
Abstract
Background and aims
As an essential mineral element, selenium (Se) plays a critical role in human health. Given the low concentrations (<100 mg Se kg–1) of Se in staple crops, the identification of genetic resources with enriched Se, as well as the genes controlling Se concentration, is valuable for the marker-assisted selection of Se-rich varieties.
Methods
We determined the chromosomal quantitative trait (QTL) for Se concentration over two consecutive plant growth cycles using recombinant inbred lines (RILs) treated with two different concentrations of Se under both field-grown and hydroponic conditions.
Results
Several QTL for Se concentration were detected across the different treatments. Significant genotypic variation in the tissues of the RIL was found at Se-deficicencycondition. Notably, a QTL located on 3D (interval 214.00–218.00, Qse.sau-3D) affected root length and Se concentration in the leaves and grains, suggesting the existence of the same allele with distinctly different functions. However, the QTL for the agronomic traits measured (plant height, flowering time, and tillering number) and Se concentration were not found to be located on the same chromosomal regions, suggesting that marker-assisted selection for both traits is feasible. Se concentrations in the grains were primarily determined by the mineral transport efficiency of the lines, and the line with the highest Se concentration in the grains always possessed larger, more fibrous root systems. The concentrations of Se in the plant tissues were in the order of: root > stem > grain.
Conclusions
This is the first study to document a Se-rich synthetic wheat line, and root structure and Se grain concentration was strongly affected by QTL located on 3D.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
http://www.ers.usda.gov/data-products/wheat-data.aspx
Introduction
Humans require more than 22 essential mineral elements, all of which can be supplied by an appropriate diet (Broadley et al. 2010). Selenium (Se) incorporates into proteins to make selenoproteins, which are important antioxidant enzymes that help prevent cellular damage from free radicals. Se deficiency can result in the development of chronic diseases, such as cancer and heart disease (Zhao et al. 2016). However, it is estimated that more than 15% of the human population suffers from Se deficiency due to the low concentrations of Se present in staple crops (White 2016a, b). As humans consume a wide variety of foods, primarily plant-derived, the elemental quality of grains, including Se concentration, may influence human health. As the second largest food crop in the world, the yield of wheat was 723 million tons in 2015 (USDA) and accounted for approximately 20% of calorie intake in humans (FAO 2016). The Se content of wheat is thus an important consideration in human health. It is for this reason that the biofortification of crops, including the application of mineral fertilizers and improvements to the ability of seeds to acquire mineral elements, is advocated as an immediate strategy to not only increase mineral concentrations in edible crops, but also improve yields from infertile soils.
Despite the importance of carbohydrates and minerals, such as Se, for human and plant health for genetic studies,previous review highlights the possible genetic basis of differences in plant species (White 2016a, b), in this study, a recombinant inbred line (RIL) population was developed from two wheat varieties that display distinct differences in Se concentration to identify the chromosomal loci that might enable the development of wheat with greater Se concentrations in kernals.. As the capacity of the roots to acquire Se from the soil is controlled by major gene loci in rice and wheat (Norton et al. 2010; Pu et al. 2014), we chose to measure root characters in order to evaluate this function. We phenotyped the wheat under treatment with Se in both field and hydroponic conditions, and genotyped using diversity arrays technology (DArT) and simple sequence repeat (SSR) markers. Novel genes or loci associated with high Se concentration were identified using quantitative trait loci (QTL) mapping.
Materials and methods
Plant material and field experimental conditions (T1)
The two lines used in this study include the synthetic hexaploid line, SHW-L1, and the Chinese wheat cultivar Chuanmai32; SHW-L1 is derived from tetraploid wheat AS2255 (Triticum turgidum) and Aegilops tauschii AS60. AS2255 is a unique tetraploid landrace in China. The population (SHW-L1/Chuanmai32) was composed of 171 F11/F12 lines and was generated by single seed descent in the field at Sichuan Agricultural University, Dujiangyan District, Sichuan Province, China.Flowering time was recorded for each plant when the first florets flowered, measured from the time of sowing.
