
Abstract
In response to herbivory, Capsicum annuum leaves adapt their specialized metabolome that may protect the plant against herbivore feeding either directly or indirectly through volatile metabolites acting as cues for natural enemies of the herbivore. The volatile blend of spider-mite infested leaves differs from non-challenged leaves predominantly by a higher contribution of mono- and sesquiterpenes. In addition to these terpenoids released into the headspace, the terpenoid composition of the leaves alters upon herbivory. All this suggests an important role for terpenoids and their biosynthetic machinery in the defence against herbivory. Here, we show that the C. annuum genome contains a terpene synthase (TPS) gene family of 103 putative members of which structural analysis revealed that 27 encode functional enzymes. Transcriptome analysis showed that several TPS loci were differentially expressed upon herbivory in leaves of two C. annuum genotypes, that differ in susceptibility towards spider mites. The relative expression of upstream biosynthetic genes from the mevalonate and the methylerythritol phosphate pathway also altered upon herbivory, revealing a shift in the metabolic flux through the terpene biosynthetic module. The expression of multiple genes potentially acting downstream of the TPSs, including cytochrome P450 monooxygenases, UDP-glucosyl transferases, and transcription factors strongly correlated with the herbivory-induced TPS genes. A selection of herbivory-induced TPS genes was functionally characterized through heterologous expression and the products that these enzymes catalysed matched with the volatile and non-volatile terpenoids induced in response to herbivory.
Key message
Annotation of the Capsicum annuum terpene synthase gene family links terpene metabolism to the formation of volatile and non-volatile terpenoids important for direct and indirect defence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Terpenoids represent a large class of chemical compounds with an extensive diversity in chemical structures and a wide range of functions. They are produced by plants, fungi, bacteria, and insects. In plants, terpenoids harbour various phytohormones involved in growth regulation, including gibberellins, abscisic acid, strigolactones and cytokinins as well as metabolites that are involved in important physiological processes such as membrane stabilization (sterols), photosynthesis (carotenoids, the phytol side chain of chlorophyll and plastoquinone) and respiration (ubiquinone) (Tholl and Lee 2011). However, a substantially larger number of terpenoids contribute to broader physiological and ecological functions, including plant defence and plant communication (Singh and Sharma 2015).
Terpenoids are formed from C5 isoprene precursors, isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP) that are produced from the mevalonate pathway (MVA) in the cytosol/peroxisomes or from the methylerythritol phosphate pathway (MEP) in the plastids (Vranová et al. 2013). The condensation of DMAPPs with varying numbers of IPP units is catalysed by prenyl diphosphate synthases to synthesize geranyl diphosphate (GPP) (trans-configuration), or neryl diphosphate (NPP) (cis-configuration), depending on the prenyl diphosphate synthase, trans- or cis-farnesyl diphosphate (FPP) and geranylgeranyl diphosphate (GGPP) (Takahashi and Koyama 2006; Wang and Ohnuma 2000). These prenyl diphosphates are substrate for plant terpene synthases (TPSs)—monoterpene synthases, sesquiterpene synthases and diterpene synthases—generating monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20), respectively (Singh and Sharma 2015).
Part of the structural diversity in the terpenoids is obtained through variation in cyclization and rearrangement reactions of the prenyl diphosphate substrates. After formation of the carbocation usually a multitude of carbocation attacks and/or quenching through water, double bond rearrangements and hydride shifts occur, which is why a single TPS enzyme can sometimes generate multiple products from one substrate (Degenhardt et al. 2009). The structural diversity in these TPS products is often further increased through the activity of cytochrome P450s (Bathe and Tissier 2019), reductases, dehydrogenases methyl transferases (Piechulla et al. 2020; Lemfack et al. 2021) and glycosyl transferases (Boutanaev et al. 2015; Kurze et al. 2022) to yield an enormous variety of different terpenoid products. The myriad of terpenoid structures mirrors the complexity of properties and functions.
TPSs of flowering plants consist of 550–850 amino acids, with a molecular mass of 50–100 kDa and are encoded by gene members of the structurally related TPS gene family that probably has a common phylogenetic origin (Aubourg et al. 2002). The number of TPS genes within plant species ranges from a single functional terpene synthase gene in the moss Physcomitrella patens (Hayashi et al. 2007) to about 20 to 150 in flowering plant species (Chen et al. 2011). Structurally the TPS gene family can be divided into seven subfamilies, TPS-a, TPS-b, TPS-c, TPS-d, TPS-e/f, TPS-g and TPS-h. The TPS-a, b and g subfamilies are angiosperm specific while the TPS-d subfamily is gymnosperm specific and TPS-h is specific to the lycopod Selaginalla moellendorffi (Li et al. 2012; Chen et al. 2011). TPS-c and e/f members are present in both angiosperms and gymnosperms; TPS-c members encode copalyl diphosphate synthases and TPS-e/f members encode kaurene synthases. Most of the TPS-a genes encode sesquiterpene synthases while TPS-b genes mostly encode monoterpene synthases (Falara et al. 2011). The TPS-g subfamily is closely related to TPS-b and its members may function in producing acyclic mono-, sesqui-, and diterpene products (Chen et al. 2011).
Terpenoids often play a role in plant–insect interactions, either as attractants of pollinators or as defensive compounds against various herbivores (Kappers et al. 2008). For instance, conifers release volatile mono- and sesquiterpenoids that insects use to distinguish suitable hosts from non-hosts (Keeling and Bohlmann 2006). The colonization by arbuscular mycorrhiza altered terpenoid composition in plants which reinforced both direct and indirect defence against herbivorous insects (Sharma et al. 2017). The non-volatile sesquiterpene lactone argophyllone B reduces the larval mass of the sunflower moth Homeosoma electellum in Helianthus spp. (Prasifka et al. 2015). In Nicotiana attenuata, 17-hydroxygeranyllinalool diterpene glycosides were demonstrated to be effective defensive metabolites to herbivores (Heiling et al. 2010). Besides a direct effect on herbivores, terpenoids play an important role in attracting natural enemies of herbivores. Overexpression of a strawberry nerolidol synthase in Arabidopsis resulted in emission of two new terpenoids (3S)-(E)-nerolidol and its derivative (E)-4,8-dimethyl-1,3,7-nonatriene (DMNT), which resulted in enhanced attraction of Phytoseiulus persimilis predatory mites (Kappers et al. 2005). Transgenic Arabidopsis expressing maize TPS10 produced more sesquiterpenes and attracted more female Cotesia mariniventris parasitoids (Schnee et al. 2006).
