
Abstract
This study investigated the flavonoid compounds in Actinidia chinensis and Actinidia arguta fruits. A total of 125 flavonoids, including 9 anthocyanins, 12 catechins, 17 flavanones, 48 flavones (including 14 flavone C-glycosides), 29 flavonols, 6 isoflavones, and 4 proanthocyanidins, were identified in “Hongyang” kiwifruit (red flesh), “Jintao” kiwifruit, “Mini Amethyst” kiwiberry (purple flesh), and “Kuilv” kiwiberry. Thirty-nine metabolites showed significantly different contents between “Hongyang” and “Jintao,” and 38 of them showed higher content in “Hongyang,” whereas 39 metabolites showed significantly different contents between “Mini Amethyst” and “Kuilv,” and 31 of them showed higher content in “Mini Amethyst.” This result indicates the superior nutritional value of the pigmented kiwi cultivars in terms of flavonoids. Multivariate statistical analysis indicates that the variation in flavonoid profiles contributes to the pigmentation phenotypes of “Hongyang” and “Mini Amethyst.” Further comparative transcriptomic analysis revealed that structural genes in the anthocyanin synthesis pathway (AcF3H, AcF3′H, AcDFR, AcUFGT) and transcription factors (AcMYB10, AcbHLH5) may be involved in the pigmentation of the red-fleshed A. chinensis, whereas AaF3H, AaF3GT, and AaMYB110 may play important roles in the pigmentation of the purple-fleshed A. arguta. This study provides broader insight into the variation in flavonoid profiles among kiwifruit/berry, evaluates the flavonoid nutrition of the four cultivars, and provides additional evidence for the correlation between the genes and metabolites involved in flavonoid synthesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kiwifruit (Actinidia chinensis) is a high-value fruit crop that is recognized for its nutrient content and attractive appearance (Li et al. 2007). Kiwiberry (Actinidia arguta), also called “hardy kiwifruit,” a semiwild kiwifruit relative, has attracted increasing attention from breeders and customers due to its smaller fruit, glabrous peel, and distinct flavor (Wang et al. 2018; Latocha and Jankowski 2011). Most kiwifruit and kiwiberry varieties have green or pale-yellow flesh and are rich in vitamin C, dietary fiber, and flavonoids that can contribute to human diet (Richardson et al. 2018). Meanwhile, few colored varieties, such as the red-pigmented A. chinensis “Hongyang” and purple-pigmented A. arguta var. arguta, have exhibited great commercial potentials in recent years due to their gorgeous color and antioxidant function provided by accumulated anthocyanins (Jaeger and Harker 2005). These qualities all share at least a partial basis in the flavonoid pathway, which gives rise to fruit colors, flavors, and healthful compounds (Winkel-Shirley 2001). Thus, previous works have highlighted anthocyanin metabolites and pigmentation mechanisms of pigmented kiwifruit and kiwiberry (Montefiori et al. 2005; Montefiori et al. 2009). However, large-scale evaluation of anthocyanin-related flavonoid compounds has seldom been reported despite colorless/pale-color or yellow flavonoids predominating among the total flavonoids of kiwi species, influencing fruit pigmentation and supporting health functions as well.
Flavonoids represent one of the largest classes of plant secondary metabolites and are involved in performing various physiological functions, including UV protection, insect attraction, and pathogen defense as well as leading to variation in plant color (Winkel-Shirley 2001). Flavonoids not only contribute to the flavor of fruit crops but also act as a powerful antioxidant in the human diet (Crozier et al. 2009). Flavonoids are synthesized via the flavonoid branch of the phenylpropanoid metabolic pathway. Based on the substituents, flavonoids can be subclassified into anthocyanidins, flavanones, flavones, isoflavones, flavanols, and flavonols (Tohge et al. 2017), which are derived through the flavonoid biosynthetic pathway using a set of common enzymatic reactions. Anthocyanin is a kind of end-product of the flavonoid pathway and is synthesized by a series of enzymes, including chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), flavonoid 3′-hydroxylase (F3′H), flavonoid 3′5′-hydroxylase (F3′5′H), dihydroflavonol 4-reductase (DFR), leucoanthocyanidin dioxygenase (LDOX), and UDP-glucose flavonoid 3-O-glucosyltransferase (UFGT). Since anthocyanin is the most important water-soluble pigment in plants (He and Giusti 2010), members of the anthocyanin chemical family, including pelargonidin, cyanidin, delphinidin, peonidin, petunidin, and malvidin, have been well characterized in many important fruit crops (Jaakola 2013). However, according to anthocyanins biosynthesized through the specific branched pathway of the flavonoid pathway (Zhang et al. 2014), the metabolism of anthocyanin was also dynamically correlated with several other flavonoids located upstream or on other branches of the metabolic pathways (Nabavi et al. 2018). Thus, only highlighting anthocyanidins or a few anthocyanin-related flavonoids, such as dihydromyricetin, bracteatin, luteolin, and (−)-epigallocatechin (Li et al. 2018b), is limiting to reveal the dynamic metabolic pattern underlying the pigmentation of kiwifruit and kiwiberry and is also not sufficient to pave the way for further gene function study and genetic improvement of these taxa.
The genetic mechanisms underlying anthocyanin accumulation have been repeatedly studied in many horticultural crops via transcriptome analysis (El-Sharkawy et al. 2015; Cao et al. 2018; Lin et al. 2018; Bai et al. 2017). Although transcriptomic studies were conducted on kiwifruit and kiwiberry, and some candidate genes were screened out (Li et al. 2018a; Li et al. 2018b; Li et al. 2015b; Wang et al. 2019; Peng et al. 2019), these results were mainly based on limited numbers of anthocyanidin-related metabolites. Furthermore, pigmented kiwifruit and kiwiberry also showed distinct pigmentation patterns. For example, the pigmentation of red kiwifruit always centered around the fruit core and showed a bright-red color, whereas the pigmentation of purple kiwiberry showed a dark-red or purple color with a global pattern. However, due to the different genetic backgrounds of these two species, the pigmentation mechanisms of these two species have not been discussed together despite the previous studies having selected analogous candidate genes (Li et al. 2018a; Li et al. 2018b; Li et al. 2015b). Currently, a combination of metabolic and transcriptomic profiles has provided important information for understanding gene-to-metabolite relations, which have been successfully applied in many plants, such as potato (Cho et al. 2016; Stushnoff et al. 2010), tea (Li et al. 2015a), and fig (Wang et al. 2017). Hence, more comprehensive flavonoid identification together with transcriptomic analysis would not only deepen our understanding of kiwifruit and kiwiberry pigmentation mechanisms but also lay the foundation for the evolutionary study of anthocyanidin biosynthesis in the Actinidia genus.