SHW-L1 was previously found to exhibit 5–8 fold greater Se concentrations than Chuanmai32 under identical growth conditions in a Se-deficient field (Pu et al. 2014). Phenotypic evaluation of the populations was conducted in the field at the Sichuan Agricultural University over two plant growth cycles (2014 and 2015). Each RIL and their parents were planted in pots (Φ = 30 cm), with five plants per pot. These pots were placed in the field using a randomized block design with three replications. The natural Se concentration of the field soil was 0.22 μg/g, and this soil was used in the control treatment (CK1). For the Se treatment, sodium selenite (Na2SeO3) was added into the soil at a concentration of 0.50 μg/g. The Se-rich soil was subjected to three wetting (50% of maximum water-holding capacity [MWHC]) and drying (30 °C) cycles 4 weeks prior to the growth experiment, then the Se-rich soil was put into pots for the treatment (T1).
Hydroponic experimental conditions (T2)
In order to assess the response of the roots to elevated Se concentration, the populations were also cultivated hydroponically in a greenhouse under a 16-h light: 8-h dark photoperiod with a temperature range of 18 °C to 25 °C and humidity maintained at 75% over two consecutive plant growth cycles. A randomized complete block design with three replications was used. The seeds of each genotype were sterilized for 5 min with 10% H2O2 and then washed with distilled water. The endosperms were removed after germination and placed on moist filter paper in Petri dishes. Each RIL and parent were divided into two groups for the hydroponic experiment, and treated with either 0.22 mg/L (CK2) or 0.50 mg/L (T2) Na2SeO3 for the duration of the plant growth cycle. Uniform seedlings from CK2 and the treatment were selected and transferred with sponges into hydroponic planting box (610 × 420 × 360 mm), with six rows and 10 columns such that each treatment group consisted of six homogenous wheat seedlings from each RIL. The hydroponic planting boxes were filled with 25 L Hoagland nutrient solutionat (Hoagland and Arnon 1950) either 0.22 mg/L or 0.50 mg/L Se concentrations, which was replaced entirely once a week to prevent nutrient depletion and to expose the seedlings to a constant Se concentration. A pumped aeration system was used to ensure a constant supply of oxygen. The pH was adjusted daily to 6.5 using a dilute sodium hydroxide (NaOH) solution (0.5%).
The plants were grown for 42 (tillering stage) and 65 (elongation stage) days to analyze the changes in the roots. An Epson XL (11,000 ×) scanner with the WinRHizo Pro 2008a image analysis system was used to measure the morphological variables (root diameter, root length, root surface area, root volume, tips, branching angles, branch roots, root forks, and root length density). The roots and stems were oven dried at 110 °C until constant weight to obtain dehydrated weight measurements.
Statistical analysis
One-way analysis of variance (ANOVA) and least significant difference (LSD) tests of the Se concentrations of each RIL, and Pearson’s correlation coefficients between Se concentration and agronomic characters, were computed using SPSS 19.0 (IBM SPSS, Chicago, IL, USA). Broad sense heritability was calculated using the formula: h2 = VG/(VG + VGE/y + VGE/l + VE/y.l), where, VG = genotypic variance, VGE = genotype × environment variance, y = number of years, l = number of plant location, and VE = environmental variance (Smith et al. 1998).
Map construction and QTL mapping
QTL analysis was conducted in IciMapping version 4.1 with BIN implemented to remove markers with high missing rates. MAP was used for the genetic map construction (Meng et al. 2015). Genetic map data of the population was reported in a previous study (Yu 2013). The genetic maps contained 1862 markers (SSR and DArT) covering a genetic distance of 3766.9 cM, with a density of 2.0 cM/marker. The ICIM-ADD model was used to identify the QTL. For each trial, a test of 1000 permutations was performed to identify the logarithm of the odds ratio (LOD) threshold corresponding to a genome-wide false discovery rate of 5% (P < 0.05). Permutation tests confirmed that an LOD threshold of 2.5 was adequate to declare the presence of a QTL.
Comparative genomics analysis
Assembled whole genome sequences of Brachypodium distachyon and rice (Oryza sativa) were obtained from Phytozome (http://www.phytozome.org/). Whole genome shotgun sequences of Aegilops tauschii were downloaded from NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/). The coding sequences (CDSs) of A. tauschii were downloaded from GIGA_DB (http://gigadb.org/); B. distachyon from PlantGDB (http://www.plantgdb.org/BdGDB); and rice from Phytozome. DArT sequences of wheat were retrieved online (http://www.diversityarrays.com/). Sequence alignments of wheat, B. distachyon, A. tauschii, and rice were conducted with BLASTN with an E-value cutoff of 10−5.