The Capsicum genus belongs to the Solanaceae nightshade family and consists of approximately 35 species (Carrizog 2013). Capsicum is native to the Americas and currently cultivated worldwide for their sweet or spicy fruits, which are used as vegetable or spice. Various generalist arthropod species can infest Capsicum resulting in serious yield losses, including western flower thrips (Franklinella occidentalis) (Macel et al. 2019), aphids (Myzus persicae (Vaello et al. 2018), Macrosiphum euphorbiae (Sun et al. 2020) and two-spotted spider mites (Tetranychus urticae) (Zhang et al. 2020). The biotic stress-related phytohormone jasmonic acid (JA) is the master regulator of plant defence responses against herbivores (Verma et al. 2016) by mediating the expression of defence-related genes (Rehrig et al. 2014). This includes the biosynthesis of secondary metabolites (Ferrieri et al. 2015; Machado et al. 2017), especially terpenoids, in multiple plant species (Zhang et al. 2009; Hong et al. 2012; Ninkuu et al. 2021). Previously, we showed that JA plays a major role in the early signalling events of spider-mite induced defences in Capsicum and that exogenous application of JA results in notably higher emission of various terpenoids (Zhang et al. 2020). The availability of the genome sequence of a Capsicum inbred line (C. annuum var Zunla) allows us to predict the Capsicum TPS (CaTPS) gene family, which will help to understand the mechanism of terpenoid formation and metabolism under herbivory in Capsicum.
Here, we report the genome-wide identification of the TPS gene family and the annotation of 27 putative functional TPS genes in the C. annuum genome. To study how herbivory in general, and specifically spider-mite infestation, affects terpenoid production and emission in Capsicum, we treated plants with JA or spider mites and studied the changes in CaTPS gene expression. Several CaTPS genes of which the expression was induced by spider mites and/or by JA were subsequently isolated and functionally characterized. Analysis of transcriptional and metabolic changes upon herbivory revealed co-expression patterns between TPS genes and upstream biosynthetic genes from the MVA and MEP pathway as well as with genes downstream of the TPSs that may be involved in the creation of the structural diversity in herbivory-related Capsicum terpenoids.
Materials and methods
Identification of the terpene biosynthetic modules
To identify putative Capsicum TPS genes, a BLAST search against the C. annuum (Zunla-1) genome database (Qin et al. 2014) was performed using the protein sequence of known terpene synthases from A. thaliana, S. lycopersicum, Medicago truncatula, Vitis vinifera and Picea glauca (blast hits with e-value lower than e−5). In addition, using the approach described by Hofberger et al. (2015) which combines multiple lines of evidence provided by gene synteny, sequence homology and Hidden Markov Modelling, the C. annuum Zunla genome was screened for proteins associated with terpene biosynthesis, thereby covering the complete terpene biosynthetic module. The functional annotation we use in the present study is based on homology with annotated Arabidopsis and tomato genes.
Phylogenetic tree reconstruction and gene model analysis
Deduced CaTPS genes were aligned using ClustalX2.1 with standard parameters (www.clustal.org/clustal2) and a phylogenetic tree was built with Maximum Likelihood method in MEGA11 (Tamura et al. 2021). The intron–exon structure of CaTPS genes was determined by comparing the coding sequences and genomic sequences using the Gene Structure Display Server (GSDS2.0) (Hu et al. 2015). Putative plastid transit peptides were predicted using the TargetP1.1 server (Emanuelsson et al. 2000). Motifs were visualized using Jalview (https://www.jalview.org/).
Plants and arthropods
Capsicum plants [C. annuum, varieties Vania (Inbred line, Genotype 8) and Ta Pien Chiao (Land race, Genotype 29)] were grown from seed for 5 weeks in a greenhouse with a photoperiod of 16 h (23/18 °C, 50–60% relative humidity). Plants were still in the vegetative stage when used for experiments. Nicotiana benthamiana plants were grown from seed in the same greenhouse. Four-week-old, non-flowering plants were used for experiments and plants were not watered for 2 days prior to infiltrations. Two-spotted spider mites Tetranychus urticae were originally obtained from Koppert BV (Berkel en Rodenrijs, The Netherlands) and maintained on Phaseolus vulgaris bean plants for multiple generations.
JA treatment and spider-mite infestation
Five-week-old, non-flowering plants were infested with approximately 300 spider mites per plant by placing similar pieces of heavily-infested bean leaves on top of the Capsicum leaves. For jasmonic acid treatment, plants were sprayed with 100 µM methyl-JA (0.01% Tween-20). Mock-treated plants were sprayed with 0.01% Tween-20. For metabolite and transcript analysis, the 4th and 5th leaf were harvested after different periods of infestation/treatment, immediately flash-frozen and powdered in liquid nitrogen. For each treatment, five biological replicates were generated to allow for transcript and metabolite analysis from the same material.
Transcript data analysis
Assembly of full length transcripts of genes in the terpene biosynthetic module was performed using the Trinity method (Grabherr et al. 2011) on previously constructed transcriptome libraries from Capsicum plants that were infested with spider mites for 3 days, treated with JA for 6 h and 24 h, or were left untreated using the Illumina HiSeq™ 2500 platform (Zhang et al. 2020). Expression levels as RPKM values were calculated for each sample and the relative contributions of genes in the terpene biosynthetic module were calculated for each experimental condition to estimate shifts in flux through the MEP and MVA pathway. Pearson correlation coefficients were calculated between 17 expressed TPSs and transcripts assembled from the RNA sequencing. Correlation coefficients above a threshold of 0.90 (either positive or negative) were used to create a co-expression network visualized using the yGraph Organic layout in Cytoscape v.3.9.1 (https://cytoscape.org/).
