
Abstract
Purpose
Push–pull is an intercropping technology that is rapidly spreading among smallholder farmers in Sub-Saharan Africa. The technology intercrops cereals with Desmodium to fight off stem borers, eliminate parasitic weeds, and improve soil fertility and yields of cereals. The above-ground components of push–pull cropping have been well investigated. However, the impact of the technology on the soil microbiome and the subsequent role of the microbiome on diverse ecosystem benefits are unknown. Here we describe the soil microbiome associated with maize—Desmodium intercropping in push–pull farming in comparison to long-term maize monoculture.
Methods
Soil samples were collected from long-term maize—Desmodium intercropping and maize monoculture plots at the international centre for insect physiology and ecology (ICIPE), Mbita, Kenya. Total DNA was extracted before16S rDNA and ITS sequencing and subsequent analysis on QIIME2 and R.
Results
Maize—Desmodium intercropping caused a strong divergence in the fungal microbiome, which was more diverse and species rich than monoculture plots. Fungal groups enriched in intercropping plots are linked to important ecosystem services, belonging to functional groups such as mycorrhiza, endophytes, saprophytes, decomposers and bioprotective fungi. Fewer fungal genera were enriched in monoculture plots, some of which were associated with plant pathogenesis and opportunistic infection in humans. In contrast, the impact of intercropping on soil bacterial communities was weak with few differences between intercropping and monoculture.
Conclusion
Maize—Desmodium intercropping diversifies fungal microbiomes and favors taxa associated with important ecosystem services including plant health, productivity and food safety.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Push–pull technology is an ecological habitat management strategy for the control of major pests of cereals, particularly maize and sorghum. Since its inception in the 90’s, the technology has spread to smallholder farmers across southern and eastern Africa who use it to manage stem-borers (Busseola fusca and Chilo partellus) and fall armyworm (Spodoptera frugiperda) attacks on cereal crops thus increasing yield (Midega et al. 2018). The technology exploits the chemical ecology of a leguminous intercrop belonging to the genus Desmodium, which ‘pushes’ stem-boring insects from the main crop reportedly through its volatile compounds that signal an unfavourable egg-laying environment. At the same time, a grass trap crop such as Brachiaria spp. or Cenchrus purpureus, commonly known as napier grass, is planted as border vegetation to ‘pull’ the insects towards itself without supporting their development (Khan et al. 2003, 2010).
Over the years several additional benefits of the Desmodium-based intercropping system have been uncovered and indicate diverse soil-based mechanisms that warrant further study. Desmodium employs allelopathic mechanisms that dramatically reduce infestation of the parasitic weed Striga hermonthica to cereal crops, further adding to yield increases (Khan et al. 2002). Moreover, the technology provides other ecological benefits contributing to improved cereal crops yield, including nitrogen fixation by Desmodium, soil structure improvement and a mulching effect in the fields. Finally, the technology reportedly reduced the incidence of human pathogenic fungal toxins in maize kernels (Njeru et al. 2020; Owuor et al. 2018). In addition, both Desmodium and the trap crops are a reliable source of animal fodder, particularly in drought periods, as farmers do not uproot them between farming seasons (Khan et al. 2010). The overall impact is increased cereal yield with minimum chemical inputs. The technology is effective and, importantly, affordable for smallholder farmers in Sub-Saharan Africa.
Whereas the components of the cereal—Desmodium push–pull farming system and their underlying mechanisms have been well investigated, one of the areas that has received no attention is its interaction with the soil microbiome. Intercropping is increasingly adopted as a sustainable alternative to monoculture production systems. The cropping practice provides diverse ecosystem services, some of which are immediate and pronounced (such as productivity, pollinator support, pest and disease reduction, nitrogen fixation (Bybee-Finley and Ryan 2018; Nourbakhsh et al. 2019), whereas others are acquired over a longer time. In push–pull cropping systems the effects on and impact of the soil microbiome fall in the latter category and have been, in part for that reason, little studied.
Soil microorganisms promote plant health and productivity through direct and indirect mechanisms mediated through root systems (van der Heijden et al. 2008). Plants use their roots exudates to actively influence the microbial assemblages in the rhizosphere often favouring those that offer survival benefits (Liu et al. 2021). Therefore, it is of interest to explore the impact of maize—Desmodium on soil microbial profiles as the first step to understand their contribution on the effectiveness of the farming system. Microbiome studies are increasingly used to discern potential impacts of farming practices such as intercropping on abundance, structure and diversity of soil microbiota, which in turn provide plants with other benefits such as higher mineral nutrients availability (Johansen & Jensen 1996; Tang et al. 2014). Studies in cereal—legume intercropping systems have shown changes in soil microbial structures as well as benefits on plants mediated by soil microbes. For instance, in a study by Li et al. (2018), an increase in yield as well as overall diversity of soil bacteria was observed in maize—peanut intercropping systems. The study observed a higher abundance of beneficial soil bacteria in intercropping systems, where belowground interactions were either complete or partial when compared to monoculture. Increases in soil microbial biomass as well as nutrient availability, especially N, P and C have been observed in multiple cereal—legume intercropping systems, such as that of wheat (Triticum aestivum), maize (Zea mays), and faba bean (Vicia faba) intercropping (Song et al. 2007), and durum wheat (Triticum turgidum durum) intercropped with either chickpea (Cicer arietinum) or lentil (Lens culinaris) (Tang et al. 2014).