Results
QTL of agronomic characters
Plant height, tillering number, and flowering time were measured in the control (CK1) and the treatment (T1) under field experimental conditions. The average heights of the treatment plants were approximately 4 cm greater than CK1, and the flowering period was earlier by about 3 d. This indicates that the Se treatment increased the height of the RILs and induced earliness. Even under the same Se conditions, the differences in agronomic characters were significant (Table 1), implying that the interaction between soil Se concentration and genotype influences height and tillering number.
The QTL for tillering number were consistently detected in both the control and treatment, such as Qse.sau-3A (position 46.81). Three QTL located on 2D (positions 54.01, 56.61, and 64.11) involved in the control of flowering time exhibited a fixed expression pattern regardless of Se treatment (Table 2).
Root characters and relative QTL under hydroponic experimental conditions
The parameters of the nine root traits, including root diameter, root length, root surface area, root volume, tips, branching angles, branch roots, root forks, root length density, and dehydrated weight of the of the seedlings were analyzed in the tillering and elongation periods of the roots in the Se treatment and control (Table 1). Changes in Se concentration in the different root samples were analyzed during growth and development. The branch roots, root volume, and root surface area were significantly increased in comparison to the control conditions, and the root length and root length density of CK2 was significantly higher than the Se addition condition, suggesting that the addition of Se altered the root structure. This trend did not vary between the growth periods.
A total of 14 QTL for root characters were identified during the growing period (Table 3). A QTL on 2A was identified for the number of branch roots and root forking at the tillering stage in the Se treatment with an LOD of 4.20, and explained 9.86% of the phenotypic variance. Another QTL was identified for tillering and root forking with an LOD of 3.41, explaining 8.42% of the phenotypic variance. The QTL located on 3D (position 216.61) was associated with the number of branch roots and root forks at the elongation stage in CK2. Branch roots and root length shared the same QTL located on 3D (214.61, in the region of wPt-73,344~wPt-667,315) with an LOD score of 4.67, explaining 8.94% of the phenotypic variance.
The RIL population grown in the field
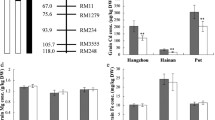
The Se concentrations of the grains (T1), roots (T2) and stems (T2) were measured (Table 1). At the tillering stage, the Se concentration in the aboveground and belowground parts of the Se addition treatment was 6.85- and 5.82-fold greater than CK2, respectively. This decreased to 2.05- and 2.10-fold, respectively, at the jointing stage. We hypothesize that Se has a higher transport rate from the belowground to aboveground parts at the tillering stage, but that the transport rate decreases with seedling growth. The concentrations of Se in the tissues between the treatment and CK differed significantly (Table 1). When Se was supplied abundantly, the concentration differences between the different genotypes disappeared. Se concentration tends to increase the most during seedling growth, and then declines before, or upon, jointing, and continues to decline until flowering.
The Se concentration of SHW-L1 ranged from 0.0891 to 0.1271 (0.1081 ± 0.019) μg/g, and the concentration of Chuanmai32 ranged from 0.0103 to 0.0699 (0.0401 ± 0.0298) μg/g with 3.63% broad-sense heritabilities. At maturity, the Se concentrations differed across the tissues in the order of root > stem > grain.
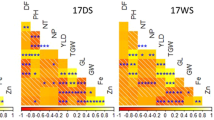
QTL for Se concentration were identified in the roots, leaves, and grains of the SHW-L1/Chuanmai32 RIL population. Most of the 24 QTL for Se concentration in the grains, roots, and leaves were detected on 1B, 3D, and 7D (Table. 4). Of these, six, six, and 12 QTL were found for grain, root, and leaf, respectively. An individual QTL explained between 4.38% (root Se concentration in T2) and 24.38% (grain Se concentration in T1) of the phenotypic variation. The highest LOD value of a single QTL was 12.44 (grain Se concentration in T1) and this QTL was also located on 3D. Sixteen and eight QTL were detected in T1 and T2, respectively. QTL located on 3D were detected in both CK and the treatment (Table 3), and most of the QTL on 3D were detected in the Se treatment in the field experiment. The QTL on chromosome 7D were detected in the Se treatment (T2), and the QTL located on chromosome 5A might be novel. The results also demonstrated that different Se treatments result in the expression of different QTL.