In the present study, a large-scale flavonoid characterization was carried out on red-fleshed A. chinensis “Hongyang” (AcH), A. chinensis “Jintao” (AcG), purple A. arguta “Mini Amethyst” (AaP), and A. arguta “Kuilv” (AaG) (Fig. 1a). Then, multiple statistical analyses were conducted to address the differences in flavonoid accumulation between pigmented and nonpigmented individuals and between kiwifruit and kiwiberry. Finally, the transcriptomic differences of pigmented and nonpigmented kiwifruit and kiwiberry were investigated respectively according to the differential flavonoid metabolites.

a Phenotype of “Hongyang” (AcH), “Jintao” (AcG), “Mini Amethyst”(AaP), and “Kuilv” (AaG). b Global results of flavonoid metabolic profiling. The bar chart at the left bottom shows the number of flavonoids in each sample and the number of each kind of flavonoids. The bar chart on the right shows the number of the same kind of flavonoids distributed in different samples
Results and Discussion
The Flavonoid Metabolic Profiling of Four Kiwi Cultivars
To profile the flavonoid metabolites and evaluate the nutritional values of the four kiwi cultivars with different flesh colors, a newly developed LC-MS-based, widely targeted metabolomics method was employed to identify and relatively quantify the flavonoid metabolites.
A total of 125 flavonoids, including 9 anthocyanins, 12 catechins, 17 flavanones, 48 flavones (including 14 flavone C-glycosides), 29 flavonols, 6 isoflavones, and 4 proanthocyanidins, were identified in the fruits of the four cultivars (Fig. 1b; Table S1); this represents the broadest characterization of the flavonoids in kiwi species to our knowledge. Because of the complex glycosylation pattern of each individual flavonoid, previous reports did not identify modified flavonoids (Liu et al. 2018; Li et al. 2018b), or only identified limited numbers of modified flavonoids (Wojdylo and Nowicka 2019; Wojdylo et al. 2017) or only identified anthocyanin derivatives (Montefiori et al. 2005; Montefiori et al. 2009). Of the 125 flavonoids in our study, 103 were identified for the first time in kiwi species, mainly in glycosylated forms such as 3-O, 5-O, and 7-O glycosylation. Among the four cultivars, AaP showed the greatest number of flavonoids (111) among the four cultivars, followed by AcH (109), AaG (99), and AcG (92) (Fig. 1b), indicating a more abundant flavonoid component in pigmented kiwi than that in unpigmented ones. In addition, 19 flavonols were shared by the four cultivars (19), followed by flavones (18) and flavone C-glycosides (12) (Fig. 1b). Focusing on the species level, the two kiwiberry cultivars specifically shared five flavonols (quercetin 5-O-malonylhexosyl-hexoside, quercetin 7-O-malonylhexosyl-hexoside, isorhamnetin O-acetyl-hexoside, quercetin O-acetyl-hexoside, and myricetin 3-O-galactoside) and three flavones (apigenin O-hexosyl-O-pentoside, tricin O-glucuronic acid, and tricin 7-O-hexoside). In the two A. chinensis cultivars, two isoflavones (glycitin and 2′-hydroxydaidzein), one flavone (tricin) and one flavonol (kaempferin) were detected specifically (Table S1).
Intriguingly, all four cultivars shared four anthocyanidins (cyanidin 3-O-glucoside, malvidin 3-O-galactoside, malvidin 3-O-glucoside, and cyanidin 3-O-rutinoside) (Fig. 1b; Table S1), indicating that the unpigmented kiwifruit/berry can also synthesize anthocyanins and suggesting that their accumulation may be limited, which is in accordance with a previous literature (Li et al. 2018b). Three anthocyanins (delphinidin 3-O-glucoside, cyanidin O-syringic acid, and pelargonidin 3-O-beta-D-glucoside) were only detected in red-fleshed AcH and purple-fleshed AaP (Fig. 1b; Table S1), suggesting a key role in the fruit pigmentation of these kiwi species. This result is in agreement with previous reports that cyanidin derivatives have been found in all Actinidia species that accumulate anthocyanins (Montefiori et al. 2009). However, delphinidin 3-O-glucoside was detected both in AcH and AaP (Fig. 1b; Table S1), which differs from the results of a previous study that reported that delphinidin derivatives only existed in A. melanandra and A. arguta var. purpurea (Montefiori et al. 2009). In addition, pelargonidin 3-O-beta-D-glucoside was detected in AcH and AaP (Table S1), which have not been reported in the genus Actinidia. In addition, cyanidin O-acetylhexoside was only detected in AaP (Table S1), suggesting its contribution to the red color of AaP. Collectively, the above results not only indicate that kiwifruit and kiwiberry are abundant in flavonoid components but also suggest a wider range of nutritional flavonoid-derived than anthocyanin-derived nutritional compounds.
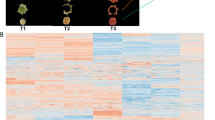
To further assess the differences in metabolic profiles among the four cultivars, hierarchical cluster analysis (HCA) was performed on the above profiles. As shown in Fig. 2a, the metabolite profiles fall into four main clusters: clusters I, II, III, and IV represent the highly accumulated flavonoids in AaG, AaP, AcG, and AcH, respectively. In addition, principal component analysis (PCA) was performed on the relative content of flavonoids in the four cultivars to address the internal structure of flavonoid variation. As shown in Fig. 2b, quality control (QC) samples (“mix01,” “mix02,” and “mix03”) grouped closely together, indicating the integrity and accuracy of the following analysis. The two main PCs (PC1 and PC2) accounted for 37.13% and 24.75% of the total system variability, respectively, and the four cultivars grouped into three main groups. AcH (red fleshed) and AaP (purple fleshed) were distinguished from the other three, whereas AcG and AaG clustered fairly close together. These results suggest that the nature and the variation in flavonoids in both species may contribute to the pigmentation of AcH and AaP. However, the variation in flavonoid profiles did not clearly separate between AcG and AaG but did clearly separate between AcH and AaP, suggesting that distinct genetic mechanisms underlie the metabolite profiles of AcH and AaP.

