
Abstract
This paper describes a journey which started with the question: why do farmers reapply phosphate? This led to understanding how phosphate and other specifically sorbed ions, both cations and anions react with soil. Under most soil conditions, they do not precipitate to give specific compounds. Rather they are specifically adsorbed onto the surface of soil particles and then penetrate the particles via diffusion mechanisms. The pathways by which they move differ between ion species; for example nickel penetrates the lattice of iron oxides but phosphate penetrates defects between particles. Such reactions change important properties of the soil. Reaction with phosphate conveys negative charge to the reacting surface and so decreases the buffering capacity for phosphate; this makes subsequent phosphate applications more effective. Nor does the diffusive penetration continue unabated; as phosphate builds up, diffusive penetration slows and then virtually stops. These effects mean that on long-term fertilised soils, phosphate fertiliser is much more effective and annual doses (if they are needed at all) should be much smaller. I suggest that failure to appreciate this is the cause of phosphate losses from farmland and consequent pollution of water bodies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
I started doing research in 1954 when, as a new graduate, I was employed by CSIRO. Since then, I think I have learned quite a bit about the interface between soil chemistry and plant nutrition, specifically how increasing knowledge about soil chemistry can help understand plant nutrition and ultimately fertiliser practice. In this article I want to take you along part of that journey to show you how ideas slowly developed. This will therefore be a rather unusual format with, inevitably, quite a bit of self-citation. I hope you do not find it too self-indulgent and that you enjoy the journey.
I first started work on the northern tablelands area of New South Wales, Australia. This area is well removed from sources of sulfur such as the sea and from industry; sulfur deficiency was widespread and severely limiting. We knew even less about sulfur then than we do now and much of my early work was concerned with its availability. When I transferred to Western Australia in 1961, it was widely held that sulfur deficiency could not occur because superphosphate, which contains slightly more sulfur than phosphorus, was almost universally used. However, in the agricultural areas of south-western Australia, winter rainfall is high, and many of the soils have low water holding capacity. When superphosphate was applied in the autumn, its sulfate content was mostly removed by the winter rains, and sulfur deficiency occurred in spring. I showed that slowly-available sources of sulfur, such as elemental sulfur, were very effective. However, it was a hard sell; in Western Australia, P deficiency was so extreme that the effects of applying phosphate seemed magical; further, superphosphate was heavily subsidised at that time so it was quite cheap. Farmers were therefore reluctant to change their practices. If phosphate was king, better learn more about it. The purpose of this preamble is to explain that I did not commence work on phosphate until about 1970.
The first question I asked was: why do farmers reapply phosphate? The short answer is that phosphate effectiveness decreased with time and it is therefore necessary. That brings up the next question: why does effectiveness decrease with time? That is the main topic of the rest of this article.
The constraints of the terminology
The words we use to describe a phenomenon may limit our ability to appreciate it. This seems to have been the case with much phosphate research. Agronomists know that they have to reapply phosphate (at least in the early stages; see later); there must therefore be some sort of slow process reducing its effectiveness. Yet when chemists mix soil with phosphate solutions for say 24 h, they almost inevitably write that soil and phosphate were “equilibrated”. If they draw a graph using such results, the label is “equilibrium concentration”. They therefore exclude a slow process because of the terminology they use.
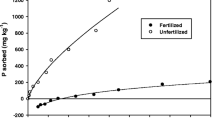
The vertical axes of such graphs are, almost always, labelled “adsorbed phosphate”. Adsorption is a reversible reaction; it does not go to completion; there is always some reactant left in solution; that is why such plots can be drawn. However there is a problem if desorption is also measured. In a typical experiment, after the initial step, the supernatant solution is removed and replaced with a solution containing no phosphate in order to induce desorption. The results are often as shown in Fig. 1; desorption does not follow the same track. The conclusion that adsorption is not reversible is, as shown above, not possible. The only possible conclusion is that something in addition to adsorption is involved. It is that “something” that causes the effectiveness of phosphate fertiliser to decrease with time and is also an important reason why farmers reapply phosphate fertilisers. That is why it is better to restrict the word “adsorption” to the situation when we really mean on the surface, but when we mean the whole process it is better to use the non-specific word “sorption”. Some further terminology; the extent to which the desorption curve and the sorption curve differ is usually called hysteresis and the amount of hysteresis is one measure of the slow process that follows adsorption. Hysteresis can be shown by simple adsorption/desorption experiments; soil is mixed for a specified period with solutions containing increasing concentrations of phosphate; from the change in concentration, sorption is calculated; the supernatant solution is then removed and replaced with a solution containing zero phosphate; after the same period, the increase in concentration is measured and desorption calculated. The usual experience is that the desorption curves do not follow the same track as sorption curves. The extent to which they differ is called hysteresis.

Results from a typical sorption-desorption experiment. Soil was mixed with 10 solutions containing different concentrations of phosphate then the supernatant solution was removed and replaced with a solution containing no P. Plots have been linearised by plotting against concentration raised to the indicated fractional power. The ratio of the slopes is 4.5; this is taken as a measure of the hysteresis. (Drawn using data also used by Barrow and Debnath (2014))
Method of measuring the rate of decline in effectiveness
How should we measure the decline in effectiveness with time? In other words, how do we measure the residual value of previously-applied fertiliser? The term “residual value” is sometimes used quite loosely; for example, it may be expressed as the number of years that may elapse before another dose is needed. That is not a very useful definition if we want to measure the rate of decline. Instead, I have measured the relative effectiveness. Suppose we designate the response to phosphate, whether as yield or as phosphate uptake, as “y” and that there are two sources of phosphate, x1 and x2. Then, if the Mitscherlich equation describes the results, we get:
where α, β, and γ are parameters.
This could also be written as:
where r is the effectiveness of x2 relative to that of x1. If x1 is freshly-added phosphate and x2 is phosphate added some time ago, then r measures the change in relative effectiveness that has occurred and thus the residual value. This approach can be made quite general and can be applied to any measure of the response and to any equation that describes it.