Four QTL alleles could be detected in different tissues and environments, three of which were clustered on 3D (positions 222.11, 216.61, 214.61) and influenced the branch roots, root length, and Se concentration of the leaves and grains, suggesting that 3D may be existent different distinct functional alleles to control Se accumulation or uptake(Table 5).
Collinearity analysis and comparative genetics
Based on the fact that 50% of the QTL for grain Se concentration in this study were mapped on 3D (position 214.61~222.11) suggests that this chromosome plays a key role in regulating Se metabolism. For further fine mapping of these QTL, comparative genomics were conducted with A. tauschii, B. distachyum, and O. sativa. As shown in Table 6, the interval where the majority of QTL were mapped had collinearity with chromosome 2 and 1 of B. distachyum and rice, respectively.
Most of the rice genes were functionally annotated: LOC_Os01g02800.1 is a receptor-like kinase ARK1AS; LOC_Os01g02770.1 is a resistance-related receptor-like kinase; LOC_Os01g02390.1 is a protein kinase domain containing protein; LOC_Os01g02020.3 may be an acetyl-CoA acetyl-transferase in the cytosol; LOC_Os01g01840.1 is a helix-loop-helix DNA-binding domain containing protein; and LOC_Os01g01780.1 belongs to the exostosin family protein. The functions of LOC_Os01g01800.2, LOC_Os01g01790.1, and LOC_Os01g01790.2 could not be determined.
Discussion
Breeding Se-enriched crops may constitute a potential solution for improving nutrition in Se-deficiency regions of the world. Improving mineral concentrations this way is widely accepted, and suitable germplasm resources are important to this end. Wheat is a non-accumulator and is Se-sensitive (White 2016a, b), thus a Se-enriched wheat genetic resource would be highly valuable. Identifying the QTL for Se concentration and applying it to wheat breeding is necessary to realize this. In this study it was established that genotypic enhancement can improve the acquisition power, utilization, or accumulation of Se in crops under low Se conditions.
QTL of agronomic characteristics
Agronomic characters of the same wheat RILs were previously analyzed by Yu (2013). The QTL for plant height, tillering number, and flowering time were also found to be located on 2D (positions 54.01, 56.61, and 64.11) in this study. Furthermore, new QTL for flowering time were found on 1A (tPt-1755-wPt-732,113) and 2B (wPt-6199-wPt-2600) in the Se addtion treatment experiment in our study. The addition of Se altered the expression pattern of the QTL for height, which is in accordance with previous studies that have demonstrated that treatment with Se improves wheat biomass (Ahmad et al. 2016).
Root characteristics and QTL expression
Se can be taken up by plant roots as selenate, selenite, or organoselenium, and is thought to be redistributed in the plant as selenate and/or organoselenium via the phloem (White et al. 2004). Most of the genetic variation in shoot Se concentrations was previously discovered to be at the Se- deficiency condition, and no obvious differences were observed at the Se-rich condition. This indicates that Se concentrations in the shoot tissues may be evolutionarily constrained (Ortiz-Monasterio et al. 2007). This phenomenon was also observed in our study, suggesting that there is a genetic component that controls uptake under low Se levels in the soil. This implies that the factor limiting the uptake of Se in the roots at Se-deficiency condition is Se uptake capacity. It is evident that if the Se in the soil is insufficient, the presence of high Se concentrations in the grains may be dictated by genotype. There is a strong positive correlation between the ability of a wheat genotype to accumulate Se and grain Se concentration. Additionally, systematic variation in shoot Se concentrations has been previously documented (Graham et al. 2005). The conversion ratio of the accumulated Se in the grains was previously found to be lower than 50% (Keskinen et al. 2010), and our findings suggest that the nutritional efficiency of Se fertilization would change the distribution pattern of Se in plants (Wang et al. 2003).