In that light, the current study compared the diversity of soil microorganisms between long-term maize—Desmodium and maize monoculture plots. Specifically, amplicon sequencing (16S rDNA and ITS) was used to investigate the differences in soil bacterial and fungal population structures between long-term maize—Desmodium intercropping and maize monoculture practices in a context of potential ecological benefits.
The mapping of the soil microbiomes demonstrated that the fungal microbiome was particularly diversified in maize—Desmodium intercropping plots compared to maize monoculture plots. The results are discussed in the context of reported benefits around maize—Desmodium intercropping in push–pull farming by inferring known ecological functions of taxa contributing to the observed difference. This is the first step towards understanding soil microbial diversity in push–pull technology for optimal exploitation of their potential ecosystem benefits in plant health and productivity. Further studies are recommended to discern key determinants of the observed differences and their importance in ecosystem (dis) services. Knowledge and translation of this knowledge into other cropping systems could advance sustainable food production through fostering belowground microbial communities that support plant health and productivity.
Methodology
Sampling site
To compare soil microbial profiles between maize monoculture and maize—Desmodium intercropping maize farming, we obtained soil samples from long-term (14–19 years old) experimental plots at the International Centre for Insect Physiology and Ecology (ICIPE), Mbita campus, Kenya (0°25.877 S 34°12.425 E). The campus has clay-loam soil type, receives approximately 900 mm of rainfall per annum, has a mean annual temperature of 27 °C, and is located at an altitude of approximately 1200 m above sea level.
The samples were collected from three sets of plots established between 1998 and 2003. The first set of plots consisting of a maize monoculture and push–pull plots was established in 1998 (30 m by 30 m). The plots had D. uncinatum (silver-leaf desmodium) as the intercrop while Sudan grass (Sorghum sudanense) was the trap crop. The second set of plots was established in 1999 (6 × 6 m) to study the ability of Desmodium intercropping to suppress Striga. These plots were not surrounded by border/trap crops but were separated from other plots by 2 m buffer spaces. The third set of plots was established in 2003 (5 × 6 m) to compare efficiency of food legumes and Desmodium intortum (greenleaf desmodium) intercrops in Striga suppression. Phosphorus, in the form of di-ammonium phosphate (DAP), was applied in each plot at planting at the rate of 60 kg/ha. Nitrogen was applied after thinning of maize, in the form of calcium ammonium nitrate (CAN), at the rate of 60 kg/ha (Midega et al. 2014). The plots were also not surrounded by a border/trap crop but they were separated from other plots by 2 m buffer spaces. In the plots established in 1999 and 2003, only plots of maize monoculture and maize—Desmodium intercropping were selected for sampling.
In all plots, maize (medium maturing commercial hybrid 513 variety) was planted at a spacing of 0.75 m between rows and 0.3 m within rows while Desmodium was planted through drilling method within a row. Plant population (maize) was therefore the same in any set of plots.
Soil sample collection
Soil samples were collected during the cool dry season in July 2017, when the maize was mature and just before harvesting. We collected seven samples from each site; four samples from maize monoculture and three from intercropped/push–pull plots. Each individual sample was made up of three 15–18 cm deep soil cores that were collected from random spots in each selected plot away from the edges. For monoculture, sampling was done between rows of maize plots while in intercropped plots, it was done close to Desmodium spp. roots system (growing in rows between maize rows). Afterwards, each soil sample was homogenized and sieved through a 4 mm wire mesh. About 200 g of soil sub-sample was collected and stored at − 20 °C until further processing.
DNA extraction and sequencing
DNeasy Powersoil kit (Qiagen, Manchester, UK) was used for total DNA extraction from the soil samples following the manufacturer’s protocol. Briefly, 0.25 g soil was added to PowerBead Tubes containing a lysis buffer and vortexed for a few seconds. The resulting mixture was centrifuged at 10,000g for 30 s before discarding the pellet and centrifugation of the supernatant in spin columns. Tris–HCl solution was used to wash off DNA from the spin column. A Nanodrop spectrophotometer and gel electrophoresis were used to assess the quality of the extracted DNA. The DNA samples were stored at − 20 °C until further processing.
DNA sequencing was done at Inqaba Biotechnical Industries (Pty) Ltd (Pretoria, South Africa). Primers targeting the V1-V3 region of 16S rDNA gene of the bacteria (27F and 518R primer pairs) were used to amplify DNA under the following PCR conditions: initial denaturation at 95 °C for 2 min, followed by 30 cycles of denaturation at 95 °C for 30 s, primer annealing at 60 °C for 30 s, and extension at 72 °C for 30 s, with a final elongation at 72 °C for 5 min. For fungi, ITS1F and ITS2 primer pairs targeting ITS1 were used for PCR amplification under the following conditions: 95 °C for 2 min, followed by 30 cycles of denaturation at 95 °C for 30 s, primer annealing at 50 °C for 30 s, and extension at 72 °C for 1 min. Final elongation was held at 72 °C for 5 min.