Measurements of the rate of decline in effectiveness
There have been several field experiments in which the decrease in effectiveness of phosphate over time has been measured. For example Arndt and Mcintyre (1963), Devine et al. (1968). However, for my purposes, field experiments have several disadvantages. Inevitably they take a long time; usually, in order to simulate practical agriculture some crops are grown and therefore the reaction, of itself, is not studied; they are less precise because of all the factors that can affect response; they are subject to the vagaries of the seasons; and there is no opportunity to investigate the effect of different treatments. I therefore decided to see whether the decrease in effectiveness could be studied using a pot trial. I applied superphosphate to a series of pots at levels designed to produce response curves. I started the first series in late spring and started a new series every four weeks or so. The pots were kept moist and stored in a potting shed. In late autumn I measured the response by growing plants. The results were a mess; they did not seem to make any sense – until I remembered that it is very hot in Perth in summer. There seemed to be a large effect of temperature. Nothing for it except to do it again this time with temperature controlled. It was not practical to keep a whole pot full of soil at constant temperature. Fertiliser was therefore applied to 100 g samples. I begged space in colleagues’ CT rooms, incubators and refrigerators and ultimately applied the fertilised soils as a layer in pots.
This time it worked beautifully (Fig. 2). The results showed me that the process was very slow; at 25° C decreasing relative effectiveness to 0.5 took about 80 days. This compares with chemical reactions which usually require seconds or minutes. Further, if rates had been first-order, a further halving would have taken a further 80 days, but extrapolating using the other temperatures showed that it would have taken 320 days. Instead, rates were proportional to a fractional power of time with the index term having a value of 0.476. There was also a large effect of temperature. On the one hand this suggests that some kind of physicochemical process is involved such that the molecules had to jump over a high-energy barrier. On the other hand, this result produced a useful tool for further work; you don’t have to wait several years to see the long-term effects; you can induce them at high temperature and a short period.

The effect of time and temperature of reaction on the relative effectiveness of phosphate for growth of subterranean clover (Trifolium subterraneum). The estimate of activation energy was 83.1 kJ mol− 1. This was used to calculate the rates at different temperatures. (Drawn from the data of Barrow (1974a))
These results were obtained using subterranean clover (Trifolium subterraneum). Would it have made a difference if some other plant were used? Plants differ in their ability to utilise phosphate; in terms of Eq. (2), they differ in the value of γ, but are there any plants that differ in their ability to utilise residual P? In terms of Eq. (2) do they differ in their value of r? I reviewed this aspect (Barrow 1980b) and concluded that there was no evidence for this. I also investigated it directly three times; the first occasion was a comparison of Wimmera ryegrass (Lolium rigidum) with subterranean clover (Barrow and Campbell 1972); the second was a comparison of plants with vesicular arbuscular mycorrhizas with the same plants without mycorrhizas (Barrow et al. 1977); and the third occasion was a comparison of the cluster-rooted Lupinus cosentinii with the non cluster-rooted L luteus, L. angustifolius and subterranean clover (Barrow and Mendoza 1990). In all cases, the first-mentioned was better able to utilise phosphate, but there no evidence of better access to residual P. In terms of Eq. (2), the values of r did not differ significantly. Hence, it seems that using a different plant would have made little difference. More importantly, it does not suggest any easy way to gain special access to residual phosphate.
Measurement of the rate of reaction
Growing plants is always the best way to measure the effects of treatments on availability. However, experiments such as the one described above take a lot of time and resources. To investigate, for example, differences between soils, it would be convenient to have methods that demanded fewer resources. There are two ways to measure the rate of any reaction; one is to measure the rate at which the product accumulates. I do not know how to measure the product and therefore this method is not possible. The other ways to measure the rate of decrease in concentration of the reactants, indicated in such cases by the concentration of phosphate in solution. This seemed better.
One way in which one might go about such an experiment is to mix the soil with a relatively large volume of solution, say 5 or 10 mL g− 1, and mix for various periods. There are several disadvantages with this approach. One is that most scientists use quite vigourous mixing. I do not understand why; it can hardly be because the reaction is rapid and therefore vigourous mixing is needed to keep the solution concentration uniform. For many soils, the mutual abrasion of particles exposes new surfaces and as a result, the rate of reaction is increased (Barrow 2020). In order to avoid this problem, when we used this method in other contexts, we mixed soil and solution using a machine which slowly rolled the containers. Another disadvantage is that it is difficult to use such methods if one wishes to measure the rates at different temperatures because space is limited to that available in incubators or refrigerators. A third disadvantage is that describing the rate at which concentration changes is somewhat complex because sorption and concentration both change during the course of the experiment. The changes in the solution concentration are therefore not only a consequence of the changes in period of reaction but also the changes in sorption (Barrow 2020).
The other method is to incubate soil with just sufficient phosphate solution to bring the soil to near field capacity so there might be say 0.2 to 0.3 mL g− 1. The reaction is therefore with soil in a more realistic state; little space is required; and only a small amount of the reactant is in the solution phase and decreases in the concentration have little effect on the amount sorbed. Changes in concentration are therefore measured at almost constant sorption, making the interpretation of the results much simpler. However, measuring the concentration provides some difficulties. We did not wish to simply extract the soil with some volume of solution because that would involve some desorption. Instead we used the “null-point” method. Explaining that method has sometimes provided complications and I therefore repeat verbatim the explanation from Barrow and Shaw (1975a). “After the indicated periods, 5 g subsamples of the incubated soils were brought to 25 °C. For each phosphate level, four subsamples were shaken for 15 min at 25 °C with 50 mL of solution which were 0.01 M with respect to calcium chloride and which contained phosphate concentrations on either side of the expected concentrations. The solution was separated by centrifuging and filtering and the phosphate concentration was measured. The concentration which would not have changed on shaking with the soil was interpolated. “.