Molecular marker utilization
When QTL are used in breeding, the relationships among trace elements, yield, and quality are important. QTL for Fe and Zinc concentration have been identified, and the corresponding molecular markers have been used in breeding programs (Ghandilyan et al. 2006). Previous studies have indicated a weak correlation between yield and mineral element concentration (including Fe, Zn, Ca, and Mg) in sorghum grain (Ng'uni et al. 2016), wheat (Pu et al. 2014), and common bean seeds (Cichy et al. 2005). Conversely, other researchers have reported no significant relationships between the concentrations of particular mineral elements in the grain and the yield of wheat and maize (Ortiz-Monasterio et al. 2007). Furthermore, the fact that there is a lack of coincidental QTL influencing seed yield and Fe or Zn concentrations at the same time (Vreugdenhil et al. 2004; Cichy et al. 2005; Stangoulis et al. 2007) suggests that these microelements and yield are controlled by different genes. Thus, increased concentrations of Fe/Zn in rice and bean breeding programs have been reported (Norton et al. 2010; Ates et al. 2016). In previous studies on lentils, rice, and wheat (Norton et al. 2010; Pu et al. 2014; Ates et al. 2016), the QTL for agronomic characteristics were not found to be located in the same chromosomal regions as those for Se concentration. This suggests that breeders could use molecular markers in breeding and selection for modified Se levels without affecting the grain. For instance, existing evidence shows that increasing Se concentrations in durum wheat grains via foliage dressing does not alter the nutritional profile of pasta during processing and cooking (De Vita et al. 2017).
Notably, Qse.sau-3D was detected in multiple tissues over two consecutive plant growth cycles in this study, and the associated QTL were continuously expressed during the growth period at Se-deficiency conditions. We therefore attach great importance to the QTL located on 3D, which were found to be associated with both root characteristics and grain Se concentration. As plants uptake mineral elements from the soil via the roots, we believe that Qse-3D improves Se concentration by controlling the root morphology and architecture, or Se phytoavailability (White 2016a). Future work should thus aim to determine the candidate genes that might control Se concentrations.
The gene denotation of the candidate interval
Compared with other available genomes, the 3D interval possessed several denoted genes, most of which were related to kinases or protein binding. To our knowledge, several of the kinases are associated with disease resistance, including a receptor-like protein kinase gene that plays important roles in rice blast disease resistance (Li et al. 2009). Though the synthetic wheat line SHW-L1 possesses the characteristic of high resistance to stripe rust, we did not identify a direct connection between kinase and Se, although the elements that control the interactions of cytoplasmic signaling proteins and kinase activity have been previously reported (Koch et al. 1991). It is possible that Se regulates the signaling pathways that control root architecture and cell metabolism in stripe rust resistance.
Potential use of synthetic wheat
Associations between genotypic variation and mineral element concentrationn wheat have been reported (Chao et al. 2014, White 2016a, b), and improving the concentration in minerals is achievable through the utilization of germplasm resources (Lyons et al. 2005). We found higher genotypic variation in the synthetic hexaploid lines at low Se conditions, implying that Se concentrations in the tissues are associated with the wheat genotype. Conversely, relatively little variation was found in the shoots, stems, and grains at grain Se concentrations at the Se-rich condition, which is in accordance with the findings of White et al. (2004).
The SHW line has been widely used as a bridge for transferring major disease-resistance genes (stripe rust, powdery mildew, and spot blotch) from tetraploid wheat and A. tauschii to common wheat. This synthetic line also exhibits higher Fe, Mn, K, and P uptake than other cultivars (Calderini and Ortiz-Monasterio 2003; Wang et al. 2011). We first discovered that SHW-L1 is a Se-rich genetic resource with higher uptake efficiency when all the positive loci, including Qse.sau-3D, emanated from SHW-L1. Additionally, the lines derived from SHW also demonstrated high Se concentration and genetic variability, and the harvest index of Se was increased in the synthetics and their offspring. This demonstrates that synthetics constitute a potentially valuable germplasm source for increasing Se concentration in the grains, as well as for increasing Fe, Mn, K, and P concentrations (Calderini and Ortiz-Monasterio 2003).