Resulting amplicons were gel purified, end repaired and Illumina specific adapter sequence were ligated to each amplicon (NEBNext Ultra II DNA library prep kit). Following quantification, the samples were individually indexed (NEBNext Multiplex Oligos for Illumina Dual Index Primers Set 1), and another AMPure XP bead-based purification step was performed. Amplicons were then sequenced on Illumina’s MiSeq platform, using a MiSeq v3 kit with 600 cycles (300 cycles for each paired read and 12 cycles for the barcode sequence) according to the manufacturer’s instructions. Demultiplexed 300 bp paired-end reads were obtained.
Bioinformatics and statistical analysis
FASTQC (Wingett and Andrews 2018) was used to assess the quality of raw sequence reads after which QIIME2 v2020.8 was used for quality control, construction of a feature table, taxonomic classification and diversity analyses (Bolyen et al. 2019). Briefly, the dada2 plugin (Callahan et al. 2016) was used to trim and truncate poor regions of both the 16S and ITS raw reads. The truncation and trimming were set to –p-trim-left-f 8, –p-trim-left-r 8; and –p-trunc-len -f 290, –p-trunc-len-r 260, for the 16S; while for the ITS, parameters used were p-trim-left 22, –p-trunc-len 299. Bacterial taxonomic assignment was done using feature-classifier classify-sklearn (Bokulich et al. 2018; Pedregosa et al. 2011), only including reference genes that were classified to at least genus level, by using SILVA v.138 97% database (Quast et al. 2013) pre-trained to V1-V3 region of 16S. For ITS, we used UNITE v8.2 reference database (Nilsson et al. 2019) pre-trained to ITS1.
The resulting feature table was converted into biom format (using QIIME2’s export tool), and then imported into R (R Core Team 2020) using “qiime2R” (Bisanz 2018). For visualising the number of amplicon sequence variants (ASVs), genera, families and orders present in the dataset we filtered out everything that was present only once at each level and then Venn diagrams were produced using function vennCounts from package “limma” (Ritchie et al. 2015). Then, everything that was unassigned at family level was filtered out.
For constructing dendrograms, primary component analysis (PCA) and heatmap data was transformed using CSS (cumulative sum scaling) by using a package “metagenomeseq” (Paulson et al. 2013). To perform a principal component analysis (PCA), we used package “recipes” (Kuhn and Wickham 2020), and annotated ellipses using a Khachiyan algorithm from package “ggforce” (Pedersen 2020). Dendrograms were constructed using a jaccard index from package “vegan” (Oksanen et al. 2020), with a presence absence standardization, and plotted using “ggtree” (Guangchuang et al. 2017).
Species diversity (Shannon) and richness (chao1) were calculated on untransformed and unfiltered data using “vegan” through the package “phyloseq” (McMurdie and Holmes 2013), while evenness was calculated as the Shannon index divided by the natural logarithm of the total number of species. All indices were tested for significance using a two tailed Student’s t-test.
Differential expression analysis was done on untransformed but filtered data based on a negative binomial distribution through “DESeq2” (Love et al. 2014). The resulting log2fold changes were shrunken using the adaptive shrinkage estimator from package “ashr” (Stephens et al. 2020). Genera were deemed to significantly impact treatments if they had an adjusted p-value smaller than 0.05 (Wald test), and an absolute log2fold change of over one, which was then visualised on a volcano plot modified from package “EnhancedVolcano” (Blighe et al. 2020). The result from the differential expression analysis also was used to group data in the heatmap and label significant genera in the PCA. All data was manipulated using “tidyverse” (Wickham et al. 2019) and visualised using “ggplot2” (Wickham 2016).
Results
Composition and abundance of soil microbiome in maize monoculture and maize—Desmodium intercropping plots
When considering the total number of ASVs (taxonomic units), a moderate divergence was observed between maize monoculture and intercropping plots. The difference becomes less pronounced at the order, family and genus levels with a high degree of overlap observed (Fig. 1). The number of fungal ASVs was higher than that of bacteria, indicating a higher richness of soil fungal communities. More bacteria ASVs (1934) were identified from monoculture plots than maize—Desmodium intercropping plots (1333 ASVs). For fungal communities however, the number of ASVs was higher in intercropping (1262 ASVs) than monoculture plots (1085 ASVs). At the genus level, monoculture plots were composed of more bacteria than fungal taxa (195 vs 162 genera), whereas the fungal genera made the larger proportion in intercropping plots than bacteria (284 vs 225 genera).

Venn diagrams showing common and unique taxonomic units as well as the degree of overlap at the genus, family and order levels for a bacteria and b fungi communities in monoculture and maize—Desmodium intercropping plots
In spite of considerable/strong overlap, the two cropping systems separated clearly based on Jaccard dissimilarity index (dendrograms on Fig. 2a, b). Furthermore, differential abundance analysis revealed several genera that were enriched in either monoculture or intercropping plots. Bacterial taxa showed few differences in abundance between the cropping systems, whereas the abundance of fungal taxa showed stark contrasts. Fungal taxa were more enriched in intercropping than monoculture plots, whereas an opposite trend was observed for bacterial taxa (Fig. 2, for a full heatmap of all bacterial and fungal taxa, see supplementary Figs. 1 and 2).