I also investigated direct analysis of the soil solution (Barrow 1982). Samples of the moist soil were centrifuged with a dense immiscible liquid (tetrachloroethylene). The aqueous phase comes to the top and is separated using phase-separating paper. The pH is measured before the liquid is passed through a membrane filter before analysis. The ionic strength of the soil solution was lower than that of 0.01 M calcium chloride and the solution concentration of phosphate was consequently lower than that observed using the null-point method (Fig. 3). However, because phosphate was added as the potassium salt, the concentration of potassium in the soil solution increased with level of phosphate application and the slope of the line in Fig. 3 is therefore slightly steeper than that for the null-point method. When 1% gypsum (CaSO4.2H2O) was mixed with the soil, the calcium concentration in the soil solution was about 0.013 M, and the results were then comparable with those obtained using the null-point method (0.01 M). The rate of change with time was a similar for both methods.

Effects of three methods of measurement on the relationship between sorbed phosphate and solution concentration, for a soil that had been incubated with four levels of phosphate for one day at 60 °C. “Soil solution” indicates that the soil solution had been extracted using the immiscible displacement method. Adding gypsum (CaSO4.2H2O) to the soil increases the calcium concentration
Observations of the rate of reaction of phosphate with soils
We ran two experiments in which we measured differences between soils in the rate at which they reacted with phosphate; one experiment involved surface soils collected from the south-west of Western Australia, the other involved soils collected from a wide range of locations both within Australia and overseas. None of the soils had ever been fertilised with P and all were reported to be phosphate deficient. We could therefore assume there was little desorbable phosphate and no need for an intercept term in the equation used to describe the results. The equation used may be written as:
where S is the amount of phosphate sorption, c is the solution concentration, t is time (all in appropriate units), T is temperature (K), E is an activation energy, R is the gas constant, and the remaining symbols are parameters. This equation is a form of the Freundlich equation modified to include the effects of period of reaction and with a term to describe the effects of temperature on the rate. It may be rearranged with c as its subject, and after taking natural logs, an equation which is linear in its terms results.
For the Western Australian surface soils, the phosphate required to give a solution concentration of 1 mg L− 1 after one day ranged from 78 to 1565 mg kg− 1 soil. There was therefore a very large range in buffering capacity. The values for the activation energy ranged from 67.2 to 90.7 kJ mol− 1 with the highest value being for the same soil as used in the pot experiment (and for which the estimate from the pot trial was 83.1 kJ mol− 1). Given the inherent errors in estimating individual terms in regression equations this is sufficient correspondence to indicate that the same process was involved.
Soils of the south-west of Western Australia may be considered as the ultimate Ultisols. Many have not been rejuvenated either by glacial or volcanic activity since at least the Permian. Others have been partially rejuvenated as a result of uplift between 3 and 5 million years ago. Further, this area has the largest concentration of plants in the Proteaceae family anywhere in the world. The roots of members of this family periodically release organic acids, especially citric acid. These dissolve iron and aluminium oxides and their subsequent oxidation results in re-precipitation. Consequently particles of iron and aluminium oxides accumulate in soils (Pate et al. 2001) Soils differ in the amount of reacting material present but the material reacting is similar for all soils. There was, therefore, a much smaller range in the index term b1 (Fig. 4).

Values of the parameters b1 and b2 when the following equation was fitted to three groups of soils: S=α cb1 tb2where S is the amount of phosphate sorption, c is the solution concentration, t is time (all in appropriate units), and the remaining symbols are parameters. The parameter b1 describes the curvature in the concentration direction; the parameter b2 describes the curvature in the time direction – low values means that the reaction is slow. (Drawn from the data of Barrow (1980) and Barrow and Shaw (1975a))
It seemed important to explore the behaviour of soils from other regions. Thanks to the cooperation of colleagues, I obtained samples of 36 phosphate-deficient soils from New Zealand, Thailand, Alabama, California and Hawaii and from four widely-spaced locations in Australia. I also included three sub-soils from Western Australia. The only thing in common was that the soils were known to be phosphate deficient and that they had never been fertilised. The experiment is described in Barrow (1980a). There was now a much wider range in sorption and in the rate of sorption (Fig. 4). Importantly, there was an increased range in the parameter that describes the rate of reaction: b2. This parameter reflects the decline in effectiveness of phosphate fertiliser with time (Fig. 5). It is therefore an important soil property, strongly influencing fertiliser requirement and one would expect it to be regularly measured and its value incorporated into fertiliser policy. Sadly, this is not the case.

Relationship between soil properties as measured in the laboratory and the decline in effectiveness of phosphate with time. The meaning of the parameters b1 and b2 are as indicated in Fig. 4. The formula used on the horizontal axis was chosen on the basis that it would better reflect the effect of soil properties on the rate of diffusion of phosphate through the soil solution to the plant root. The curve fitted to the data has a value of unity when b2 equals zero because reaction between soil and phosphate would be instantaneous and thus no decline in effectiveness with time. (Drawn using the data of Barrow (1980))
Comparison with other reactants
An important precept is that, if we want to understand that the behaviour of phosphate, we need to know how it is different from, or similar to the behaviour of other reactants. To misquote Kipling: what do they know of phosphate who only phosphate know (Kipling 1881).
For several other reactants, the effects of solution concentration, period of reaction, and temperature were similar to those of phosphate as described in Eq. 3. The reactants studied were: molybdate (Barrow and Shaw 1975b); fluoride (Barrow and Shaw 1977a); sulfate (Barrow and Shaw 1977b); selenate and selenite (Barrow and Whelan 1989); zinc (Barrow 1986); cadmium, zinc, nickel and cobalt (Barrow 1998). Thus similar behaviour occurs for all so-far tested reactants; it is not confined to anions; cations behave similarly. However the magnitude of the effects differs; among the anions, sulfate and selenate react more slowly and have lower activation energies; similarly cadmium reacts more slowly and has lower activation energy than the other cations. It is relevant to note that the behaviour of fluoride is similar to that of divalent anions. As will be discussed later, one of the suggested mechanisms for the slow reaction is that it involves the conversion of a single link to the surface to a double link. Such an explanation cannot apply to fluoride and therefore cannot be comprehensive.
Further properties of reaction
Any explanation of the behaviour should be able to include all observations. There are three diverse behaviours to be considered.