Multivariate statistics of flavonoids in four cultivars. a Heat map visualization. Each metabolite is represented by a single row. Red indicates relatively high abundance, whereas metabolites with relatively low abundance are shown in green (color key scale right of the heat map). The content of each metabolite comes from the average content of three biological replicates. b Score plots for principle components 1 and 2. “Mix” corresponds to the quality control sample. The same color represents three biological replications
To further assess the differential flavonoid metabolites that induced variation in metabolite profiles, partial least squares discriminant analysis (PLS-DA) was performed on the flavonoid profiles between AcH and AcG, AaP and AaG, AcH and AaP, and AcG and AaG. As shown in Fig. 3a, high predictability (Q2) and strong goodness of fit (R2X, R2Y) of the PLS-DA models were observed in the comparisons between AcH and AcG (Q2 = 0.994, R2X = 0.845, R2Y = 0.999), between AaG and AaP (Q2 = 0.995, R2X = 0.9, R2Y = 0.999), between AcH and AaP (Q2 = 0.998, R2X = 0.944, R2Y = 1), and between AcG and AaG (Q2 = 0.986, R2X = 0.846, R2Y = 997). Screening of differential metabolites was performed based on fold change (FC) analyses, and variables determined to be important in the projection scores (VIP) were identified (VIP ≥ 1.0 and fold change ≥ 2 or ≤ 0.5 were set as thresholds). As shown in Fig. 3b, between AcH and AcG, a total of 39 differential metabolites (including 6 anthocyanins, 2 catechins, 10 flavanones, 8 flavones, 2 flavone C-glycosides, 9 flavonols, and 2 isoflavones) showed significantly different content, 38 of which showed higher content in AcH than that in AcG, and only syringetin showed higher content in AcG, suggesting a superior nutritional value of AcH in terms of flavonoids (Table 1). According to the previous reports, anthocyanins have antioxidant, angiocardiopathy, and cancer prevention capacities and are recognized as natural health ingredients with beneficial effects on humans (Pojer et al. 2013). In our study, anthocyanin content varied greatly between AcH and AcG. For example, delphinidin 3-O-glucoside, cyanidin O-syringic acid, and pelargonidin 3-O-beta-D-glucoside were only detected in AcH, and the contents of cyanidin 3-O-glucoside, cyanidin 3-O-rutinoside, and pelargonidin 3-O-beta-D-glucoside were 1753-, 9-, and 371-fold higher in AcH than those in AcG (Table 1). These results agree with the finding that cyanidins are the predominant pigment in kiwifruit with red colors (Montefiori et al. 2009; Liu et al. 2018).

PLS-DA model and volcano plot. a PLS-DA model plots and loading plots. The same color represents three biological replications. b Volcano plot of the identified flavonoids. The DEGs are shown in red and green, while black indicates genes that were not differentially expressed
For kiwiberry, 39 differential metabolites were identified between AaP and AaG. Among them, 31 showed higher content in AaP (6 anthocyanins, 5 catechins, 1 flavanone, 6 flavones, 4 flavone C-glycosides, 8 flavonols, and 1 isoflavone), whereas 8 showed higher content in AaG (1 flavanone, 6 flavones, and 1 flavone C-glycosides) (Table 2), suggesting a more complicated regulation of metabolic flux in kiwiberry compared with that in kiwifruit. Among the six differentially accumulated anthocyanins, the content of cyanidin 3-O-glucoside in AaP was 1358-fold higher that that present in AaG, and the other five anthocyanins (delphinidin 3-O-glucoside, cyanidin O-syringic acid, cyanidin O-acetylhexoside, cyanidin 3,5-O-diglucoside, and pelargonidin 3-O-beta-D-glucoside) were not detected in AaG (Table 2). Collectively, the content level of flavonoids was higher in pigmented kiwifruit/berry than that in unpigmented kiwifruit/berry either in kind or in quantity, which suggests that these cultivars may differ in the expression of flavonoid biosynthetic or regulatory gene expression.
Between AaP and AcH, 43 differential metabolites were identified. In addition, 21 of them (2 catechins, 3 flavanones, 7 flavones, 3 flavone C-glycosides, 6 flavonols, and 1 isoflavone) showed higher content in AaP, whereas 22 (1 anthocyanin, 6 flavanones, 8 flavones, 1 flavone C-glycoside, 2 flavonols, and 3 isoflavones) showed higher content in AcH (Table S1). Between AcG and AaG, 37 metabolites (1 anthocyanin, 3 catechins, 5 flavanones, 12 flavones, 2 flavones C-glycosides, 11 flavonols, and 3 isoflavones) showed significantly differential content, among which 13 showed higher and 24 showed lower content in AcG (Table S1). These results indicate a great difference in flavonoid composition in pigmented kiwifruit/kiwiberry and unpigmented kiwifruit/kiwiberry, suggesting the distinct flavonoid synthesis and regulation mechanism of A. chinensis and A. arguta.
Transcriptome Analysis and Candidate Genes Involved in Anthocyanin Accumulation of Red-Fleshed Kiwifruit
To investigate the molecular mechanisms underlying pigmentation in red-fleshed kiwifruit, cDNA libraries of AcH and AcG were subjected to RNA-seq based on the Illumina HiSeq 2000 platform, and a total of ~ 43.07 Gb clean data was obtained. These clean reads were mapped to the “Red5 kiwifruit genome” (Pilkington et al. 2018) with 72.13–78.09% of mapping rates and 90.11–91.16% of Q30 (Table 3). For differential expression analysis, a total of 9641 genes showed differential expression between AcH and AcG, and 4916 of them showed higher expression in AcH and 4725 showed lower expression (Table S2). Gene ontology analysis (GO) categorized all the differentially expressed genes (DEGs) into 51 functional groups, including 20 groups in “Biological processes,” 15 groups in “Cellular components,” and 16 groups in “Molecular functions” (Fig. S1a.). “Metabolic process,” “Cell part,” and “Catalytic activity” were the most abundant terms in “Biological processes,” “Cellular components,” and “Molecular functions,” respectively. To further understand the enriched pathways of these DEGs, KEGG analysis was performed using BLASTX. As shown in Fig. S1b, “Metabolism” was the most enriched category, followed by “Genetic information processing”. In the present study, we were especially interested in the anthocyanin accumulation of AcH. Thus, we focused on the 37 DEGs enriched in “Phenylpropanoid biosynthesis (ko00940)” and the 17 DEGs enriched in “Flavonoid metabolism (ko00941),” which may possibly be involved in fruit pigmentation. Among these DEGs, two 4CL (Acc23502 and Acc24655), one CHI (Acc03848), one F3H (Acc01070), two F3′H (Acc18331 and Acc25698), one F3′5′H (Acc28896), one DFR (Acc01005), four 3GT (Acc15351, Acc21776, Acc26546, and Acc26544), four 5GT (Acc30748, Acc20257, Acc20256, and Acc01677), three FLS (Acc26709, Acc05091, and Acc24372), one ANR (Acc17426), and one HCT (Acc28989) gene were remarkably upregulated in AcH. In contrast, three PAL (Acc08481, Acc08530, and Acc20944), one C4H (Acc00871), two CHS (Acc08970 and Acc24927), three F3H (Acc11906, Acc03241, and Acc02579), one F3′H (Acc00389), one 3GT (Acc06090), one FLS (Acc00668), two COMT (Acc11227 and Acc07400), and two HCT (Acc17914 and Acc29568) genes were downregulated in AcH (Fig. 4; Table S2).