Differential abundance of bacteria genera (A) and fungal genera (B) in monoculture or maize—Desmodium intercropping plots. The abundances were normalized by cumulative sum scaling (CSS). The dendrogram on the left was produced by using a Jaccard dissimilarity index, with a presence-absence standardization; each node corresponds to one sample
Statistical analysis confirmed that bacteria contributed little to the microbial divergence between the two treatments: only four bacterial genera were significantly abundant in monoculture while only one genus was significantly more abundant in maize—Desmodium intercropping plots (Fig. 3a). Among fungal genera, the trend was reversed, with more genera being enriched in maize—Desmodium intercropping (52 genera) than monoculture plots (16 genera) (Fig. 3b).

Volcano plots showing bacterial (A) and fungal (B) genus level features that are differentially and significantly abundant in monoculture and maize—Desmodium intercropping plots. Red dots represent genus entities that are significantly abundant in each group with log2 fold change greater than 1. The grey and green dots represent the genus features whose abundance is similar between the two farming systems and the blue dots represents values where the p—value is significant between the treatments, but where the log2 fold change is smaller than one
Fewer bacterial taxa were classified at the genus level due to limited information in classification databases, limiting further analysis and dissection of the findings. In contrast, a large proportion of fungal genera were identified. In maize monoculture plots, several fungal genera were enriched including plant pathogens Curvularia, Parastagonospora and Tetracladium as well as human opportunistic pathogens such as Aspergillus and Exserohilum. Only a few of the fungal genera enriched in monoculture plots are known for beneficial ecosystem services, notably the mycorrhizal genus Glomus and endophytic Laetisaria (Fig. 3b). In maize—Desmodium intercropping plots, noteworthy abundant fungal genera include saprophytic fungi like Pithya, Albifimbria, Acremonium, Pseudorobillarda and Cristinia, mycorrhizal and endophytic fungi like Edenia, Acrocalyma and Colletotrichum, as well as fungal groups known for plant bio-protection such as Talaromyces, Penicillin, Clonostachys and Trichoderma. A few pathogenic genera were also enriched in intercropping plots, for example, Mycoleptodiscus, a genus of fungi reported to cause disease in legumes (Fig. 3b).
The impact of maize—Desmodium intercropping on diversity of soil microbial populations
Comparing overall diversity of soil microbial populations, no statistically significant difference was found among bacteria genera between monoculture and maize—Desmodium intercropping plots (Shannon index p = 0.246, Fig. 4). In contrast, fungal genera in maize—Desmodium intercropping were significantly more diverse compared to monoculture plots (Shannon index p = 0.047). Likewise, the richness of bacterial genera did not significantly differ between the two farming systems (Chao1 estimator p = 0.238), whereas that of fungal genera was significantly higher intercropping plots (Chao1 estimator p = 0.012). Evenness of both fungal and bacterial communities was not significantly different in both treatments (Fig. 4).

Diversity index (shannon), richness (chao1) and evenness (shannon/ln[number of species]) of bacterial (a) and fungal (b) genera across monoculture and maize—Desmodium intercropping (push–pull) plots. The box indicates the interquartile range (25–75%), whereas points are deemed to be outliers to the whiskers when they exceed 1.5 times the interquartile range in either direction of the hinges of the box
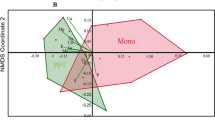
The impact of maize—Desmodium intercropping on the soil microbiome is also reflected in beta diversity measures. As noted above, the impact on the two farming practices on bacterial populations communities is weaker compared to that on fungi. Although the PCA plots for both bacterial and fungal communities show clear separation based on cropping practice, the separation was much stronger in fungal taxa (Fig. 5b). Cropping practises contributed to a major extent to the variation observed, with fungal taxa showing a clear non-overlapping clustering pattern between monoculture and intercropping plots along PC1, which contributed to a total of 30% of the variation (Fig. 5b). In bacterial taxa the separation was clearest again on PC1, but the total contribution of PC1 to the variation was only 19% and did not fully separate the cropping practises (Fig. 5a).