One is that if, after prolonged prior reaction with phosphate, the reaction of newly-added phosphate is measured using sorption curves, the slopes of the curves decreases. That is, the buffering capacity is decreased (Barrow 1974b).
The second is that reaction continues even in dry soil albeit more slowly. This is reflected in changes in the null-point concentration (Bramley et al. 1992) and in changes in the effectiveness of phosphate fertilisers to plants (Bramley and Barrow 1992).
The third is concerned with the effects of temperature on desorption. Chemical reactions involve molecules jumping over an energy barrier. Normally the energy levels on the two sides of this barrier differ. The activation energy for the forward reaction therefore differs from that for the back reaction. However, this is not the case for sorption and desorption of phosphate; the activation energies are similar (Barrow 1979).
Explaining the decrease in effectiveness with time
Diverse mechanisms have been proposed. An early proposal (Kafkafi et al. 1967) was that it involved the conversion of a monodentate link to the surface to a bidentate link. A similar explanation appeared more recently (Shi et al. 2008) where it was proposed that for zinc there was a fast reaction associated with monodentate binding sites and a slow reaction associated with bidentate binding sites. However, as shown above, many ions behave similarly. A comprehensive explanation should apply to all ions and should be applicable to fluoride – for which such explanations are not possible. Further, such surface reactions would be expected to be fairly rapid. It is difficult to envisage them continuing for periods measured in years.
Multi-reaction models have also been published. In the model of Zhang and Selim (2007), adsorbed ions react with sites that are described as “equilibrium sites”, “kinetic sites”, “consecutive irreversible sites”, and “concurrent irreversible sites”. Movement between the solution and the appropriate sites, and between sites, involves five rate constants describing first-order reactions. Models with this many rates are very flexible and can often describe overall rates at least over limited periods, but may require more terms to fit over longer periods. It is difficult to understand the purpose of such models. They cannot be regarded as mechanistic because no physical evidence is provided that such sites exist. They have not been shown to apply to any other conditions and therefore cannot be regarded as comprehensive. Their virtue appears to be that they can be combined with differential equations that describe the movement of water through columns thus allowing the emergence of reactants from such columns to be described.
We require a mechanism that is comprehensive. It should be able to explain that the reaction occurs even in dry soil; the forward and back reactions should have similar activation energies; it should result in decreased buffering capacity; and it should occur for all adsorbed reactants. A mechanism that meets the criteria involves the postulates that an initial adsorption of ions is followed by solid state diffusion into the adsorbing material. Solid state diffusion was proposed as early as 1972 by Probert and Larson (1972). They wrote: “exchange between the solution and the surface of the solid-phase (is) followed by deeper penetration of the tracer into the lattice“. This does not mean that the pathways for diffusion are the same in all cases. Sulfate and selenate react more slowly and have lower activation energies. This suggests that they penetrate via major defects. Strauss et al. (1997) showed that phosphate can only diffuse into goethite when there are defects between crystal domains. When goethite was `healed’ by hydrothermal treatment, diffusion was prevented. In contrast, Brümmer et al. (2013) showed that Ni could diffuse into healed goethite. This indicates that Ni, but not phosphate, could penetrate directly into the crystal lattice of goethite. Cd differed from Ni in that its diffusion into healed goethite was very slow, but diffusion into poorly crystalline goethite was appreciable. This suggests that it was less able to penetrate the crystal lattice and this is consistent with its larger ionic radius. The suggestion that phosphate diffuses into defects between crystal domains is consistent with the observation that reaction with phosphate decreases the rate of dissolution of goethite by acid (Strauss et al. 1997). It is also consistent with the observations of Bramley et al. (1992). They showed that reaction was slower in dry soil, but when the soil was re-moistened, there was a faster drop in concentration (Fig. 6). This suggests that during the dry phase, phosphate was unable to move across the surface, but when the soil was re-moistened, movement across the surface was again possible.

Effect of the indicated conditions of incubation on the null-point concentration of phosphate. (Drawn from the data of Bramley et al. (1992))
Development of comprehensive models
In order to test whether the adsorption-penetration hypothesis can really describe data, it is necessary to have models of reaction into which this hypothesis can be incorporated. Such models exist at two levels. At one level there are models that apply to well-defined surfaces such as that of goethite. At another level those detailed models are simplified and applied to the less well-defined surfaces of soils.
I suggest that development of such models has been hindered by the existence of an apparent incongruity. How can negatively charged phosphate ions react with negatively charged surfaces? Perhaps it is such a consideration that led Penn and Camberato (2019) to write that phosphate adsorption “is not dependent on surface charge of the mineral”. In fact, the surface charge, or more precisely, surface electric potential, plays an essential role in adsorption. This can be deduced from the interaction between pH and the ionic strength of the background solution in which phosphate is measured. Figure 7 shows that at high pH increasing NaCl concentration increases phosphate sorption; this is because the increased concentration of sodium ions near negative surfaces make the potential less negative and so increase sorption. At low pH increasing NaCl solution decreased sorption; this is because increasing concentration of chloride ions near the positively charged surfaces made the potential less positive and so decreased sorption. That is, the initial reaction is adsorption on variable-charged surfaces.

Effect of the indicated concentrations of sodium chloride on the amount of phosphate sorbed by a soil at a solution phosphate concentration of 10 µg mL− 1. (Drawn from the data of Barrow and Ellis (1986))
It was proposed by Bowden et al. (1977) that sorption should be related not to concentration but rather to the surface activity function (Sa). This is given by:
where Ki is the binding constant for the reacting ion (i), zi is its valency, ψ is the electrical potential in the plane of adsorption, α is the dissociation coefficient for the appropriate ion, γ is the activity coefficient, F is the Faraday, R the gas constant and T (K) the temperature. Negative values for the potential term (ψ) do not mean that reaction is impossible; they mean that the value of the exponential function is small; reaction can occur provided that the affinity of the ion for the surface, as indicated by Ki, is high, and the concentration of the reacting ion as determined by α and by the pH, is also high.