Gene expression
Total RNA was isolated from the same samples as used for endogenous metabolite analysis using the RNeasy Plant Mini Kit (Qiagen, USA). One µg of each genomic-digested total RNA sample was reverse transcribed with SuperScript II Reverse Transcriptase (Invitrogen) to synthesize complementary DNA. Quantitative real time-PCR (qRT-PCR) was performed using the iQ SYBR-Green Supermix (Biorad). A linear standard curve was constructed using a serial dilution of cDNA that was pooled from all plants, and generated by plotting the threshold cycle (Ct) against the log10 of the dilution factors. The relative transcript levels of the genes of interest were determined according to the standard curve. C annuum ACTIN and GADPH genes were used to normalize cDNA concentrations. The primers used are listed in Supplementary Table S1. Data are the means of five biological replicates ± SD.
Endogenous terpenoids
Semi-polar endogenous metabolites were analysed by LC-Orbitrap-MS using an untargeted metabolomics approach (De Vos et al. 2007). Briefly, 200 mg of leaf material from each sample was extracted with 2 ml 75% MeOH:25% water (HPLC-grade) containing 0.1% formic acid (FA), sonicated for 15 min (40 kHz) and centrifuged for 10 min at 5000×g. Ten µl of the supernatant was taken from each sample of the series and combined to make a quality control sample that was analysed every 20 samples to control for machine stability. Metabolites were analysed using a LC-Orbitrap-MS consisting of an Accela U-HPLC equipped with a 1.7 μm AQUITY UPLC BEH C18 column (2.1 × 150 mm; Waters), an Accela photodiode array detector and an ion trap-Orbitrap FTMS hybrid mass spectrometer (Thermo Fisher Scientific) in negative electrospray ionization mode (m/z 95–1350) in centroid mode at a source voltage of 4.5 kV. Five µL of each extract was injected and separated with a linear 20 min gradient of 5 to 35% acetonitrile in water (0.1% FA) at a flow rate of 400 µL min−1 and 40 °C. The Metalign software (Lommen 2009) was used for baseline correction, mass spectra extraction and mass signal alignment with standard settings. MSClust (Tikunov et al. 2012) was used for data reduction by unsupervised clustering and extracting of putative metabolite mass spectra from ion-wise chromatographic alignment data. Significantly altered metabolites were semi-quantified by comparing characterized ion intensity between non-treated plants and treated plants (Criteria for selection: Fold Change [Treated/Non-treated] > 1.5 or < − 1.5, two-tailed Student’s t test P < 0.05). Putative annotation of non-volatile terpenoid compounds was based on comparison of accurate masses with the KNApSAcK database (http://kanaya.naist.jp/knapsack_jsp/top.html) after correction for possible adducts.
Volatile terpenoids
Volatiles emitted by plants were collected on Tenax (25/35 mesh) cartridges using a dynamic headspace sampling system as previously described (Zhang et al. 2020). In this system, 12 plants can be sampled simultaneously, and different treated plants were randomized among multiple days while volatiles were collected at the same time of the day (11:00 to 13:00 h). After collection, volatiles were desorbed from the cartridges with a Thermal Desorber TD100-xr (Unity, MARKES International) connected to a GC/Q-ToF MS (7890B GC, Agilent Technologies) and analytes were measured as described in Zhang et al. 2020. Mass spectra were acquired by scanning from 50 to 350 m/z with a scan rate of 5 scans min−1. Metabolites were identified on basis of chromatographic and spectral comparisons against the NIST11 library (http://chemdata.nist.gov/), Wiley (http://www.sisweb.com/software/wiley-ffnsc.htm), the Adams essential oil library (http://essentialoilcomponentsbygcms.com/) and a comprehensive in-house spectral library generated with authentic standards. For semi-quantification of compounds areas under the curve were calculated and normalized for total leaf area. For each treatment, five replicates were analysed and data show mean ± SD.
Heterologous expression in E. coli
Selected full length TPS genes were amplified from cDNA obtained from TSSM-damaged or methyl-JA-treated Capsicum leaves. Primer sequence information is available in Supplementary Table S1. PCR products of open reading frames were cloned into the pACYC-Duet vector and transformed in E. coli strain BL21. Single colonies grown in 2 mL LB were induced with 50 µl 1 M IPTG and grown for another 24 h at 18 °C. Subsequently, cells were collected by centrifugation (3.000×g), re-suspended in ice-cold extraction buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 1.4 mM β-mercapto ethanol), disrupted using a sonicator (4 times 15 s) and centrifuged (14.000×g). The supernatant containing the heterologous protein was used for enzymatic assays. To determine the catalytic activity of TPSs, 150 µl of the crude protein solution was added to 1340 µl assay buffer with 5 μl 10 mM GPP, FPP or GGPP (Sigma-Aldrich) in a screw-capped vial with an overlay of 100 µl pentane and incubated for 1 h at 30 °C with gentle shaking. For analysis of the captured reaction products, the pentane layer was filtered over a Pasteur’s-pipet filled with Na2SO4 and 2 μL was analysed using GC–MS (5890 series II/5972A, Hewlett-Packard) equipped with a 30 m × 0.25-mm i.d., 0.25 μm film thickness column (5MS, Hewlett-Packard). The GC was programmed at an initial temperature of 45 °C for 1 min, with a ramp of 10 °C min−1 to 220 °C and final time of 5 min and a constant Helium column flow of 1.0 mL min−1. The injection port, interface, and MS source temperatures were 250 °C, 290 °C, and 180 °C, respectively and the ionization potential was set at 70 eV. Scanning was performed from 45 to 300 atomic mass units. As a negative control, crude protein extract from E. coli expressing the empty vector pACYC-Duet incubated with substrate, was used.
One µl of the pentane extract was injected split less at a temperature of 250 °C at an initial column temperature of 40 °C. The temperature was held for 3.5 min, then increased with 10 °C min−1 to 280 °C and a hold for 2.5 min at a helium flow of 1 ml min−1. The ion source of the DSQ quadrupole mass spectrometer was directly coupled with the analytical column and operated in the 70 eV EI ionization mode. Masses 45 to 350 m/z were scanned. Compound identification was performed as described above. For each TPS enzyme and substrate combination, assays were repeated at least once but most often three times.