References
Ahmad R, Waraich EA, Nawaz F, Ashraf MY, Khalid M (2016) Selenium (Se) improves drought tolerance in crop plants – a myth or fact? J Sci Food Agric 96(2):372–380. https://doi.org/10.1002/jsfa.7231
Ates D, Sever T, Aldemir S, Yagmur B, Temel HY, Kaya HB, Ahmad Alsaleh A, Kahraman A, Ozkan H, Albert V, Bahattin T (2016) Identification QTLs Controlling Genes for Se Uptake in Lentil Seeds. PLoS One 11(4):e0154054. https://doi.org/10.1371/journal.pone.0149210
Broadley MR, Alcock J, Alford J, Cartwright P, Foot I, Fairweather-Tait SJ, Hart DJ, Hurst R, Knott P, McGrath SP, Meacham MC, Norman K, Mowat H, Scott P, Stroud JL, Tovey M, White TM, Philip J, Young SD, Zhao FJ (2010) Selenium biofortification of high-yielding winter wheat (Triticum aestivum L.) by liquid or granular Se fertilization. Plant Soil 332:5–18. https://doi.org/10.1007/s11104-009-0234-4
Calderini DF, Ortiz-Monasterio I (2003) Are synthetic hexaploids a means of increasing grain element concentrations in wheat? Euphytica 134:169–178. https://doi.org/10.1023/B:EUPH.0000003849.10595.ac
Chao DY, Baraniecka P, Danku J, Koprivova A, Lahner B, Luo HB, Yakubova E, Dilkes B, Kopriva S, Salt DE (2014) Variation in sulfur and selenium accumulation is controlled by naturally occurring isoforms of the keysulfur assimilation enzyme ADENOSINE 5’-PHOSPHOSULFATEREDUCTASE2 across the Arabidopsis species range. Plant Physiol 166:1593–1608. https://doi.org/10.1104/pp.114.247825
Cichy KA, Forster S, Grafton KF, Hosfield G (2005) Inheritance of Seed Zinc Accumulation in Navy Bean. Crop Sci 45(3):864–870. https://doi.org/10.2135/cropsci2004.0104
De Vita P, Platani C, Fragasso M, FiccoDBM CSA, Nobile MAD, Padalino L, Spartaco DG, Petrozza A (2017) Selenium-enriched durum wheat improves the nutritional profile of pasta without altering its organoleptic properties. Food Chem 214:374–382. https://doi.org/10.1016/j.foodchem.2016.07.015
Doerge R, Churchill G (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Food and Agriculture Organisation of the United Nations. FAOSTAT (2016) http://faostat3.fao.org/browse/Q/QC/E
Ghandilyan A, Vreugdenhil D, Aarts MGM (2006) Progress in the genetic understanding of plant iron and zinc nutrition. Physiol Plant 126:407–417. https://doi.org/10.1111/j.1399-3054.2006.00646.x
Graham HL, James CRS, Robin DG (2005) Tolerance of wheat (Triticum aestivum L.) to high soil and solution selenium levels. Plant and Soil 270(1):179–188 https://doi.org/10.1007/s11104-004-1390-1
Hoagland DR, Arnon DI (1950) The water-culture method for growing plants without soil. Circ Calif Agr Exp Sta 347:1–39
Keskinen R, Turakainen M, Hartikainen H (2010) Plant availability of soil selenate additions and selenium distribution within wheat and ryegrass. Plant Soil 333(1–2):301–313. https://doi.org/10.1007/s11104-010-0345-y
Koch CA, Anderson D, Moran MF, Ellis C, Pawson T (1991) SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 252(5006):668
Li H, Zhou SY, Zhao WS, Su SC, Peng YL (2009) A novel wall-associated receptor-like protein kinase gene, OsWAK1, plays important roles in rice blast disease resistance. Plant Mol Biol 69(3):337–346. https://doi.org/10.1007/s11103-008-9430-5
Lyons G, Ortiz-Monasterio JI, Stangoulis GR (2005) Selenium concentration in wheat grain: Is there sufficient genotypic variation to use in breeding? Plant Soil 269(1–2):369–380. https://doi.org/10.1007/s11104-004-0909-9
Meng L, Li HH, Zhang LY, Wang JK (2015) QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283. https://doi.org/10.1016/j.cj.2015.01.001