Phosphate forms bidentate links to oxide surfaces and reaction can therefore be related to the concentration of divalent ions in solution (Barrow 1999). The value of zi is therefore − 2. The effects of increasing pH can be seen as a balance between increasing proportion of divalent ions and increasingly unfavourable electric potential. Up to pH values a little below 7, the concentration of divalent ions increases about 10 fold for unit increase in pH. Above this value the fraction present as divalent ions approaches unity, the decreasing potential then dominates, and sorption decreases rapidly with increasing pH.
Of the several published models, the charge distribution model (CD) of Hiemstra and Van Riemsdijk (1996) is easily the most detailed. In it, the charge on a phosphate molecule is distributed between protonated surface oxygen atoms and atoms in the so-called Stern layer. The model of Bowden et al. (1980) can be regarded as a simplification with the charge on the phosphate molecule treated as a point charge located at a position between the two planes specified by the CD model. It is nevertheless an effective model describing the effects of pH on adsorption of: phosphate, selenite, and citrate Bowden et al. (1980); copper lead and zinc (Barrow et al. 1981); and four organic phosphates (Barrow et al. 2015). With the addition of solid-state diffusion into imperfectly crystallised samples of goethite particles, it describes the effect of time and of pH on sorption of phosphate (Strauss et al. 1997) and of 10 metals including copper and zinc (Fischer et al. 2007).
Adapting models to soil
There are two main problems in adapting detailed models to apply to soils. One is allocating a value to the electric potential. In the detailed models, the structure of the surface is well-known and theories about the dissociation of surface water molecules are used to calculate the effects of pH on charge and from this the electric potential is calculated. Similarly the charge conveyed to the surface by adsorption of ions can be calculated from the amount of adsorption and from the charge on the ions, again leading to calculations of the potential. Neither of these calculations is possible for soil. The effects of pH on potential are therefore regarded as parameters the values of which are chosen to fit the data. In order to calculate the effects of ion adsorption on the electrical potential, a simplification suggested by Alan Posner is utilised. The simplification is to assume that the change in potential is linearly related to the amount of phosphate adsorption. Unfortunately, Alan died before this work could be completed and I wrote it up on his behalf (Posner and Barrow 1982).
The other problem is dealing with heterogeneity. The reacting surfaces in soils cannot be expected to be uniform. This is reflected by the widespread use of the Freundlich equation. This equation can be derived if an appropriate distribution of affinity terms is used. I chose to assume a normal distribution of electric potentials mainly because I felt that the distribution had to be two-tailed with anions reacting at the more -positive end and cations reacting at the more-negative end. In the model, the distribution was divided into 30 slices and within each slice a modified Langmuir equation was used. The modifications were that the surface activity function was used rather than concentration; adsorption was assumed to decrease electric potential as calculated using the Posner simplification; the surface concentration of adsorbed molecules was assumed to be the driving concentration for diffusive penetration; and the penetrated molecules were also assumed to decrease the electric potential.
Fortunately, the development of these theories coincided with the availability of the early personal computers. They were laughably slow by modern standards but far better than writing programs in Fortran using punch cards and working in batch mode using a distant computer via a landline.
The model was published (Barrow 1983) and republished as a Landmark Paper (Barrow 2015). Figure 8 shows that it quantitatively described the effects of time and of temperature on sorption of phosphate. Over several years after the original publication, I showed that the model could also quantitatively describe the effects of time and temperature on sorption of molybdate, zinc, fluoride, selenite, selenate and 32P as well as the effects of pH of those ions and also on borate. I also showed that it could describe the effects of time on competition between selenite and phosphate. These several papers are summarised in Barrow (1999).

Observed and modelled effects of time, temperature and concentration on sorption of phosphate. (Drawn from the data of Barrow (1983))
The use of a normal distribution turned out to be serendipitous. This does not produce a Freundlich equation for which log-log plots of sorption against concentration are linear. Instead it produces gentle curves. In most work, solution concentration is only measured over a limited range and such curvature cannot be detected. However if the range of concentrations is greatly increased the curvature becomes obvious. This can be achieved by regarding selenate and selenite as ersatz phosphate molecules with the advantage that they can be measured at very low concentrations (Fig. 9).

Observed and modelled effects of equivalent concentrations of anions on anion sorption. The concentrations of selenite and selenate have been multiplied by the indicated factors. (Drawn from the data of Barrow and Whelan (1989))
In summarising this work (Barrow 1999), I claimed that the model was: efficient, effective, realistic, comprehensive, and predictive.
Long-term P fertilised soils
It is understandable that much phosphate research is conducted on soils that are P deficient. Papers that report such research emphasise how many soils of the world are seriously phosphate deficient. However, there are many soils, especially in developed countries, that have been fertilised with phosphate for many decades and indeed in some cases for more than a century. I suggest the scientific community has paid insufficient attention to such soils; they are now rather different. You cannot reapply phosphate to the same soil. After each application it is a different soil.
I have indicated above that reaction with phosphate decreases the buffering capacity. This means that phosphate is more effective; less is needed to achieve the same yields. However there is an even more important effect. The diffusive penetration does not continue at the same rate for ever; as phosphate status of soils is increased by repeated fertilisation it becomes slower and eventually stops. This can be shown by hysteresis as indicated earlier. When hysteresis is measured on soils that have been fertilised for a long time hysteresis decreases to unity. Figure 10 shows this for a soil of high phosphate status; this is illustrated by the high concentrations in solution when there is no sorption (the null-point) and by the large negative values when the sorption curves are extrapolated to zero solution concentration.

Results from a typical sorption-desorption experiment on a soil of high phosphate status, measured under the same conditions as those of Fig. 1. As in that figure, plots have been linearised by plotting against concentration raised to the indicated fractional power. The ratio of the slopes is 1.0: that is there is no hysteresis. (Drawn using data of Barrow and Debnath (2014))
There are therefore three ways in which long-term phosphorus fertilisation affects the amount of phosphate required for subsequent crops. One is that some of that phosphate remains effective and I would expect this to be strongly reflected in soil tests.