Schematic representation of the phenylpropanoid/flavonoid pathway. This pathway was constructed based on the KEGG pathway and literary references. And the heat map visualization of the mean expression levels of biosynthetic genes for A. arguta and A. chinensis, respectively. The content of each metabolite was normalized (log2(FPKM + 1)) to complete linkage hierarchical clustering. Each example is visualized in a single column, and each metabolite is represented by a single row. The color key scale is beside the heat map. PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumaroyl:CoA-ligase; CHI, chalcone isomerase; CHS, chalcone synthase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-monooxygenase; FLS, flavonol synthase; DFR, dihydroflavonol-4-reductase; ANR, anthocyanidin reductase; ANS, anthocyanidin synthase; LAR, leucoanthocyanidin reductase; 3GT, anthocyanidin 3-O-glucosyltransferase; 5GT, anthocyanidin 5,3-O-glucosyltransferase; COMT, caffeoyl-CoA O-methyltransferase; HCT, hydroxycinnamoyl-CoA shikimate
Among these DEGs involved in anthocyanin biosynthesis, F3H, F3′H, DFR, and UFGT showed significantly upregulated expression in AcH. Combined with the metabolome results, although cyanidins are the major anthocyanin category in red-fleshed kiwifruit, only the contents of dihydrokaempferol and the final cyanins were significantly higher in AcH than those in AcG (Table S1). Since F3H catalyzes the conversion of naringenin to dihydrokaempferol and UFGT glycosylates anthocyanidins, these results suggest a regulatory scenario: the upregulated expression of F3H promotes the accumulation of dihydrokaempferol, providing sufficient substrates for the biosynthesis of downstream metabolites, and UFGT increases the hydrophilicity and stability of cyanidins, mediating the flux of flavonoid intermediates towards anthocyanin biosynthesis. Thus, we speculate that F3H and UFGT perform key functions in the pigmentation of red-fleshed kiwifruit. In addition, F3′5′H showed low expression levels in AcH and no expression in AcG (Fig. 4; Table S2). To our knowledge of anthocyanin biosynthesis, dihydrokaempferol would be hydroxylated by F3′H to produce dihydroquercetin leading to the accumulation of cyanidins, which are responsible for red color (Montefiori et al. 2009); alternatively, dihydrokaempferol could be hydroxylated by F3′5′H to produce dihydromyricetin leading to accumulation of delphinidins, which are responsible for the purple color (Dixon et al. 2013). Since delphinidin 3-O-glucoside was also detected in AcH, and F3′H showed a higher expression level than F3′5′H in AcH, we speculate that F3′H also plays an important role in cyanidin accumulation in AcH by competing with F3′5′H. In addition, DFR and FLS showed higher expression in AcH, and flavonols and anthocyanins also showed higher content in AcH (Table 1). Coupled with the fact that FLS and DFR compete for common substrates to direct biosynthesis to flavonols or anthocyanins, we speculate that the anthocyanin accumulation of AcH is also influenced by DFR.
In addition, it has been proven that transcription factors (TF), such as R2R3-MYB, basic helix-loop-helix (bHLH), and WD40, are involved in flavonoid biosynthesis through regulating the transcription of the above structural genes (Zhao et al. 2013; Lin-Wang et al. 2010). In our transcriptome dataset, 82 MYB TFs were identified in the DEGs (Table S2), and nine of the TFs belonged to the R2R3-MYB subfamily. Among the nine R2R3-MYB, five (Acc29865, Acc00493, Acc07258, Acc10925, and Acc02217) showed upregulated expression in AcH, and Acc00493 (AcMYB10) showed the greatest fold change (~ 32-fold) between AcH and AcG, suggesting that this gene regulates the activity of the above structural genes involved in anthocyanin biosynthesis (F3H, F3′H, DFR, and UFGTs). In addition, 47 bHLH TFs showed markedly different expression between AcH and AcG; 11 of these TFs were MYC-like bHLH TFs, and eight of them showed higher expression levels in AcH (Table S2). Intriguingly, one bHLH5 (Acc19563) was reported to be the partner protein of AcMYB10 and AcMYB110 (Brendolise et al. 2017), which play an important role in the regulation of anthocyanin synthesis. In addition, thirteen WD40 TFs were also differentially expressed between AcH and AcG (Table S2).
To further confirm the results of the transcriptomic analysis, the expression of the 16 structure genes, 4 MYBs, and 7 bHLHs was measured by qRT-PCR in the two kiwifruit cultivars. The qRT-PCR results showed that most of the differences in expression were consistent with the expression levels obtained by RNA-seq (Fig. 5). The qRT-PCR data for 5GT (Acc20256) and FLS (Acc24372), however, did not differ significantly between AcH and AcG. As expected, the expression levels of F3H (Acc01070), F3′H (Acc25698), DFR (Acc01005), and 3GT (Acc15351, Acc21776, Acc26546, and Acc26544) were ~ 5.2-, ~ 28.8-, ~ 3.8-fold, and > 15-fold higher in AcH than those in AcG (Fig. 5), respectively, and the expression levels of AcMYB10 (Acc00493) and bHLH5 (Acc19563) were ~ 7.9- and ~ 15.8-fold higher in AcH than those in AaG, respectively. The above results indicated the reliability of the transcriptomic analysis and suggested the involvement of these candidate genes in anthocyanin biosynthesis.

Real-time RT-PCR was used to validate the transcript levels of 27 selected DEGs from AcH and AcG. Values are means ± SDs of three independent biological replicates. Statistical significance: *P < 0.05, **P < 0.01
Transcriptome Analysis and Candidate Genes Involved in Anthocyanin Accumulation of Purple-Fleshed Kiwiberry
A total of 44.11 Gb clean data was obtained from the RNA-seq libraries prepared from AaP and AaG, with 89.70–90.81% of Q30 rate (Table 4). Since the mapping rate of A. arguta reads was not satisfactorily high (~ 20% to the two A. chinensis kiwifruit genomes (Huang et al. 2013; Pilkington et al. 2018)), we de novo assembled A. arguta transcripts. A total of 129,537 unigenes were assembled with an N50 of 861. Then, all the assembled unigenes were annotated by public databases (Nr, Nt, Swiss-Prot, GO, and KEGG), and a total of 35,407 unigenes were annotated by using BLAST (E-value ≤ 1e-5) and HMMER (E-value ≤ 1e-10). A total of 7686 DEGs were detected, with 3721 upregulated and 3965 downregulated in AaP (Table S4). These DEGs were then subjected to GO analysis, and all the DEGs were classified into 54 functional groups, including 20 groups in “Biological processes,” 17 groups in “Cellular components,” and 17 in “Molecular functions.” Among them, “Metabolic process,” “Cell,” and “Binding” were the most abundant terms under the above three categories (Fig. S1c). KEGG analysis revealed that “Metabolism” was also the most enriched category, followed by “Genetic information processing” (Fig. S1d). There were 29 DEGs enriched in “Phenylpropanoid biosynthesis (ko00940)” and 14 DEGs enriched in “Flavonoid metabolism (ko00941).” Almost all the structural genes in anthocyanin biosynthesis showed higher expression in AaP, such as one PAL, one C4H, two 4CLs, two CHSs, two CHIs, one F3H, three F3′H, one DFR, one LDOX (ANS), two 3GTs, two 5GTs, one FLS, one COMT, one ANR, one LAR, and one HCT gene (Fig. 4; Table S3).