Principal component analysis of genus level communities of soil bacteria (A) and fungi (B) in long term maize monoculture and maize—Desmodium intercropping/push–pull farming plots. Solid ellipses around each treatment were drawn using the Khachiyan algorithm. Dotted ellipses represent samples from the same experiment in each treatment. In the background are scaled up eigenvectors sized according to average abundance and color-coded according to bacterial and fungal families
Discussion
Abundance and differential abundance of taxa and their potential functional significance
A large proportion of the fungal taxa that were abundant in maize—Desmodium intercropping plots appear to fulfil a saprophytic role, including Talaromyces, Trichoderma, Penicillium and Colletotrichum (see supplementary Table II). Presence of these taxa may indicate higher carbon sequestration in intercropping plots that is enhanced by the perennial intercropping system (Cong et al. 2015). Other enriched fungi genera likely confer more distinct ecosystem services, such as forming mycorrhizal (Ravnskov et al. 2006;) and endophytic associations with plant roots that promote plant growth activities through increased nutrient supply (Díaz-González et al. 2020; Macías-Rubalcava et al. 2008; Munasinghe et al. 2017). The fungi may also directly or indirectly stimulate production of beneficial secondary metabolites and protection against pathogens and insect pests (Hiruma et al. 2016; Zin and Badaluddin 2020). Indeed, effects can be indirect and intricate, for example,T. atroviride was shown to promote growth and herbivory resistance of maize against Spodoptera frugiperda, possibly linked to induction of the jasmonic acid pathway leading to heightened induced defence (Contreras-Cornejo et al. 2018). In general, a positive correlation between soil microbe composition and productivity of plants above ground has been reported in most systems (Schnitzer et al. 2011), with positive effect on above ground biodiversity and biological control.
How direct and indirect microbial and plant interactions in the rhizosphere contribute to the diverse ecosystem services observed in push–pull intercropping needs further study. For instance, a recent study showed that maize grown in soil from push–pull plots displayed a higher induced-defence response, including higher release of induced volatiles and lower herbivore damage compared to that growing on soil from monoculture (Mutyambai et al. 2019). Soil microbiota may be a missing link explaining the observed differences in maize direct and indirect defence pathways. The increased abundance of several soil fungal groups noted in intercropping plots in this study, such as Edenia and Clonostachys species, is particularly noteworthy in this context. Species belonging to these genera are associated with increased plant health, biocontrol of plant diseases and increased resistance against herbivore damage on plants (Iqbal et al. 2018; Macías-Rubalcava et al. 2008; Poveda et al. 2020).
Recent papers reported lower incidences of maize ear rot and associated mycotoxins (aflatoxins and fumonisins) (Owuor et al. 2018) as well as lower rate of infection of maize kernels with Fusarium verticillioides and Aspergillus flavus (Njeru et al. 2020) in smallholder farmers’ push–pull plots compared to monoculture. Push–pull thus appears to promote food safety by reducing the risk of mycotoxins entering the human food chain, although the mechanisms remained unclear. Interestingly, in the current study, a lower relative abundance of A. flavus was indeed associated with maize—Desmodium intercropping cropping. However, no association was found for F. verticillioides, a mycotoxin producing fungus in maize. The earlier reported lower incidence of ear rot infections may thus be partially explained by the shift in relative abundance of key species in intercropping/push–pull plots, causing competition between taxa and lowering mycotoxin incidence levels. Suppression of some taxa through fungal competition or biocontrol is a common phenomenon. Sarrocco et al. (2019) found that Fusarium graminearum, a plant pathogen and mycotoxin producer, was controlled by competition from other fungi, including Clonostachys, and Trichoderma, both of which were found in higher abundance in maize—Desmodium intercropping plots than monoculture in this study. Further research on how mycotoxin incidence in maize kernels can be reduced by interactions between mycotoxin producing fungi and other soil microbes in maize—Desmodium intercropping would help in devising strategies to increase food safety through more healthy plant production systems.
Diversity of cropping systems links to diversity in soil microbiome
In this study, long-term maize—Desmodium was associated with a higher diversity of soil microbial communities, with a stronger shift observed in fungal populations. Other studies have reported a similar trend where cereal—legume intercropping increases overall diversity of soil microorganisms. Such observations have been made in wheat—soybean intercropping (Bargaz et al. 2017), maize/wheat—faba bean intercropping (Wang et al. 2020) and millet—mung bean intercropping (Dang et al. 2020). While intercropping with annual legumes may cause a temporary shift in the soil microbial profiles, the impact of perennial crops and intercrops, such as Desmodium spp., on soil microbial diversity is likely to be stronger and more resilient.
Diversifying cropping systems, often by using legumes as an intercrop, were originally for purposes other than increasing biodiversity, such as food security, pest control (push–pull), green manure, or to avoid negative plant-soil feedback and soil legacy (Stagnari et al. 2017). However, ripple effects on biodiversity and ecosystem services have become apparent and maize—Desmodium intercropping and/or push–pull farming is a good example of this. The system was initially designed to combat stem-borers of maize and sorghum, but additional ecosystem services gradually emerged to include combating parasitic weeds of cereals (such as Striga spp.), increase soil nitrogen and carbon, and even reducing incidence of mycotoxins in maize (Balaso et al. 2019; Cook et al. 2007; Xu et al. 2018; Owuor et al. 2018; Njeru et al. 2020). This study adds to these benefits by describing a diversification of the soil microbial communities, with a particularly strong shift in the composition of fungal taxa. By itself diversity in ecosystems is generally regarded as increasing stability, resilience and productivity (Prieto et al. 2015), mostly as a consequence of resource complementarity and functional redundancy (Cleland 2011; Rosenfeld 2002).