Transient expression in leaves of N. benthamiana
Plant binary plasmids containing open reading frames of selected TPS were constructed as previously described (Van Herpen et al. 2010). The final and intermediate vectors were checked by enzymatic restriction analysis and sequencing and introduced in Agrobacterium tumefaciens AglI by electrotransformation. A. tumufaciens cells were infiltrated into the abaxial side of the leaf of N. benthamiana plants for transient heterologous expression of CaTPS according to Van Herpen et al (2010). An A.tumefaciens strain encoding the TBSV P19 protein was added to suppress gene silencing. In addition, the pIVA_HMGR vector was co-infiltrated to enhance the flux toward terpenoid production. Three days after infiltration, infiltrated leaves were analysed for their volatile emissions and endogenous terpenoid content as described above.
Statistical analysis
All quantitative data were analysed by Student’s t test and one-way ANOVA using MetaboAnalyst (Version 4.0; www.metabolanalyst.ca). Statistical significance was determined using a Tukey’s honest significant difference.
Results
Capsicum annuum harbours a mid-size TPS gene family
We identified 103 loci with sequence homology to known TPS genes using version 2 of the C. annuum genome. The predicted TPS proteins contain one or more TPS characteristic domains, IPR008930, IPR001906, IPR008949, IPR005630 as described in the InterPro database (www.ebi.ac.uk/interpro/) and encode putative proteins with sizes ranging from 102 to 852 amino acids (Supplementary Table S2). Out of the 103 gene models, 27 encode proteins with more than 500 amino acids and are considered to encode functional TPS enzymes.
Forty-four of the 103 detected putative CaTPS genes had transcripts present in leaves of which 27 were considered non-functional and 17 putatively functional (Supplementary Table S3). The 103 TPS loci are distributed across all 12 chromosomes of Capsicum (Fig. 1), and the majority of these loci are physically located close to each other and show high sequence similarity with each other, suggesting multiple tandem and segmental duplication and recombination events. Such sequential repeat TPS gene clusters are located on chromosomes III, VIII, XI and XII, while chromosomes IX, IV, I, VI and VII contain respectively 9, 9, 7, 5 and 2 TPS loci which are more scattered across the chromosome. Chromosome V and X have a single TPS locus. Eight TPS genes could not be anchored to any specific position and were assigned to an artificial chromosome. In general, CaTPS genes are located compactly on the chromosomes with a high tandem repeat structure. Most tandem repeated genes show high similarity in sequence but with diverse gene sizes, suggesting a recent evolutionary tandem duplication of which multiple copies are not complete (anymore).

Distribution of CaTPS loci in the Capsicum annuum genome. The location of all CaTPS genes is indicated at the relative position in twelve artificial chromosomes (grey bars). Colour coding is indicative for the TPS sub families: TPS-a, orange; TPS-b, blue; TPS-c, red; TPS-e/g, green and TPS-g, purple
Six of the generally recognised eight TPS subfamilies are present in the Capsicum genome, with the exceptions being the gymnosperm-specific TPS-d subfamily and the S. moellendorffii-specific TPS-h subfamily (Fig. 2). Most (24 out of 27) CaTPS genes belong to class-III type (TPS-a: 18 members; TPS-b: 5; and TPS-g: 1) subfamilies, while class-I CaTPS genes comprise a single member for TPS-c, TPS-e and TPS-f each. Gene members from class-III type subfamilies have 6 to 7 exons while the exon number of class-I type subfamily members ranges from 11 to 15, consistent with TPS exon numbers found in other species (Trapp and Croteau 2001) (Supplementary Fig. S1).

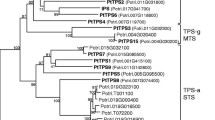
Phylogenetic relationship of full length CaTPS (indicated with *) and selected TPS from other species, representative for each of the TPS sub families. Ag Abies grandis, At Arabidopsis thaliana, Cb Clarkia breweri, Cm Cucumis maximus, Crs Croton sublyratus, Cs Cucumis sativus, Ml Mentha longifolia, Mp Mentha piperita, Mt Medicao trunculata, Pg Picea glauca, Sh Solanum habrochiates, Sl Solanum lycopersicum, Vv Vitis vinifera. For amino acid sequences and accession numbers, see Supplementary Table S7
N-terminal chloroplast transit peptides are predicted for TPS-b subfamily members, CaTPS61-63, TPS-c members CaTPS42 and TPS-g member CaTPS76 (Supplementary Table S4). The presence of a plastid transit peptide in the TPS-b members further supports their role as monoterpene synthases. In contrast, no signal peptide was found in the other TPS-b members, CaTPS29 and CaTPS66. The putative function as diterpene synthase of the TPS-c member CaTPS42 is supported by the presence of a plastid transit peptide. The TPS-f member CaTPS39 is also predicted to be a diterpene synthase, but lacks a chloroplast signal peptide. TPS-g member CaTPS76 could be either a monoterpene or diterpene synthase.
The conserved RR(X)8W motif near the N-terminus, involved in the initiation of the isomerization cyclization reaction or stabilizing the protein through electrostatic interactions (Martin et al. 2010; Aubourg et al. 2002) was present in three out of five putative monoterpene synthases of the TPS-b subfamily, while most (16 out of 18) TPS-a subfamily members showed variation as (R/P)(S/A/Y/V/I/E)(X)8W (Supplementary Fig. S2). The RXR motif was found to be conserved in most CaTPS members and in CaTPS35, CaTPS49, CaTPS53, CaTPS39, CaTPS76 with variation as RX(R/K/Q) but was not conserved in TPS-c member CaTPS42. The aspartate-rich DDXXD motif, located about 35 amino acids downstream of the RXR motif and responsible for the divalent metal ion-assisted binding and cleavage of the prenyl diphosphate substrate (Davis and Croteau 2000), is highly conserved in the CaTPS family except for the TPS-c member, CaTPS42, a putative copalyl diphosphate synthase (CPS). In Arabidopsis, CPS initiates ionization of GGPP by protonation rather than by diphosphate ester cleavage (Aubourg et al. 2002) (Bohlmann et al. 1998). The aspartate-rich motif (DIDDTA) initiates cyclization by protonation of GGPP (Peters et al. 2001) and is indeed present in CaTPS42 (aa384-389). The (N/D)DXX(S/T/G)XXXE (NSE/DTE) motif is present in all TPS-b, TPS-e/f, TPS-g members and most of the TPS-a members but not in TPS-c member CaTPS42, nor in three of the TPS-a members CaTPS98, CaTPS91 and CaTPS37. Together with the DDXXD motif, the NSE/DTE motif is also required for metal ion dependent ionization of the prenyl diphosphate substrate which is not present in a monofunctional CPS (Chen et al. 2011).