In the flavonoid biosynthesis network, naringenin acts as a junction point: it can be either catalyzed by F3H to produce dihydrokaempferol leading to anthocyanin biosynthesis or catalyzed by flavone synthase (FNS) to produce flavone derivatives (Fig. 4) (Zhang et al. 2014). It is worth noting that F3H (c96449.g0) showed ~ 600-fold higher expression in AaP than that in AaG; the metabolites downstream of dihydrokaempferol, especially anthocyanidin derivatives, showed higher accumulation in AaP, whereas one flavanone and seven flavone derivatives showed higher accumulation in AaG (Table 2). The above results indicate that more naringenins were allocated to the synthesis of flavone and flavanone in AaG, whereas more naringenins were allocated to synthesis of dihydrokaempferol and anthocyanins in AaP, suggesting that F3H (c96449.g0) can switch the metabolic flux towards anthocyanin biosynthesis in AaP. On the other hand, in our metabolite profile, delphinidin 3-O-glucoside showed the highest content in AaP among all four cultivars. Delphinidin has been reported to be responsible for purple color in the plant kingdom (Dixon et al. 2013). In our transcriptomic dataset, the expression of F3′5′H (c109457.g0) in AaP was higher than that in AaG, indicating its contribution to the unique purple color of AaP. In addition, the unigene (c77921.g0) encoding a protein that is highly homologous with the reported AcF3GT1 and AcF3GT1 has been proven to be a key enzyme that regulates the accumulation of anthocyanin in red-fleshed kiwifruit cultivars (Montefiori et al. 2011). This gene (c77921.g0) showed ~ 270-fold higher expression in AaP than that in AaG. Thus, we designate c77921.g0 as AaF3GT and speculate that it plays an important role in the anthocyanin biosynthesis of A. arguta.
Furthermore, 54 MYB TFs were differentially expressed between AaP and AaG, and 32 of them were upregulated in AaP (Table S3), of which 10 belong to R2R3 MYB-like genes. Thirty-one bHLHs showed differential expression between AaP and AaG, and four of them (c115933.g0, c103501.g2, c110375.g0, and c115920.g0) were MYC-like bHLH TFs and showed higher expression in AaP (Table S3). Only one WD40 was upregulated in AaP (Table S3). It is worth noting that one of these, R2R3-MYB, AaMYB (c94087.g0), showed an ~ 300-fold higher expression in AaP than that in AaG. AaMYB (c94087.g0) encodes a protein that is highly homologous to AcMYB110, which has been reported to regulate anthocyanin production in the petals of A. chinensis and A. eriantha (Fraser et al. 2013). Thus, we designated this gene as AaMYB110, and we speculate that it plays an analogous role in the anthocyanin accumulation of A. arguta.
To confirm the results of the transcriptomic analysis, 25 candidate genes were selected from DEGs and were subjected to qRT-PCR. The expression trend was representative of all the selected genes (except DFR), and these findings were consistent with the transcriptomic analysis results (Fig. 6). As expected, AaF3H, AaF3GT, and AaMYB110 showed ~ 148-, ~ 308-, and ~ 1418-fold higher expression in AaP than that in AaG, respectively.

RT-PCR was used to validate the transcript levels of 25 selected DEGs of AaP and AaG. Values are means ± SDs of three independent biological replicates. Statistical significance: *P < 0.05, **P < 0.01
Conclusion
We identified 125 flavonoid metabolites in A. chinensis “Hongyang” (AcH) and “Jintao” (AcG), purple A. arguta accession “Mini Amethyst” (AaP), and A. arguta “Kuilv” (AaG); this study represents the broadest characterization of the flavonoids in kiwi species to date. Combined with flavonoid metabolic profiles, transcriptome analysis screened out candidate genes, AcF3H, AcF3′H, AcDFR, AcF3GT, AcMYB10, and bHLH5, which may be responsible for the anthocyanin accumulation in “Hongyang,” and AaF3H, AaF3GT, and AaMYB110, which may be responsible for anthocyanin accumulation in A. arguta “Mini Amethyst.” These findings improved our understanding of the flavonoid metabolism and pigmentation mechanism in kiwi species and laid the foundation for related gene function studies in the future.
Materials and Methods
Plant Materials
The red-fleshed A. chinensis “Hongyang” (AcH) and “Jintao” (AcG), purple A. arguta accession “Mini Amethyst” (AaP), and A. arguta “Kuilv” (AaG) were collected from the germplasm field of Wuhan Botanical Garden. Three vines of each accession were treated as three biological replicates, and 15–20 fruits were sampled from each vine. All the fruits were sampled at a ripe-for-eating stage. The endocarp of A. chinensis (AcH and AcG) and the flesh of A. arguta (AaP and AaG) were sampled. The fleshes of sampled fruits were first cut into pieces, immediately frozen in liquid nitrogen, and then stored at − 80 °C until RNA and metabolite extraction.
Metabolite Extraction and Identification
The freeze-dried samples were crushed to a uniform powder using a mixer mill (1.5 min, 30 Hz; MM 400, Retsch). A measure (100 mg) of powder from each sample was suspended in 1.0 mL 70% aqueous methanol, kept at 4 °C for 12 h in the dark, and centrifuged at 10,000 rpm for 10 min at 4 °C. Finally, the supernatant solution was absorbed (CNWBOND Carbon-GCB SPE Cartridge, 250 mg, 3 mL; ANPEL, Shanghai, China) and filtered (SCAA-104, 0.22-μm pore size; ANPEL) before LC-MS analysis. LC-MS analysis was performed on an LC-ESI-MS/MS system (HPLC, Shim-pack UFLC SHIMADZU CBM30A system; MS, Applied Biosystems 4500 Q TRAP). Five microliters of the extracted solution was injected into an HPLC system (Shimpack UFLC SHIMADZU CBM30A) equipped with a C18 column (Waters ACQUITY UPLC HSS T3, 1.8 μm, 2.1 mm × 100 mm). The binary solvent system was ultrapure water containing 0.04% acetic acid as the mobile phase A and acetonitrile containing 0.04% acetic acid as the mobile phase B. The A:B (v/v) gradient was 95:5 at 0 min, 5:95 at 11.0 min, 5:95 at 12.0 min, 95:5 at 12.1 min, and 95:5 at 15.0 min. The flow rate was kept at 0.40 mL/min, and the column temperature was maintained at 40 °C.