The second is the decrease in buffering capacity. When differences in buffering capacity are caused by differences in the amount and nature of reacting surface present, soil tests such as the Olsen test reflect the differences (Barrow and Shaw 1976). However, I question whether this would be the case when decreases in buffering capacity are caused by increased negative charge. I think the differences in surface charge brought about by the marked change in pH involved in the Olsen test would swamp these effects. A similar argument would apply to other soil tests. Separate measurement of buffering capacity seems desirable.
Especially in Australia, the importance of buffering capacity has been recognised since the early work of Ozanne and Shaw (1967) and soil tests are adjusted using a measure of phosphate buffering capacity (Moody and Bolland 1999). Single-point estimates of buffering capacity have been developed and the method of Burkitt et al. (2002) is widely used. Its use should be effective provided it is accepted that buffering capacity is not a fixed property of soil like for example the clay content and that it changes with fertiliser use.
The third is the decrease in diffusive penetration. This can be assessed by measuring the rate of reaction, but this is rather tedious and unlikely to be undertaken by soil testing laboratories. Measuring hysteresis is simpler but still somewhat complex; my colleagues and I are therefore testing a single-point method for estimating hysteresis.
Full circle
Earlier in this article, I pointed out that there were large differences amongst soils in the rate at which they continue to react with phosphate. I did not know why these differences occurred. However, it is now apparent that the phosphate status of the soils must have been important. This is why the rate of reaction is so large for Western Australian topsoils; the soils are world champions in the degree of phosphate deficiency.
Organic P in soils
Especially after the second world war, large areas of temperate Australia were converted to legume-based pastures fertilised with superphosphate. There was interest in the consequent accumulation of soil organic matter and of organic P. Measurements of the amount accumulated varied from 1.5 to 2.5 kg P per year (reviewed by Barrow 1980b). While organic matter increased, this could be considered as a sink for P, and another reason why it was necessary to reapply it.
However, to me, the most interesting aspect of organic P is that it occurs in soil at all. Organic phosphates appear to be readily decomposed; even inositol phosphate was a good source of P for lupins (Adams and Pate 1992) – provided it was supplied as sand culture. They concluded that the differences in availability of P sources depended on the proportion in solution, not on their susceptibility to phosphatases. These results did not receive adequate attention and in some cases were ignored or misinterpreted and the assumption that phosphatase activity was the limiting step was (wrongly) preferred (Gerke 2015).
There is only a small proportion in solution because they react strongly with variable-charge surfaces. The reaction is similar to that of inorganic phosphates, but differs in that the charge conveyed to the surface by the reaction seems to be repelled to the “outside” of the reaction complex (Barrow et al. 2015). Their reaction with soil components is one reason for their stability in soil; the phosphate component of the molecule is protected from phosphatases by reaction with the soil components. But I wonder whether it is a sufficient explanation. As far as I am aware, no one has investigated whether organic phosphates also diffuse into the reacting materials. This would help explain their stability.
The further puzzle about organic P is that much of it has not yet been identified. It is thought to be associated with high molecular mass humic substances (Gerke 2015).
Implications
This work has implications for some of the widely held theories in soil science. These theories are largely based on the notion that soil phosphate exist in discrete compounds with iron, aluminium and calcium. The work summarised above indicates that such compounds do not exist under most common soil conditions.
One of these theories holds that phosphate availability to plants is greatest near pH 7 because at this pH there is “valley” between aluminium compounds at medium pH and calcium compounds at higher pH (Price 2006, Penn and Camberato 2019). This theory was so widely held that that it was not tested experimentally until 2020 (Barrow et al. 2020) and it was then found that, in fact, lowest availability occurred near pH 7. The most important influence above pHCaCl2 of about 5 was the effects of lower pH in increasing the rate of uptake by plant roots. Below that pH the most important effect was that caused by aluminium toxicity.
Much effort has been devoted to measuring the supposed discrete phosphate fractions present in soil. The seminal paper for this work was that of Chang and Jackson (1957). Their logic appears to have been: soil phosphate exists as discrete iron aluminium and calcium compounds; this sequence of extractions dissolves such compounds; when we apply this sequence to soil each extraction does dissolve phosphate; therefore we have measured these compounds. There are several variants of the procedure; more recent ones do not identify specific compounds but nevertheless purport to measure discrete compounds. We thought it of interest to apply such methods to more realistic substrates: iron oxide and aluminium oxide that had been reacted with phosphate (Barrow et al. 2021). There were no discrete compounds present and there was no calcium. Yet the two fractionation procedures both detected discrete compounds. The Chang and Jackson procedure measured appreciable calcium phosphate though none existed and both iron and aluminium phosphate where none existed. We concluded that such procedures did not produce realistic results.
Much effort has also been devoted to find and use phosphate solubilising organisms. In a typical experiment, organisms are cultivated in a Petri dish in which the medium contains a sparingly soluble phosphate compound, usually tricalcium phosphate. Organisms which produce a halo around their colonies are selected. They produce this halo by secreting acid and it is implied that this will dissolve soil phosphate. There are problems in expecting such organisms to establish and persist in competition with the native microorganisms. However my concern here is that if they do establish and do decreased the pH, any beneficial effect on soils above pHCaCl2 of about 5 is an increased rate of uptake by plant roots rather than a release of phosphate. I suspect that phosphate solubilising organisms don’t! This is discussed further by Barrow and Lambers (2022).
Where to from here?
The notion that phosphate fertiliser is inefficient and that large amounts of applied phosphorus are ineffective because they are locked up or “fixed” is firmly established. It is therefore thought to be necessary to always supply more phosphate than plants need for good growth. One consequence is large costs; another is depletion of a limited resource. When pollution of water bodies occurs, this seems to be regarded as an unfortunate consequence of an unavoidable practice. My work indicates that long-term fertilised soils no longer lock up or fix applied P; it is only necessary to feed the plants not the soil.
It is a major task to convince the soil science community to put these ideas into practice. This community is very conservative; it is reluctant to abandon old ideas. Recently, Penn and Camberato (2019) staunchly defended the theory that phosphate is precipitated in soil as iron and aluminium compounds. This theory has never been shown to describe observations; if precipitation were to occur, one would expect a sharp “kick-up” in sorption curves such as those in Figs. 1 and 10 as the phosphate concentration increased so that the solubility product was exceeded; this is never been observed. Formation of such precipitates involves a reversal of the soil formation process – and requires that entropy decreases. Adherence to it may be one reason for the idea that large amounts of phosphate must always be supplied.