Looking at bacterial populations, the discussion is limited by two factors; fewer taxa that are significantly enriched in either of the farming systems and limited classification (identification) at the genus level. Nevertheless, the genus Nitrospira is one of the identified genera that was enriched in maize—Desmodium intercropping plots. Species of this genus are known for their ability to perform the complete nitrification process (oxidation of ammonia) during nitrogen fixation, unlike other nitrifying bacteria in which the process occurs in two different organisms (Koch et al. 2015). An enriched presence of Nitrospira spp. in intercropping plots suggests involvement in nitrogen fixation, potentially contributing to increased nitrogen supply in the soil and in turn leading to a higher maize yield as previously reported (Khan and Pickett 2008).
Concluding remarks
This study has shown that long-term maize—Desmodium intercropping causes a complex shift in composition of the soil microbiome compared to maize monoculture. Many functions of soil microbial communities arise through complex interactions and ecosystem services may therefore not be readily attributed to a single taxon, but arise as an emergent property of system, although exceptions exist (Reva et al. 2019). Given the increasing accessibility of sequencing technologies, metagenomics and other DNA-based analyses should be included as an integral part of intercropping studies for improvement of crop health and productivity. Metagenomics data can facilitate interpretation of complex community structure and composition in the light of plant productivity, plant health, and more broadly, ecosystem health.
Availability of data and material
The datasets generated during and analysed during the current study are available in the Genbank Sequence Read Archive (SRA) repository under project number PRJNA667690 linked here https://www.ncbi.nlm.nih.gov/bioproject/PRJNA667690/.
Change history
15 August 2022
Missing Open Access funding information has been added in the Funding Note.
References
Balaso GM, Nalini C, Yankit P, Thakur P (2019) Push-pull strategy: novel approach of pest management. J Entomol Zool Stud 7(5):220–223
Bargaz A, Noyce GL, Fulthorpe R, Carlsson G, Furze JR, Jensen ES, Dhiba D, Isaac ME (2017) Species interactions enhance root allocation, microbial diversity and P acquisition in intercropped wheat and soybean under P deficiency. Appl Soil Ecol 120:179–188
Bisanz JE (2018) qiime2R: Importing QIIME2 artifacts and associated data into R sessions. R package https://github.com/jbisanz/qiime2R. Accessed 01 Oct 2020
Blighe K, Rana S, Lewis M (2020) EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.8.0 https://github.com/kevinblighe/EnhancedVolcano. Accessed 01 Oct 2020
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu Y-X, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857
Bybee-Finley KA, Ryan MR (2018) Advancing intercropping research and practices in industrialized agricultural landscapes. Collect FAO Agric 8:80
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583
Cleland EE (2011) Biodiversity and ecosystem stability. Nature Educ Knowl 3:14
Cong W-F, Hoffland E, Li L, Six J, Sun J-H, Bao X-G, Zhang F-S, van der Werf W (2015) Intercropping enhances soil carbon and nitrogen. Glob Change Biol 21(4):1715–1726
Contreras-Cornejo HA, Macías-Rodríguez L, del-Val E, Larsen J, (2018) The root endophytic fungus Trichoderma atroviride induces foliar herbivory resistance in maize plants. Appl Soil Ecol 124:45–53
Cook SM, Khan ZR, Pickett JA (2007) The use of push-pull strategies in integrated pest management. Annu Rev Entomol 52:375–400
Dang K, Gong X, Zhao G, Wang H, Ivanistau A, Feng B (2020) Intercropping alters the soil microbial diversity and community to facilitate Nitrogen assimilation: a potential mechanism for increasing proso millet grain yield. Front Microbiol 11:2975
Díaz-González S, Marín P, Sánchez R, Arribas C, Kruse J, González-Melendi P, Brunner F, Sacristán S (2020) Mutualistic fungal endophyte Colletotrichum tofieldiae Ct0861 colonizes and increases growth and yield of maize and tomato plants. Agronomy 10:1493
Guangchuang Y, David S, Huachen Z, Yi G, Ggtree TT-YL (2017) ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8:28–36
Hiruma K, Gerlach N, Sacristán S, Nakano RT, Hacquard S, Kracher B, Neumann U, Ramírez D, Bucher M, O’Connell RJ, Schulze-Lefert P (2016) root endophyte Colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent. Cell 165:464–474
Iqbal M, Dubey M, McEwan K, Menzel U, Franko MA, Viketoft M, Jensen DF, Karlsson M (2018) Evaluation of Clonostachys rosea for control of plant-parasitic nematodes in soil and in roots of carrot and wheat. Phytopathology 108:52–59
Johansen A, Jensen ES (1996) Transfer of N and P from intact or decomposing roots of pea to barley interconnected by an arbuscular mycorrhizal fungus. Soil Biol Biochem 28(1):73–81