Ng'uni D, Shargie NG, Andersson SC, Biljon AV, Labuschagne MT (2016) Genetic variation and trait associations of yield, protein and grain micronutrients for identification of promising sorghum varieties. Cereal Res Commun 44(4):681–693. https://doi.org/10.1556/0806.44.2016.033
Norton GJ, Deacon CM, Xiong LZ, Huang SY, Meharg AA (2010) Price Adam H. Genetic mapping of the rice ionome in leaves and grain: identification of QTLs for 17 elements including arsenic, cadmium, iron and selenium. Plant Soil 329:139–153. https://doi.org/10.1007/s11104-009-0141-8
Ortiz-Monasterio JI, Palacios-Rojas N, Meng E, Pixley K, Trethowan R, Peña RJ (2007) Enhancing the mineral and vitamin content of wheat and maize through plant breeding. J Cereal Sci 46:293–307. https://doi.org/10.1016/j.jcs.2007.06.005
Pu ZE, Yu M, He QY, Chen GY, Wang JR, Liu YX, Jiang QT, Li W, Dai SF, Wei YM, Zheng YL (2014) Quantitative Trait Loci Associated with Micronutrient Concentrations in Two Recombinant Inbred Wheat Lines. J Integr Agric 13(11):2322–2329. https://doi.org/10.1016/S2095-3119(13)60640-1
Smith SE, Kuehl RO, Ray IM, Hui R, Soleri D (1998) Evaluation of simple methods for estimating broad-sense heritability in stands of randomly planted genotypes. Crop Sci 38:1125–1129. https://doi.org/10.2135/cropsci1998.0011183X003800050003x
Stangoulis JCR, Huynh BL, Welch RM, Choi EY, Graham RD (2007) Quantitative trait loci for phytate in rice grain and their relationship with grain micronutrient content. Euphytica 154(3):289–294. https://doi.org/10.1007/s10681-006-9211-7
Vreugdenhil D, Aarts MGM, Koornneef M, Nelissen H, Ernst WHO (2004) Natural variation and QTL analysis for cationic mineral content in seeds of Arabidopsis thaliana. Plant Cell Environ 27(7):828–839. https://doi.org/10.1111/j.1365-3040.2004.01189.x
Wang CX, Mo Z, Wang HX, Wang ZJ, Cao ZH (2003) The transportation, time-dependent distribution of heavy metals in paddy crops. Chemosphere 50(6):717–723. https://doi.org/10.1016/S0045-6535(02)00211-4
Wang SW, Yin LN, Tanaka H, Tanaka K, Tsujimoto H (2011) Wheat-Aegilops chromosome addition lines showing high iron and zinc contents in grains. Breed Sci 61(2):189–195. https://doi.org/10.1270/jsbbs.61.189
White PJ (2016a) Selenium accumulation by plants. Ann Bot 117(2, 1):217–235. https://doi.org/10.1093/aob/mcv180
White PJ (2016b) Biofortification of Edible Crops. Wiley, Chichester. https://doi.org/10.1002/9780470015902.a0023743
White PJ, Bowen HC, Parmaguru P, Fritz M, Spracklen WP, Spiby RE, Meacham MC, Mead A, Harriman M, Trueman LJ et al (2004) Interactions between selenium and sulphur nutrition in Arabidopsis thaliana. J Exp Bot 55:1927–1937. https://doi.org/10.1093/jxb/erh192
Yu M (2013) Genetic map construction and QTL mapping for important agronomic traits in synthetic hexaploid wheat (D)(Chinese). Sichuan Agricultural University, Ya’an
Zhao JX, Xing HJ, Liu CP, Zhang ZW, Xu SW (2016) Effect of Selenium Deficiency on Nitric Oxide and Heat Shock Proteins in Chicken Erythrocytes. Biol Trace Elem Res 171(1):208–213
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31301318).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Philip John White.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pu, Z., Pei, Y., Yang, J. et al. A QTL located on chromosome 3D enhances the selenium concentration of wheat grain by improving phytoavailability and root structure. Plant Soil 425, 287–296 (2018). https://doi.org/10.1007/s11104-017-3556-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-017-3556-7