The HPLC effluent was connected to an electrospray ionization (ESI)-triple quadrupole-linear ion trap-MS/MS system (Applied Biosystems 4500 Q TRAP). The ESI source operation parameters were as follows: ion source, turbo spray; source temperature, 550 °C; ion spray voltage (IS), 5500 V; ion source gas I (GSI), gas II (GSII), and curtain gas (CUR) were set at 55, 60, and 25.0 psi, respectively; and collision gas (CAD) was high. Instrument tuning and mass calibration were performed with 10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. The monitoring mode was set to multiple reaction monitoring (MRM). Analyst 1.6.1 was used to process the mass spectrometry data. Peak area integration was performed on all mass spectral peaks. The area of each chromatographic peak (Area) represented the relative amount of the corresponding substance. Integral calibration was performed for the peaks in the mass spectrum of the same metabolite in different samples.
Multivariate Statistical Analysis and Targeted Metabolite Identification
Qualitative analysis of primary and secondary MS data was carried out by comparing the accurate precursor ions (Q1), product ions (Q3), retention time (RT), and fragmentation patterns with those obtained by the injection of standards under the same conditions if the standards were available (Sigma-Aldrich, USA; http://www.sigmaaldrich.com/united-states.html), or by a self-compiled database MWDB (MetWare biological science and Technology Co., Ltd. Wuhan, China) and publicly available metabolite databases if the standards were unavailable. PCA and orthogonal partial least-squares discriminant analysis OPLS-DA with data from 6 samples (two cultivars × three biological replicates) were performed to evaluate the differences in metabolite composition. The reliability correlation (p(corr)) values of all metabolites from the S-plot of the OPLS-DA were obtained using the first component. The metabolites satisfying the following criteria were selected as potential markers: fold change ≥ 2 or ≤ 0.5 and VIP ≥ 1 (variable importance in project (VIP) of the OPLS-DA model). The metabolite content data were normalized using the range method, and the clustering analysis of metabolites between different samples was performed with R software. Molecular formulas and assignments of the compounds to flavonoid pathways were established through KEGG (Kyoto Encyclopedia of Genes and Genomes) (http://www.genome.jp/kegg).
RNA Extraction, Library Construction, and Illumina Sequencing
A 100 mg frozen sample was ground to a fine powder in liquid nitrogen. Then, the total RNA was extracted using TRIzol reagents (Invitrogen, USA). Each RNA sample was subjected to DNase digestion (TaKaRa, Dalian, China) to remove the remaining DNA. RNA degradation and contamination were monitored on 1% agarose gels. The purity was checked using a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). The concentration was measured using a Qubit® RNA Assay Kit with a Qubit®2.0 Fluorometer (Life Technologies, CA, USA), and its integrity was assessed using the RNA Nano 6000 Assay kit with the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
Three micrograms of total RNA from each sample was prepared for construction of a sequencing library. Sequencing libraries were generated using NEBNext®Ultra™ RNA Library Prep kit for Illumina® (NEB, USA), and index codes were added to attribute sequences of each sample. All procedures for cDNA library construction were performed following the standard Illumina sample preparation protocol. The library quality was assessed on the Agilent Bioanalyzer 2100 system. The library preparations were sequenced on an Illumina HiSeq 2000 platform, and the paired-end reads were generated.
De Novo Transcriptome Assembly and Functional Annotation
Adapter-containing, ploy-N, or low-quality reads of raw data were removed by in-house Perl scripts to generate clean data. Q20, Q30, and GC content and sequence duplication level were calculated based on the clean data. Then, transcriptome assembly was accomplished using Trinity. Gene function was annotated based on NR (NCBI nonredundant protein sequences), Pfam (protein family), KOG/COG/eggNOG (Clusters of Orthologous Groups of proteins), Swiss-Prot (a manually annotated and reviewed protein sequence database), and KEGG and GO (Gene Ontology) databases. The MEME 5.0.2 online program (http://meme-suite.org/) was used for motif identification of the annotated MYB protein sequences.
Screening and Enrichment of Differentially Expressed Genes
The gene expression levels of each sample were estimated by RSEM. Differential expression analysis of each of the two sample groups was performed using DESeq. Genes with an adjusted P value of 0.05 and abs (log2 (fold change)) of 1 found by DESeq were designated as differentially expressed genes (DEGs). Gene Ontology (GO) enrichment analysis of the DEGs was implemented by the topGO R-packages based on the Kolmogorov-Smirnov test. KOBAS software was used to test the statistical enrichment of differentially expressed genes in the KEGG pathways.
Validation of RNA-seq Results by Quantitative Real-time PCR
Genes related to anthocyanin biosynthesis were selected from the DEG results. The primers used in RT-PCR are shown in Table S4. RNA was extracted from samples transformed using HiPure Plant RNA kits (Magen, R4151-03). The cDNA libraries were constructed by using HiScript II QRT SuperMix for qPCR (Vazyme, R223-01) and were then used as the input for quantitative PCR (qPCR) experiments. The RT-PCR was performed on a Roche (Indianapolis, IN, USA) LightCycler®480, following the manufacturer’s instructions for LightCycler®480 SYBR Green I Master mix (Vazyme). The reactions were carried out with the following cycling program: 95 °C for 30 s, followed by 45 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 20 s. Melt-curve analyses were carried out as follows: 15 s at 95 °C, 1 min at 60 °C, 30 s at 95 °C, and 15 s at 60 °C. Each sample was amplified in triplicate, and Roche LightCycler 480 software version 1.5.1.62 was used to perform the data analysis. The 2−ΔΔCt method was employed with Achn107181 (kiwifruit actin gene) as an endogenous control (Petriccione et al. 2015).