Biotic stress affects the terpene biosynthetic module
Using sequence homology, Hidden-Markov-Modeling and genomic context as described by Hofberger et al. (2015), 14 prenyl transferases (PTF), four isopentenyl isomerases (IPP) as well as 18 and 13 proteins predicted to be involved in the MVA and MEP pathways, respectively, were identified in the Capsicum genome, which together with the CaTPSs form the terpenoid biosynthetic module (Supplementary Table S3). Furthermore, 15 triterpene synthases were identified.
Expression patterns of genes related to the terpenoid biosynthetic module were examined in two C. annuum genotypes that were previously found to differ in spider mite susceptibility, after spider-mite infestation and after JA treatment using a previously generated RNA-sequencing dataset (Zhang et al. 2020). Among the 103 putative CaTPS loci, at least 44 transcripts were found present in Capsicum leaves in either genotype or experimental condition. Most TPS-a and TPS-b subfamily members were induced by spider mites and/or by JA, except CaTPS49, CaTPS87, CaTPS94 (TPS-a) and CaTPS66 (TPS-b) (Fig. 3A).

Change in CaTPS transcripts as result of spider-mite herbivory or jasmonic acid treatment. A heatmap of TPS transcripts retrieved from the RNA-seq dataset (Zhang et al. 2020) in which plants were infested with spider mites for three days or treated with JA for 6 or 24 h. Colour coding indicates log2 fold change relative to non-treated leaves. B Quantitative RT-PCR analysis of changes in transcripts of selected TPS genes relative to non-treated leaves as result of spider-mite infestation for 6 h, 24 h, and three days and methyl-JA treatment for 6 and 24 h. Expression was normalized to the expression of reference genes CaACTIN and CaEF1α. Data are means of five independent biological replicates ± SD
Putative copalyl diphosphate synthase CaTPS42 (TPS-c) and kaurene synthase CaTPS53 (TPS-e) were constitutively expressed in Capsicum leaves regardless of genotype or treatment, probably due to its importance for primary metabolite (gibberellin) production, and their expression was slightly altered in opposite directions by spider mites and JA (Fig. 3A, B). Members of the TPS-f and TPS-g clade, CaTPS39 and CaTPS76, were expressed in non-treated leaves and induced by (methyl)-JA while hardly affected or even slightly repressed by spider mites, dependent of the duration of infestation (Fig. 3A, B).
To obtain insight into how herbivory affects the expression of genes encoding for the MEP and MVA pathways, which provide the substrate for terpene biosynthesis, we calculated the relative expression of all gene candidates in a particular group under each experimental condition (Fig. 4A). All four IPP isomerases, 11 out of 13 MEP pathway genes, 16 out of 18 MVA pathway genes, 12 out of 13 prenyl transferases and 9 out of 15 triterpene synthases were expressed under at least one experimental condition (Supplementary Table S3).

Biotic stress affects the terpenoid biosynthetic module. A Expression changes in genes related to the biosynthetic module of the mevalonate (MVA) pathway in the cytosol/peroxisomes and the methylerythritol phosphate (MEP) pathway in the plastids leading to terpenoid precursors; Colour coding indicates log2 fold change relative to non-treated leaves. JA6, JA24: jasmonate treatment 6, 24 h prior to harvest; TSSM: spider-mite herbivory for three days prior to harvest. Gene abbreviations: AACT acetyl-CoA acetyltransferase, HMGS 3-hydroxy-3-methylglutaryl-CoA synthase, HMGR 3-hydroxy-3-methylglutaryl-CoA reductase, MK mevalonate kinase, PMK phosphomevalonate kinase, MDD mevalonate diphosphate decarboxylase, IDI isopentenyl-diphosphate delta isomerase, FPPS farnesyl diphosphate synthase, DXS deoxyxylulose 5-phosphate synthase, DXR deoxyxylulose 5-phosphate reductoisomerase, MCT 4-diphosphocytidyl-2C-methyl-d-erythritol 4-phosphate synthase, CMK 4-diphosphocytidyl-2C-methyl-d-erythritol kinase, MCS 2Cmethyl-d-erytrithol 2,4-diphosphate synthase, HDS 2C-methylD-erytrithol 2,4-cyclodiphosphate reductase, HDR 1-hydroxy-2-methylbutenyl 4-diphosphate reductase, GPPS geranyl diphosphate synthase, GGPS geranylgeranyl diphosphate synthase, mTPS monoterpene synthases, sTPS sesquiterpene synthases, diTPS diterpene synthases, triTPS triterpene synthases. B Pearson correlation network showing positive (red) and negative (blue) corelations between genes related to the terpene biosynthetic module, based on their induction profiles in response to biotic stress. Submodule 1 (green) includes multiple genes related to the MEP pathway and putative mono- and diterpene synthases, submodules 2 (orange) and 3 (red) include TPS-a clade sesquiterpene TPS which correlate with MVA pathway genes in submodule 3. Submodule 4 (grey) includes putative triterpene synthases and various MEP and MVA members. For a high resolution version including gene identifiers see [add link upon acceptation]
While the expression of genes encoding the MEP pathway was mostly upregulated by JA, this was less pronounced for genes encoding the MVA pathway. Spider-mite infestation resulted in low induction of few genes in the MEP and MVA pathways. Expression levels of IPP isomerases were hardly affected, neither by JA nor by spider mites. Prenyl transferase genes with putative functions such as GPPS or GGPPS were upregulated by JA and spider mites, while in contrast, FPPS genes were upregulated by JA but downregulated by spider mites. Genes with homology to solanesyl diphosphate synthase which are involved in the formation of plastoquinone in the chloroplasts and ubiquinone in the mitochondria were downregulated in all treatments. Together our data suggest that biotic stresses predominantly induce the MEP pathway resulting in increased substrate availability for mono- and diterpene biosynthesis. Indeed, the expression of multiple MEP pathway genes correlated with the expression of putative monoterpene synthases located on chromosome VIII (group 1 in Fig. 4B). In addition, the expression of TPF-f subfamily member CaTPS39 correlated strongly with two putative GGPPS and multiple MEP-pathway genes, supporting their role in diterpene formation.