Nor is it easy to convince people that the major reason for the decline in effectiveness with time is penetration by diffusion into the reacting surface. Frossard et al. (2011) wrote that “on the basis of long-term sorption experiments,(we) concluded that inorganic P ions located on the solid phase of the soil are distributed along a continuum of solubility”. The proposal that phosphate ions penetrate the reacting surfaces and thereby change their properties is essential to understanding the increased effectiveness of subsequent phosphate applications.
It is probably even more difficult to convince people that soil tests are sometimes a poor indicator of phosphate status. We have known for some time that the decline in soil tests with time do not closely mirror the decline in effectiveness (Mendoza and Barrow 1987), but I am now suggesting that, for long-term fertilised soils, soil tests do not give information on two important characteristics: the decrease in P buffering and the slowing of the penetration. We need to add methods for rapidly estimating these characteristics to our toolboxes. The method of Burkitt et al. (2002) provides a simple estimate of the buffering capacity; it assumes that the b1 characteristic has a value of 0.41; this is appropriate for Australian soils (Fig. 4), but may not be so for other soils; further calibration may be needed. We have, as yet, no simple method for measuring the slowing of penetration.
We also need to reconsider our sampling procedures. When phosphate is applied to the soil surface, it is the properties of the first few centimetres that are relevant because these are the components that will react with each further addition and it is these properties that should be evaluated.
And finally
This has been a long story; I hope you have enjoyed it. And a long journey; I hope it will continue for some little time further.
References
Adams MA, Pate J (1992) Availability of organic and inorganic forms of phosphorus to lupins (Lupinus spp.). Plant Soil 145:107–113
Arndt W, Mcintyre GA (1963) The initial and residual effects of superphosphate and rock phosphate for sorghum on a lateritic red earth. Aust J Agric Res 14:785–795
Barrow NJ (1974) The slow reactions between soil and anions. I. Effects of soil, temperature and water content of a soil on the decrease in effectiveness of phosphate for plant growth. Soil Sci 118:380–385
Barrow NJ (1974) Effect of previous additions of phosphate on phosphate adsorption by soils. Soil Sci 118:82–89
Barrow NJ (1979) Three effects of temperature on the reactions between inorganic phosphate and soil. J Soil Sci 30:271–279
Barrow NJ (1980) Difference amongst a wide ranging collection of soils in the rate of reaction to phosphate. Aust J Soil Res 18:215–224
Barrow NJ (1983) A mechanistic model for describing the sorption and desorption of phosphate by soil. J Soil Sci 34:733–750
Barrow NJ (1986) Testing a mechanistic model II. The effects of time and temperature on the reaction of zinc with a soil. J Soil Sci 37:277–286
Barrow NJ (1998) The effects of time and temperature on the sorption of cadmium, zinc, cobalt and nickel by a soil. Aust J Soil Res 36:941–950
Barrow NJ (1999) The four laws of soil chemistry: the Leeper lecture 1998. Aust J Soil Res 37:787829
Barrow NJ (2015) A mechanistic model for describing the sorption and desorption of phosphate by soil. Europ J Soil Sci 66:9–18
Barrow NJ (2020) Describing and explaining the reaction of soils with phosphate using existing observations. Eur J Soil Sci 72:234–242. https://doi.org/10.1111/ejss.12968
Barrow NJ, Campbell NA (1972) Methods of measuring residual value of fertilizers. Aust J Exp Agric Anim Husb 12:502–510
Barrow NJ, Debnath A (2014) Effect of phosphate status on the sorption and desorption properties of some soils of northern India. Plant Soil 378:383c395
Barrow NJ, Ellis AS (1986) Testing a mechanistic model. V The points of zero salt effect for phosphate retention, for zinc retention and for acid/alkali titration. J Soil Sci 37:303–310
Barrow NJ, Lambers H (2022) Phosphate-solubilising microorganisms mainly increase plant phosphate uptake by effects of pH on root physiology. Plant Soil. https://doi.org/10.1007/s11104-021-05240-0.
Barrow NJ, Mendoza RE (1990) Equations for describing sigmoid yield responses and their application to some phosphate responses by lupins and by subterranean clover. Fert Res 22:181–188
Barrow NJ (1980b) Evaluation and utilisation of residual phosphorus in soils. In: Kasewnah F, Sample G (eds) The role of phosphorus in agriculture, pp 333–359
Barrow NJ (1982) An evaluation of the immiscible displacement method for studying the reaction between soil and phosphate. Fert Res 3:423–433
Barrow NJ, Shaw TC (1975) The slow reactions between soil and anions. II. Effects of time and temperature on the decrease in phosphate concentration in the soil solution. Soil Sci 119:167–177
Barrow NJ, Shaw TC (1975) The slow reactions between soil and anions. IV. Effect of time and temperature of contact between soil and molybdate on the uptake of molybdenum by plants and the molybdate concentration in the soil solution. Soil Sci 119:301–310
Barrow NJ, Shaw TC (1976) Sodium bicarbonate as an extractant for soil phosphate. III. Effects of buffering capacity of a soil for phosphate. Geoderma 16:273–283
Barrow NJ, Shaw TC (1977) The slow reactions between soils and anions. 6. Effects of time and temperature of contact on fluoride. Soil Sci 124:265–278
Barrow NJ, Shaw TC (1977) The slow reactions between soils and anions. 7. Effects of time and temperature of contact between an absorbing soil and sulfate. Soil Sci 124:347–354
Barrow NJ, Whelan BW (1989) Testing a mechanistic model. VII The effects of time and temperature of incubation on the sorption and subsequent desorption of selenite and selenate by a soil. J Soil Sci 40:29–37