Abbreviations
- FC:
-
Fold change
- VIP:
-
Variables determined to be important in the projection scores
- HCA:
-
Hierarchical cluster analysis
- PCA:
-
Principal component analysis
- PLS-DA:
-
Partial least squares discriminant analysis
- FPKM:
-
Fragments per kilobase of transcript per million mapped reads
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PAL:
-
Phenylalanine ammonia-lyase
- C4H:
-
Cinnamate 4-hydroxylase
- 4CL:
-
4-Coumaroyl:CoA-ligase
- CHS:
-
Chalcone synthase
- CHI:
-
Chalcone isomerase
- F3H:
-
Flavanone 3-hydroxylase
- F3′H:
-
Flavonoid 3′-monooxygenase
- FLS:
-
Flavonols synthase
- DFR:
-
Dihydroflavonol-4-reductase
- ANR:
-
Anthocyanidin reductase
- ANS:
-
Anthocyanidin synthase
- LAR:
-
Leucoanthocyanidin reductase
- 3GT:
-
Anthocyanidin 3-O-glucosyltransferase
- 5GT:
-
Anthocyanidin 5,3-O-glucosyltransferase
- COMT:
-
Caffeoyl-CoA O-methyltransferase
- HCT:
-
Hydroxycinnamoyl-CoA shikimate
- qRT-PCR:
-
Quantitative real-time PCR
References
Bai S, Sun Y, Qian M, Yang F, Ni J, Tao R, Li L, Shu Q, Zhang D, Teng Y (2017) Transcriptome analysis of bagging-treated red Chinese sand pear peels reveals light-responsive pathway functions in anthocyanin accumulation. Sci Rep 7
Brendolise C, Espley RV, Lin-Wang K, Laing W, Peng Y, McGhie T, Dejnoprat S, Tomes S, Hellens RP, Allan AC (2017) Multiple copies of a simple MYB-binding site confers trans-regulation by specific flavonoid-related R2R3 MYBs in diverse species. Front Plant Sci 8
Cao K, Ding T, Mao D, Zhu G, Fang W, Chen C, Wang X, Wang L (2018) Transcriptome analysis reveals novel genes involved in anthocyanin biosynthesis in the flesh of peach. Plant Physiol Biochem 123:94–102
Cho K, Cho K-S, Sohn H-B, Ha IJ, Hong S-Y, Lee H, Kim Y-M, Nam MH (2016) Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J Exp Bot 67(5):1519–1533
Crozier A, Jaganath IB, Clifford MN (2009) Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep 26(8):1001–1043
Dixon RA, Liu C, Jun JH (2013) Metabolic engineering of anthocyanins and condensed tannins in plants. Curr Opin Biotechnol 24(2):329–335
El-Sharkawy I, Liang D, Xu K (2015) Transcriptome analysis of an apple (Malus x domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J Exp Bot 66(22):7359–7376
Fraser LG, Seal AG, Montefiori M, McGhie TK, Tsang GK, Datson PM, Hilario E, Marsh HE, Dunn JK, Hellens RP, Davies KM, McNeilage MA, De Silva HN, Allan AC (2013) An R2R3 MYB transcription factor determines red petal colour in an Actinidia (kiwifruit) hybrid population. BMC Genomics 14
He J, Giusti MM (2010) Anthocyanins: natural colorants with health-promoting properties. In Annual Review of Food Science and Technology, Vol 1, Vol. 1, 163-187 (Eds M. P. Doyle and T. R. Klaenhammer)
Huang S, Ding J, Deng D, Tang W, Sun H, Liu D, Zhang L, Niu X, Zhang X, Meng M, Yu J, Liu J, Han Y, Shi W, Zhang D, Cao S, Wei Z, Cui Y, Xia Y, Zeng H, Bao K, Lin L, Min Y, Zhang H, Miao M, Tang X, Zhu Y, Sui Y, Li G, Sun H, Yue J, Sun J, Liu F, Zhou L, Lei L, Zheng X, Liu M, Huang L, Song J, Xu C, Li J, Ye K, Zhong S, Lu B-R, He G, Xiao F, Wang H-L, Zheng H, Fei Z, Liu Y (2013) Draft genome of the kiwifruit Actinidia chinensis. Nat Commun 4
Jaakola L (2013) New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci 18(9):477–483
Jaeger SR, Harker FR (2005) Consumer evaluation of novel kiwifruit: willingness-to-pay. J Sci Food Agric 85(15):2519–2526
Latocha P, Jankowski P (2011) Genotypic difference in postharvest characteristics of hardy kiwifruit (Actinidia arguta and its hybrids), as a new commercial crop part II. Consumer acceptability and its main drivers. Food Res Int 44(7):1946–1955
Li J, Li X, Soejarto D (2007) Actinidiaceae. Flora of China 12:334–360
Li C-F, Zhu Y, Yu Y, Zhao Q-Y, Wang S-J, Wang X-C, Yao M-Z, Luo D, Li X, Chen L, Yang Y-J (2015a) Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camellia sinensis). BMC Genomics 16
Li W, Liu Y, Zeng S, Xiao G, Wang G, Wang Y, Peng M, Huang H (2015b) Gene expression profiling of development and anthocyanin accumulation in kiwifruit (Actinidia chinensis) based on transcriptome sequencing (vol 10, e0136439, 2015). PLoS One 10(9)
Li Y, Fang J, Qi X, Lin M, Zhong Y, Sun L (2018a) A key structural gene, AaLDOX, is involved in anthocyanin biosynthesis in all red-fleshed kiwifruit (Actinidia arguta) based on transcriptome analysis. Gene 648:31–41
Li Y, Fang J, Qi X, Lin M, Zhong Y, Sun L, Cui W (2018b) Combined analysis of the fruit metabolome and transcriptome reveals candidate genes involved in flavonoid biosynthesis in Actinidia arguta. Int J Mol Sci 19(5)
Lin Y, Jiang L, Chen Q, Li Y, Zhang Y, Luo Y, Zhang Y, Sun B, Wang X, Tang H (2018) Comparative transcriptome profiling analysis of red- and white-fleshed strawberry (Fragaria x ananassa) provides new insight into the regulation of the anthocyanin pathway. Plant Cell Physiol 59(9):1844–1859
Lin-Wang K, Bolitho K, Grafton K, Kortstee A, Karunairetnam S, McGhie TK, Espley RV, Hellens RP, Allan AC (2010) An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol 10:50–50
Liu Y, Qi Y, Chen X, He H, Liu Z, Zhang Z, Ren Y, Ren X (2018) Phenolic compounds and antioxidant activity in red-and in green-fleshed kiwifruits. Food Res Int
Montefiori M, McGhie TK, Costa G, Ferguson AR (2005) Pigments in the fruit of red-fleshed kiwifruit (Actinidia chinensis and Actinidia deliciosa). J Agric Food Chem 53(24):9526–9530
Montefiori M, Comeskey DJ, Wohlers M, McGhie TK (2009) Characterization and quantification of anthocyanins in red kiwifruit (Actinidia spp.). J Agric Food Chem 57(15):6856–6861