Interestingly, while most MVA-pathway genes and FPPS were only moderately induced upon JA and not responsive to spider mites, expression of a number of TPS-a subfamily members, most likely encoding sesquiterpene synthases, was induced by JA and spider mites. The rate-limiting step in the MVA pathway is considered to be 3-hydroxy-3-methylglutaryl-coenzyme a reductase (HMGR) (Heuston et al. 2012), and indeed, Capana02g000059 and Capana04g002482 both encoding for HMGR are the only genes in the MVA pathway that were upregulated by JA while they were not responsive to spider mites. As the overall expression of triterpene synthases decreased upon JA treatment and spider mite herbivory, possibly substrate was rechannelled towards sesquiterpene biosynthesis resulting in enhanced FPP availability also without upregulation of the MVA pathway and FPP biosynthesis. Finally, there was a, less strong, but clearly visible, correlation between the expression of a number of triterpene synthases and the MEP and MVA pathway genes (group 4 in Fig. 4B). Triterpenes are, however, not within the scope of the present study and are therefore not discussed further.
Co-expression analysis of TPS genes reveals gene candidates for further diversification of terpenoids
To discover other genes possibly involved in Capsicum terpene biosynthesis or its regulation, the expression profiles of 17 expressed and putatively functional TPS genes were used as baits in a co-expression analysis using RNA-seq data (Fig. 5).

Gene co-expression network of transcripts correlated with Capsicum annuum TPS genes that were significantly altered in their expression upon spider-mite infestation or JA induction. Nodes (green) represent genes that show Pearson correlation |r|> 0.9 with TPS genes (magenta). Numbers indicate modules that are addressed in the text. For a detailed figure including gene identifiers based on the Pepper Genome Database (release 2.0) see [add link upon acceptation]
Among the expressed TPS genes, 13 showed strong co-expression with a series of other genes resulting in 1606 correlation pairs with 512 genes in total (Supplementary Table S5). The co-expression network showed that TPS genes segregate into distinct sub-networks containing multiple classes of genes. Five TPS-a clade genes, CaTPS6, CaTPS18, CaTPS50, CaTPS96, CaTPS98 and their co-expressed genes formed a module (1) with the highest number of nodes and edges). Within this module, CaTPS18 showed the highest correlation (0.998) with a cytochrome P450, CYP71D7. Furthermore, both CaTPS18 and CaTPS50 displayed high co-expression with multiple putative cytochrome P450s (correlation coefficients varying from 0.974 to 0.988) and HMGR2 (0.949), encoding the rate-limiting step in the MVA pathway. Co-expressed putative transcription factors included homologues of WRKY33 (0.985), WRKY72 (0.983), ERF2 (0.979), WRKY75 (0.931) and MYB4 (0.928), suggesting that CaTPS18 and CaTPS50 expression is tightly regulated. Other genes co-expressed with the TPS-a clade members mentioned above, include genes putatively encoding galacturonosyl and glutathione transferases.
The highly co-expressed TPS-b clade members, CaTPS62 and CaTPS63 (correlation coefficient 0.915, module 2), are located closely together on chromosome VIII. This could imply that these genes are regulated by the same transcription factor. Furthermore, several UDP-glycosyltransferase and glutathione S-transferase genes co-expressed with CaTPS62 and CaTPS63. No WRKY nor MYB transcription factors were found in this module, but a single ERF113 transcription factor co-expressed (0.938) with CaTPS62. In contrast to the module containing TPS-a gene members, negative correlations were found for TPS-b gene member CaTPS63 with two putative cytochrome P450s (Capana01g000486, − 0.858; Capana01g000368, − 0.862). The strongest negative correlation with both CaTPS62 and CaTPS63 was displayed by Capana08g000288, a putative RNA polymerase sigma factor, which is important to RNA biosynthesis (Ellermeier and Craig 2012).
Positive co-expression relations were found for TPS-f member CaTPS39 with multiple UDP-glycosyltransferases and with genes that likely belong to the JA biosynthetic pathway including a linoleate 13S-lipoxygenase 2-1 (LOX2.1; 0.929) and an allene oxide synthase (AOS; 0.928) in module 3. Furthermore, a putative transcription factor with homology to AtMYB4, highly correlated (0.923) with CaTPS39. In module 4, CaTPS93 was found to be correlated with a putative cytochrome P450 (Capana05g001954) and a UTG glucosyl transferase (Capana08g002046), while TPS in modules 5 to 7 correlated mostly to genes with unknown functions.
In conclusion, the co-expression network of Capsicum TPS genes that are responsive to spider-mite infestation and/or JA treatment revealed that TPS-a subfamily members predominantly co-express with multiple P450s and most of them (except CaTPS93) also with MVA pathway gene HMGR2. In addition, multiple transcription factors including WRKYs, MYBs, and EFRs correlated with these putative sesquiterpene synthases. Several Capsicum TPS-a, TPS-b and TPS-e/f gene members co-expressed with UDP-glycosyltransferases. Negative gene correlations were predominant among the putative monoterpene synthases suggesting that biosynthesis of monoterpenes is more affected by interaction with other biological processes in plants, while sesquiterpene biosynthesis is more regulated by signalling pathways or external biotic/abiotic stresses.