Barrow NJ, Malajczuk N, Shaw TC (1977) A direct test of the ability of vesicular-arbuscular mycorrhyiza to help plants take up fixed soil phosphate. New Phytol 78:269–276
Barrow NJ, Bowden JW, Posner AM, Quirk JP (1981) Describing the adsorption of copper, lead and zinc on a variable charge mineral surface. Aust J Soil Res 19:309–321
Barrow NJ, Feng XH, Yan YP (2015) The specific adsorption of organic and inorganic phosphates by variable charge oxides. Eur J Soil Sci 66:859–866
Barrow NJ, Debnath A, Sen A (2020) Measurement of the effects of pH on phosphate availability. Plant Soil 454:217–224. https://doi.org/10.1007/s11104-020-04647-5
Barrow NJ, Sen A, Roy N, Debnath A (2021) The soil phosphate fractionation fallacy. Plant Soil 459:1–11. https://doi.org/10.1007/s11104-020-04476-6
Bowden JW, Posner AM, Quirk JP (1977) Ionic adsorption on variable charge mineral surfaces. Theoretical-charge development and titration curves. Aust J Soil Res 15:121–126
Bowden JW, Nagarajah S, Barrow NJ, Posner AM, Quirk JP (1980) Describing the adsorption of phosphate, citrate and selenite on a variable charge mineral surface. Aust J Soil Res 18:49–60
Bramley RGV, Barrow NJ (1992) The reaction between phosphate and dry soil. II The effect of time, temperature and moisture status during incubation on the amount of plant available phosphate. J Soil Sci 43:759–766
Bramley RGV, Barrow NJ, Shaw TC (1992) The reaction between phosphate and dry soil. I The effect of time, temperature and dryness. J Soil Sci 43:749–758
Brümmer GW, Barrow NJ, Fischer L (2013) Effect of porosity of goethite on the sorption of six heavy metal ions. Europ J Soil Sci 64:805–813
Burkitt LL, Moody PW, Gourley CJP, Hannah MC (2002) A simple phosphorus buffering index for Australian soils. Aust J Soil Res 40:497–513
Chang SC, Jackson ML (1957) Fractionation of soil phosphorus. Soil Sci 84:133–144
Devine JR, Gunaryd D, Larsen S (1968) Availability of phosphate as affected by duration of fertilizer contact with soil. J Agric Sci 71:359–364
Fischer L, Brümmer GW, Barrow NJ (2007) Observations and modelling of the reactions of ten metals with goethite: adsorption and diffusion processes. Eur J Soil Sci 58:1304–1315
Frossard E, Achat DL, Bernasconi SM, Bunemann EK, Fardeau JC, Jansa J, Morel C, Rabeharisoa L, Randriamanantsoa L, Sinaj S, Tamburini F, Oberson A (2011) The use of tracers to investigate phosphate cycling in soil-plant systems. In: Bunemann EK, Oberson A, Frossard E (eds) Phosphorus in Action: Biological Processes in Soil Phosphorus Cycling. Springer-Verlag, Berlin, pp 59–91
Gerke J (2015) Phytate (inositol hexakisphosphate) in soil and phosphate acquisition from inositol phosphates by higher plants. A review. Plants 4:253–266. https://doi.org/10.3390/plants4020253
Hiemstra T, Van Riemsdijk WH (1996) A surface structural approach to ion adsorption: the charge distribution (CD) model. J Colloid Interface Sci 179:488–508
Kafkafi U, Posner AM, Quirk JP (1967) The desorption of phosphate from kaolinite. Soil Sci Soc Am Proc 31:348–353
Kipling R (1881) The English Flag. St. James’s Gazette and the National Observer on April 4th. https://www.kiplingsociety.co.uk/poem/poems_englishflag.htm. Accessed June 2022
Mendoza RE, Barrow NJ (1987) Ability of three soil extractants to reflect the factors that determine the availability of soil phosphate. Soil Sci 144:319–329
Moody PW, Bolland MDA (1999) Phosphorus. In: Peverill KI, Sparrow LA, Reuter D (eds) Soil analysis an interpretation manual. eBook
Ozanne PG, Shaw TC (1967) phosphate sorption by soils as a measure of the phosphate requirement for pasture growth. Aust J Agric Res 18:601–602
Pate JS, Verboom WH, Galloway PD (2001) Co-occurrence of Proteaceae, laterite and related oligotrophic soils: coincidental associations or causative inter-relationships? Aust J Bot 49:529–560
Penn CJ, Camberato J (2019) A critical review on soil chemical processes that control how soil pH affects phosphorus availability to plants. Agriculture 9:120. https://doi.org/10.3390/agriculture9060120
Posner AM, Barrow NJ (1982) Simplification of a model for ion adsorption on oxide surfaces. J Soil Sci 33:211–231
Price G (ed) (2006) Australian soil fertility manual, 3rd edn, fertilizer industry Federation of Australia and CSIRO, p 45
Probert ME, Larsen S (1972) The kinetics of heterogenous isotopic exchange. J Soil Sci 23:76–81
Shi ZQ, Di Toro D, Allen HE, Sparks DL (2008) A WHAM-based kinetics model for Zn adsorption and desorption to soils. Environ Sci Technol 42:5630–5636
Strauss R, Brümmer GW, Barrow NJ (1997) Effects of crystallinity of goethite II Rates of sorption and desorption of phosphate. Europ J Soil Sci 48:101–114
Zhang H, Selim HM (2007) Modeling competitive arsenate–phosphate retention and transport in soils: A multi-component multi-reaction approach. Soil Sci Soc Am J 71:1267–1277. https://doi.org/10.2136/sssaj2006.0422
Acknowledgements
It is a pleasure to acknowledge the many people who have contributed to this work. Some, but not all, are indicated in the citations. I want to particularly acknowledge the help of Abhijit Debnath whose cooperation over the last several years has enabled me to extend my work way beyond my expected use-by date.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
I have no relevant financial or non-financial interests to disclose. The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Additional information
Responsible Editor: Ismail Cakmak.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barrow, N.J. How understanding soil chemistry can lead to better phosphate fertilizer practice: a 68 year journey (so far). Plant Soil 476, 117–131 (2022). https://doi.org/10.1007/s11104-022-05468-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-022-05468-4