Montefiori M, Espley RV, Stevenson D, Cooney J, Datson PM, Saiz A, Atkinson RG, Hellens RP, Allan AC (2011) Identification and characterisation of F3GT1 and F3GGT1, two glycosyltransferases responsible for anthocyanin biosynthesis in red-fleshed kiwifruit (Actinidia chinensis). Plant J 65(1):106–118
Nabavi SM, Samec D, Tomczyk M, Milella L, Russo D, Habtemariam S, Suntar I, Rastrelli L, Daglia M, Xiao J, Giampieri F, Battino M, Sobarzo-Sanchez E, Nabavi SF, Yousefi B, Jeandet P, Xu S, Shirooie S (2018) Flavonoid biosynthetic pathways in plants: versatile targets for metabolic engineering. Biotechnol Adv
Peng Y, Kui L-W, Cooney JM, Wang T, Espley RV, Allan AC (2019) Differential regulation of the anthocyanin profile in purple kiwifruit (Actinidia species). Hortic Res 6
Petriccione M, Mastrobuoni F, Zampella L, Scortichini M (2015) Reference gene selection for normalization of RT-qPCR gene expression data from Actinidia deliciosa leaves infected with Pseudomonas syringae pv actinidiae. Sci Rep 5
Pilkington SM, Crowhurst R, Hilario E, Nardozza S, Fraser L, Peng Y, Gunaseelan K, Simpson R, Tahir J, Deroles SC, Templeton K, Luo Z, Davy M, Cheng C, McNeilage M, Scaglione D, Liu Y, Zhang Q, Datson P, De Silva N, Gardiner SE, Bassett H, Chagne D, McCallum J, Dzierzon H, Deng C, Wang Y-Y, Barron L, Manako K, Bowen J, Foster TM, Erridge ZA, Tiffin H, Waite CN, Davies KM, Grierson EP, Laing WA, Kirk R, Chen X, Wood M, Montefiori M, Brummell DA, Schwinn KE, Catanach A, Fullerton C, Li D, Meiyalaghan S, Nieuwenhuizen N, Read N, Prakash R, Hunter D, Zhang H, McKenzie M, Knabel M, Harris A, Allan AC, Gleave A, Chen A, Janssen BJ, Plunkett B, Ampomah-Dwamena C, Voogd C, Leif D, Lafferty D, Souleyre EJF, Varkonyi-Gasic E, Gambi F, Hanley J, Yao J-L, Cheung J, David KM, Warren B, Marsh K, Snowden KC, Lin-Wang K, Brian L, Martinez-Sanchez M, Wang M, Ileperuma N, Macnee N, Campin R, McAtee P, Drummond RSM, Espley RV, Ireland HS, Wu R, Atkinson RG, Karunairetnam S, Bulley S, Chunkath S, Hanley Z, Storey R, Thrimawithana AH, Thomson S, David C, Testolin R, Huang H, Hellens RP, Schaffer RJ (2018) A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genomics 19
Pojer E, Mattivi F, Johnson D, Stockley CS (2013) The case for anthocyanin consumption to promote human health: a review. Compr Rev Food Sci Food Saf 12(5):483–508
Richardson DP, Ansell J, Drummond LN (2018) The nutritional and health attributes of kiwifruit: a review. Eur J Nutr 57(8):2659–2676
Stushnoff C, Ducreux LJM, Hancock RD, Hedley PE, Holm DG, McDougall GJ, McNicol JW, Morris J, Morris WL, Sungurtas JA, Verrall SR, Zuber T, Taylor MA (2010) Flavonoid profiling and transcriptome analysis reveals new gene-metabolite correlations in tubers of Solanum tuberosum L. J Exp Bot 61(4):1225–1238
Tohge T, de Souza LP, Fernie AR (2017) Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J Exp Bot 68(15):4013–4028
Wang Z, Cui Y, Vainstein A, Chen S, Ma H (2017) Regulation of fig (Ficus carica L) fruit color: metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front Plant Sci 8
Wang Y, Zhao C-l, Li J-Y, Liang Y-J, Yang R-Q, Liu J-Y, Ma Z, Wu L (2018) Evaluation of biochemical components and antioxidant capacity of different kiwifruit (Actinidia spp.) genotypes grown in China. Biotechnol Biotechnol Equip 32(3):558–565
Wang L, Tang W, Hu Y, Zhang Y, Sun J, Guo X, Lu H, Yang Y, Fang C, Niu X, Yue J, Fei Z, Liu Y (2019) A MYB/bHLH complex regulates tissue-specific anthocyanin biosynthesis in the inner pericarp of red-centered kiwifruit Actinidia chinensis cv. Hongyang. Plant J
Winkel-Shirley B (2001) Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 126(2):485–493
Wojdylo A, Nowicka P (2019) Anticholinergic effects of Actinidia arguta fruits and their polyphenol content determined by liquid chromatography-photodiode array detector-quadrupole/time of flight-mass spectrometry (LC-MS-PDA-Q/TOF). Food Chem 271:216–223
Wojdylo A, Nowicka P, Oszmianski J, Golis T (2017) Phytochemical compounds and biological effects of Actinidia fruits. J Funct Foods 30:194–202
Zhang Y, Butelli E, Martin C (2014) Engineering anthocyanin biosynthesis in plants. Curr Opin Plant Biol 19:81–90
Zhao L, Gao L, Wang H, Chen X, Wang Y, Yang H, Wei C, Wan X, Xia T (2013) The R2R3-MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis. Funct Integr Genomics 13(1):75–98
Acknowledgments
This work was supported by the Natural Science Foundation of China (NSFC), project numbers 31501745, 30671433, and 31171945.
Author information
Authors and Affiliations
Contributions
Yanchang Wang proposed the research topic and revised the article; Min Yu designed the study, analyzed and interpreted the data, and drafted the article. Rui Lei collected samples of A. arguta; Yuping Man and Xuemei Lu revised the article. All authors read and approved the manuscript for publication.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlight
• A total of 125 flavonoids were identified.
• Multivariate statistical analysis indicates the variation of flavonoid profiles contributes to the pigmentation phenotypes.
• Comparative transcriptomic analysis revealed that some structural genes and key transcription factors may be involved in the pigmentation of the red-fleshed A. chinensis and purple-fleshed A. arguta.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, M., Man, Y., Lei, R. et al. Metabolomics Study of Flavonoids and Anthocyanin-Related Gene Analysis in Kiwifruit (Actinidia chinensis) and Kiwiberry (Actinidia arguta). Plant Mol Biol Rep 38, 353–369 (2020). https://doi.org/10.1007/s11105-020-01200-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-020-01200-7