Functional characterization of recombinant TPS enzymes
Transcriptional analysis suggests that multiple Capsicum TPSs are responsive to biotic stresses. To further characterize the gene functions and their ecological roles in plant defence, nine full-length TPS ORFs of genes from Capsicum that were found to be induced by spider mites or JA were expressed in E. coli and the recombinant TPS proteins analysed for enzymatic activity (Table 1, Supplementary Tables S6, S9). Consistent with sequence analysis, six TPS-a subfamily members were identified as sesquiterpene synthases as CaTPS6, CaTPS18, CaTPS37, CaTPS96 and CaTPS98 all accepted FPP as substrate to form 4,5-di-epi-aristolochene. In addition, CaTPS93 catalysed the conversion of FPP into germacrene A. None of these enzymes accepted GPP nor GGPP, earmarking them as monofunctional sesquiterpene synthases. TPS-f subfamily member CaTPS39 was characterized as a bifunctional enzyme accepting both FPP and GGPP to produce (E)-nerolidol and geranyl linalool, respectively, while no product was detected using GPP as substrate. Except for CaTPS63, expressed TPS-b subfamily members could not be cloned or characterized. CaTPS63 accepted GPP to produce β-pinene. TPS-g member CaTPS76 accepted both GPP and FPP to form linalool and (E)-nerolidol, respectively, while no product was detected using GGPP as substrate.
Transient in-planta overexpression of the abovementioned TPS in N. benthamiana confirmed the results of the in-vitro assays, although more terpenoid products were detected that were not present in the E. coli assays or in trace amounts (Supplementary Table S6).
Induced terpenoids in Capsicum in response to biotic stresses
To characterize terpenoid metabolites produced upon biotic stress, both volatile and endogenous terpenoids were analysed in (the headspace of) leaves of two genotypes in response to spider mite herbivory and methyl-JA treatment. At least 34 terpenoids were detected in the volatile blend emitted by non-infested (Mock-treated) plants, of which the dominant compounds were monoterpenes (E)-β-ocimene and β-pinene, and sesquiterpene germacrene A in both genotypes (Fig. 6). While the abundance of specific monoterpenoids increased in response to spider-mite feeding, the overall contribution of monoterpenoids decreased due to the stronger increase in abundance of multiple sesquiterpenoids and homoterpenes DMNT and (E,E)-4,8,12-trimethyltrideca-1,3,7,11-tetraene (TMTT).

Emitted volatile terpenoid metabolites from Capsicum annuum leaves that were infested with spider-mites (brown), treated with methyl-jasmonate (green) or not treated (black). Data show GC–MS area units at the relative retention time. Numbers indicate compound (Retention Index); 1, α-Pinene (933); 2, β-Pinene (976); 3, 3-carene (1006); 4, Unknown monoterpene (1015); 5, Limonene (1024); 6, (Z)-β-Ocimene (1037); 7, (E)-β-Ocimene (1045); 8, γ-Terpinene (1056); 9, Linalool (1094); 10, (E)-DMNT (1118); 11, (E)-β-Ocimene epoxide (1132); 12, Neo-allo-ocimene (1150); 13, Unknown monoterpene alcohol (1195); 14, 3-Caren-2-one (1240); 15, Geraniol (1250); 16, α-Copaene (1372); 17, β-cubebene (1389); 18, β-Longipinene (1401); 19, (Z)-α-Bergamotene (1408); 20, β-Caryophyllene (1419); 21, (E)-α-Bergamotene (1432); 22, Aromandendrene (1439); 23, γ-Muurolene (1474); 24, γ-Himachalene (1481); 25, β-Selinene (1487); 26, α-Selinene (1496); 27, Unknown sesquiterpene (1501); 28, Germacrene A (1508); 29, (Z)-Nerolidol (1533); 30, (E)-Nerolidol (1564); 31, Caryophyllene oxide (1580); 32, (E,E)-TMTT (1584); 33, Unknown sesquiterpene lactone (1688)
Furthermore, spider mites and methyl-JA altered the abundance of endogenous and mostly glycosylated terpenoids (Table 2). Abundance of tetraterpenoid carotenoids, including β-cryptoxanthin and zeaxanthin decreased in response to spider-mite feeding and methyl-JA treatment. Most endogenous terpenoids that increased in response to spider-mite infestation and/or methyl-JA are putative hemi- and diterpene glycosides. Furthermore, a number of triterpene saponins and steroid terpenoids conjugated with glycosides were found to be mostly induced in genotype 29.
Discussion
Terpenoids are the most widely occurring secondary metabolites in nature and show enormous diversity across the plant kingdom. Many of them play important roles in plant direct or indirect defence against microbes and herbivores (Gershenzon and Dudareva 2007; Pichersky and Raguso 2018). Here, we aimed to increase our understanding of how the terpenoid biosynthetic pathway in the economically important crop species Capsicum annum is affected by cell-content feeding spider mites. We used a combinatorial approach to identify the Capsicum TPS gene family and the genes responsible for the generation of precursors and those that might be involved in further terpenoid diversification. Our results provide insight into Capsicum specialized terpenoid metabolism related to biotic stress and provide a template for further exploration of genetic resources for Capsicum breeding for improved resilience against herbivores.
Compared to TPS gene families of other species, the CaTPS gene family is of moderate size and characterized by a significant number of pseudogenes and short gene fragments. For instance, in P. patens (Hayashi et al. 2007), there is one full-length gene out of four gene models, while Arabidopsis (Aubourg et al. 2002) has 32 full-length genes out of 40 gene models. Additionally, apple contains 10 full-length genes out of 55 gene models (Nieuwenhuizen et al. 2013), tomato 29 full-length genes out of 44 gene models (Falara et al. 2011; Zhou and Pichersky 2020) and grape contains 69 full-length genes out of 152 gene models (Martin et al. 2010). This is probably due to a relatively high speed of duplication accompanied by gene degradation and loss of function. TPS-a and TPS-b subfamilies form the two largest clades in the CaTPS family and most of the genes in these clades have high sequence homology and closely co-localize on the chromosomes. This clustered localization is prominent for TPS-a members on chromosome II, III, VIII, IX, XI, XII and for TPS-b members on chromosome VIII. Many TPS genes of the TPS-a and TPS-b clade have likely resulted from local duplication events (Aubourg et al. 2002). Those gene copies tend to be retained as their products are specialized metabolites that will not affect the development and survival of the plant. Hence, this high speed of duplication may not be restricted too much by selection pressure (Jiang et al. 2019; Heidel-Fischer and Vogel 2015; Pichersky and Gang 2000). The rapid evolution of TPS genes has resulted in species–specific paralogous TPS gene clusters. Despite the high frequency of duplication and gene degradation, all TPS-a family members have conserved DDXXD and RXR domains. The angiosperm-specific TPS-a subfamily is highly divergent across all the seed plants and TPSs in this clade are either sesquiterpene synthases or diterpene synthases while TPS-b family members mainly encode cyclic monoterpene and hemiterpene synthases (Chen et al. 2011).
