AM, Glownia JM, Nelson S, Sikorski M, Zhu DL, Hunter MS, Lane TJ, Aquila A, Koglin JE, Robinson J, Liang MN, Boutet S, Lyubimov AY, Uervirojnangkoorn M, Moriarty NW, Liebschner D, Afonine PV, Waterman DG, Evans G, Wernet P, Dobbek H, Weis WI, Brunger AT, Zwart PH, Adams PD, Zouni A, Messinger J, Bergmann U, Sauter NK, Kern J, Yachandra VK, Yano J (2016) Structure of photosystem II and substrate binding at room temperature. Nature 540(7633):453–457. https://doi.org/10.1038/nature20161
Zimmermann JL, Rutherford AW (1984) EPR studies of the oxygen-evolving enzyme of photosystem II. Biochim Biophys Acta 767(1):160–167. https://doi.org/10.1016/0005-2728(84)90091-4
Acknowledgements
This work was supported by the Swedish Science Foundation (Vetenskapsrådet; Grant Nr.: 2020-03809).
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
Johannes Messinger designed, supervised and provided funding for the study. Petko Chernev performed calculations and wrote the first draft of the manuscript. Johannes Messinger and A. Orkun Aydin revised the manuscript. Petko Chernev and A. Orkun Aydin prepared the figures.
Corresponding author
Ethics declarations
Competing interests
JM is Editor-in Chief of Photosynthesis Research but was not involved in the evaluation of this manuscript. PC and AOA declare they have no financial or non-financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chernev, P., Aydin, A.O. & Messinger, J. On the simulation and interpretation of substrate-water exchange experiments in photosynthetic water oxidation. Photosynth Res (2024). https://doi.org/10.1007/s11120-024-01084-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11120-024-01084-8