Khan ZR, Pickett JA (2008) Push-pull strategy for insect pest management. In: Capinera JL (ed) Encyclopedia of entomology. Springer, Dordrecht. https://doi.org/10.1007/978-1-4020-6359-6_3253
Khan ZR, Hassanali A, Overholt W, Khamis TM, Hooper AM, Pickett JA, Wadhams LJ, Woodcock CM (2002) Control of witchweed Striga hermonthica by intercropping with Desmodium spp., and the mechanism defined as allelopathic. J Chem Ecol 28:1871–1885
Khan Z, Hassanali A, Pickett J, Wadhams L, Muyekho F (2003) Strategies for control of cereal stemborers and striga weed in maize-based farming systems in eastern Africa involving “push-pull” and allelopathic tactics, respectively. Afr Crop Sci Conf Proc 6:602–608
Khan ZR, Midega CAO, Bruce TJA, Hooper AM, Pickett JA (2010) Exploiting phytochemicals for developing a “push-pull” crop protection strategy for cereal farmers in Africa. J Exp Bot 61:4185–4196
Koch H, Lücker S, Albertsen M, Kitzinger K, Herbold C, Spieck E, Nielsen PH, Wagner M, Daims H (2015) Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc Natl Acad Sci U S A 112:11371–11376
Kuhn M, Wickham H (2020) recipes: Preprocessing tools to create design matrices R package version 1.8.0 https://github.com/tidymodels/recipes. Accessed 01 Oct 2020
Li Q, Chen J, Wu L, Luo X, Li N, Arafat Y, Lin S, Lin W (2018) Belowground Interactions impact the soil bacterial community, soil fertility, and crop yield in maize/peanut intercropping systems. Int J Mol Sci 19
Liu H, Li J, Carvalhais LC, Percy CD, Prakash Verma J, Schenk PM, Singh B (2021) Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytol 229:2873–2885
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Macías-Rubalcava ML, Hernández-Bautista BE, Jiménez-Estrada M, González MC, Glenn AE, Hanlin RT, Hernández-Ortega S, Saucedo-García A, Muria-González JM, Anaya AL (2008) Naphthoquinone spiroketal with allelochemical activity from the newly discovered endophytic fungus Edenia gomezpompae. Phytochem 69:1185–1196
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217
Midega CAO, Salifu D, Bruce TJ, Pittchar J, Pickett JA, Khan ZR (2014) Cumulative effects and economic benefits of intercropping maize with food legumes on Striga hermonthica infestation. Field Crop Res 155:144–152
Midega CAO, Pittchar JO, Pickett JA, Hailu GW, Khan ZR (2018) A climate-adapted push-pull system effectively controls fall armyworm, Spodoptera frugiperda (J E Smith), in maize in East Africa. Crop Prot 105:10–15
Munasinghe MVK, Kumar NS, Jayasinghe L, Fujimoto Y (2017) Indole-3-acetic acid production by Colletotrichum siamense, an endophytic fungus from Piper nigrum leaves. J Biol Active Prod Nat 7:475–479
Mutyambai DM, Bass E, Luttermoser T, Poveda K, Midega CAO, Khan ZR, Kessler A (2019) More than “push” and “pull”? plant-soil feedbacks of maize companion cropping increase chemical plant defenses against herbivores. Front Ecol Evol 7:217
Nilsson RH, Larsson K-H, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, Kennedy P, Picard K, Glöckner FO, Tedersoo L, Saar I, Kõljalg U, Abarenkov K (2019) The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47:D259–D264
Njeru NK, Midega CAO, Muthomi JW, Wagacha JM, Khan ZR (2020) Impact of push–pull cropping system on pest management and occurrence of ear rots and mycotoxin contamination of maize in western Kenya. Plant Pathol 69:1644–1654
Nourbakhsh F, Koocheki A, Mahallati MN (2019) Investigation of biodiversity and some of the ecosystem services in the intercropping of corn, soybean and marshmallow. Int J of Plant Prod 13:35–46
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2020) vegan: Community ecology package. R package https://github.com/vegandevs/vegan. Accessed 01 Oct 2020
Owuor M, Midega C, Obonyo M, Zeyaur K (2018) Impact of companion cropping on incidence and severity of maize ear rots and mycotoxins in Western Kenya. Afr J Agric Res 13:2224–2231
Paulson JN, Stine OC, Bravo HC, Pop M (2013) Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10:1200–1202
Pedersen TL (2020) ggforce: Accelerating “ggplot2”. R package https://github.com/thomasp85/ggforce. Accessed 01 Oct 2020
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V et al (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830
Poveda J, Zabalgogeazcoa I, Soengas P, Rodríguez VM, Cartea ME, Abilleira R, Velasco P (2020) Brassica oleracea var. acephala (kale) improvement by biological activity of root endophytic fungi. Sci Rep 10:20224
Prieto I, Violle C, Barre P, Durand J-L, Ghesquiere M, Litrico I (2015) Complementary effects of species and genetic diversity on productivity and stability of sown grasslands. Nat Plants 1:15033
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available online at https://www.R-project.org/
Ravnskov S, Jensen B, Knudsen IMB, Bødker L, Funck Jensen D, Karliński L, Larsen J (2006) Soil inoculation with the biocontrol agent Clonostachys rosea and the mycorrhizal fungus Glomus intraradices results in mutual inhibition, plant growth promotion and alteration of soil microbial communities. Soil Biol Biochem 38:3453–3462
Reva ON, Swanevelder DZH, Mwita LA, Mwakilili AD, Muzondiwa D, Joubert M, Chan WY, Lutz S, Ahrens CH, Avdeeva LV, Kharkhota MA, Tibuhwa D, Lyantagaye S, Vater J, Borriss R, Meijer J (2019). Genetic, epigenetic and phenotypic diversity of four bacillus velezensis strains used for plant protection or as probiotics. Front Microbiol 10:2610. Available at: https://www.frontiersin.org/article/10.3389/fmicb.2019.02610
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl Acids Res 43:e47
Rosenfeld JS (2002) Functional redundancy in ecology and conservation. Oikos 98:156–162
Sarrocco S, Mauro A, Battilani P (2019) Use of competitive filamentous fungi as an alternative approach for mycotoxin risk reduction in staple cereals: state of art and future perspectives. Toxins 11
Schnitzer SA, Klironomos JN, Hillerislambers J, Kinkel LL, Reich PB, Xiao K, Rillig MC, Sikes BA, Callaway RM, Mangan SA, van Nes EH, Scheffer M (2011) Soil microbes drive the classic plant diversity-productivity pattern. Ecology 92:296–303
Song YN, Zhang FS, Marschner P, Fan FL, Gao HM, Bao XG, Sun JH, Li, L (2007) Effect of intercropping on crop yield and chemical and microbiological properties in rhizosphere of wheat (Triticum aestivum L.), maize (Zea mays L.), and faba bean (Vicia faba L.) Biol Fert Soil 5(43)565–574
Stagnari F, Maggio A, Galieni A, Pisante M (2017) Multiple benefits of legumes for agriculture sustainability: an overview. Chem & Biol Tech in Agric 4:2
Stephens M, Carbonetto P, Dai C, Gerard D, Lu M, Sun L, Willwerscheid J, Xiao N, Zeng M (2020) ashr: Methods for adaptive shrinkage, using empirical Bayes. R package https://github.com/stephens999/ashr. Accessed 01 Oct 2020
Tang X, Bernard L, Brauman S, Daufresne T, Deleporte P, Desclaux D, Souche G, Placella SA, Hinsinger P (2014) Increase in microbial biomass and phosphorus availability in the rhizosphere of intercropped cereal and legumes under field conditions. Soil Biol Biochem 75:86–93
van der Heijden MGA, Bardgett RD, van Straalen NM (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310
Wang G, Bei S, Li J, Bao X, Zhang J, Schultz PA, Li H, Li L, Zhang F, Bever JD, Zhang J (2020) Soil microbial legacy drives crop diversity advantage: Linking ecological plant–soil feedback with agricultural intercropping. J Appl Ecol 186:281
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer, New York
Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen TL, Miller E, Bache SM, Müller K, Ooms J, Robinson D, Seidel DP, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H (2019) Welcome to the tidyverse. J Open Source Softw 4:1686
Wingett SW, Andrews S (2018) FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res 7:1338
Xu Q, Hatt S, Lopes T, Zhang Y, Bodson B, Chen J, Francis F (2018) A push–pull strategy to control aphids combines intercropping with semiochemical releases. J Pest Sci 91:93–103
Zin NA, Badaluddin NA (2020) Biological functions of Trichoderma spp. for agriculture applications. Sci Ann Univ Agric Sci Vet Med 65:168–178
Acknowledgements
This research was made possible with funding from SIDA of Sweden through the University of Dar es Salaam, Tanzania, within the sub-programme ‘Sustainable Agricultural Productivity—Processing and Value Chain to Enhance Food Security in Tanzania (ADM, TD, SLL), and The Swedish Research Council (621-2014-4816, TD). We thank the laboratory and field staff at ICIPE Thomas Odhiambo and Duduville campuses for their generous support in sample collection and processing and DNA extraction, especially Dickens Nyagol, Levi and Maureen.
Funding
Open access funding provided by Swedish University of Agricultural Sciences. Funding for this research was provided by a Research Capacity Building Program from Swedish International Development Cooperation Agency (SIDA) to University of Dar es Salaam (UDSM)—(ADM, TD, SLL) and the Swedish Research Council (621-2014-4816, TD).
Author information
Authors and Affiliations
Contributions
Conceptualization and advice: TD, SLL, ADM; Methodology and advice: TD, SLL, SLH, ADM, BA, CM, FM; Formal analysis and investigation: ADM, SLH, KSM, SLL, TD; Writing—original draft preparation: ADM, KSM, TD, SLL Writing, review and editing: all authors. Funding acquisition: TD, SLL, FM Supervision: TD, SLL, FM, CM, BA. All authors approved the submitted version for publication.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Responsible Editor: Birgit Mitter.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mwakilili, A.D., Mwaikono, K.S., Herrera, S.L. et al. Long-term maize-Desmodium intercropping shifts structure and composition of soil microbiome with stronger impact on fungal communities. Plant Soil 467, 437–450 (2021). https://doi.org/10.1007/s11104-021-05082-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-021-05082-w