In contrast to the abovementioned subfamilies, TPS genes from the TPS-c, TPS-e/f and TPS-g clades are maintained as a single copy. The low ratio of divergence and slow speed of duplication indicate a strong selection. Sequence analysis suggested that CaTPS53, the only member in the TPS-e clade, encodes ent-kaur-16-ene synthase. Together with CaTPS42 (TPS-c) with high homology to copalyl diphosphate synthase, these genes are probably responsible for the production of gibberellin in Capsicum (Prisic et al. 2004; Xu et al. 2007).
Terpene biosynthesis depends on the activity of various independent pathways acting in balance to convert primary metabolites to specialized compounds designed to mediate interactions of plants with their surroundings. The modular organization of plant terpenoid biosynthesis includes distinct reaction modules, including genes from the MVA and MEP pathways, IPP isomerases, prenyl transferases and TPS genes encoding monoterpene, sesquiterpene and diterpene synthases (Hofberger et al. 2015). Furthermore, triterpene synthases are considered to be part of the terpenoid biosynthetic module and they share a common evolutionary origin with genes from the MVA and MEP pathway (Matsuba et al. 2013). We were able to identify gene candidates for each of the reaction modules leading to the substrates for terpene synthases, and the expression of these genes correlated with TPS expression, implying that the biosynthetic module is regulated in response to biotic stress.
The plastidial MEP pathway is the supplier for primary metabolites important in photosynthesis and hence of major relevance for growth. Herbivory is known to induce defences with often a trade-off to growth. Miltra et al. (2021) demonstrated that Spodoptera littoralis herbivory on Arabidopsis decreased flux through the MEP pathway via DXP and DXS inhibition. In Capsicum, spider-mite herbivory suppressed terpenoid biosynthetic flux through genes in both the MVA and the MEP module as well as the IPP and PTF modules, while the effect of jasmonate was mostly apparent through a decrease in gene expression in the IPP module. The down-regulation of the MEP pathway relates with a decreased abundance of metabolites derived from the plastids, including volatile monoterpenoids, and endogenous terpenoid-glycosides and carotenoids. The latter might partly explain the suppression of photosynthesis in response to herbivory (Zangerl et al. 2002) and indeed, our RNA-sequencing data showed lower transcript abundances of genes related to photosynthesis (Zhang et al. 2020). In our current work, we aim to characterize the role of MEP and MVA pathway genes in Capsicum in relation to the growth-defence trade-off, in order to predict the impact of cell-content feeders on Capsicum specialized metabolism and growth.
Out of 27 predicted full length TPS genes, we selected nine genes for heterologous expression. The terpene synthases that were selected from the TPS-a subfamily displayed high homology with 5-epi-aristolochene synthase, and indeed five of them produced 4,5-di-epi-aristolochene, an isomer of 5-epi-aristolochene. While the expression of these genes was clearly induced upon spider-mite infestation or JA-treatment, 4,5-di-epi-aristolochene was not detected in the headspace of the two genotypes. 5-Epi-aristolochene is the precursor for the production of the anti-microbial sesquiterpenoid capsidiol which is produced upon stress in several Solanaceous species (Whitehead et al. 1989; Bohlmann et al. 2002; Lee et al. 2017; Zavalapáramo et al. 2000). The oxidation of 5-epi-aristolochene to capsidiol has been shown to be mediated by elicitor-inducible cytochrome P450 hydroxylase, CYP71D in tobacco (Ralston et al. 2001). In Capsicum, the expression of CaTPS18 and CaTPS98 exhibited a high correlation with cytochrome P450s annotated as CYP71 family members, which are thus likely candidates for the oxidation of 4,5-di-epi-aristolochene. Upon spider-mite infestation and JA-treatment, a compound with a similar accurate mass as capsidiol, but different retention time, was found in genotype 8, suggesting that in Capsicum 4,5-di-epi-aristolochene is converted into a structural homolog of capsidiol (Supplementary Fig. S3).
Except CaTPS29, CaTPS-b subfamily members have the conserved RRW8R domain and three of them were predicted to have a transit peptide which would direct the proteins to the chloroplasts. Only the genes predicted to have a transit peptide were found to have transcripts in the RNA-seq data and they show high similarity to a (−)-camphene/tricyclene synthase from C. baccatum (Kim et al. 2017). The headspace of both C. annuum genotypes contained at least 14 different monoterpenoids of which the abundance of 8 altered in response to spider-mite infestation. We were able to successfully characterize CaTPS66, which accepts GPP to form the cyclic monoterpene, β-pinene in-vitro and in-planta when transiently expressed in N. benthamiana, and which shares a 92.7% similarity with a monoterpene synthase from tomato (SlTPS8) catalysing the formation of cyclic 1,8-cineole from both GPP and NPP (Falara et al. 2011).
TPS-e member CaTPS53 shows 94.4% similarity with SlTPS24 which is a kaurene synthase (Falara et al. 2011) and CaTPS39 in the TPS-f clade is closely related to AtTPS04 ((E,E)-geranyl linalool synthase), MtTPS18 ((E)-nerolidol/(E,E)-geranyl linalool synthase) and CbS-linalool synthase (Aubourg et al. 2002; Parker et al. 2014; Dornelas and Mazzafera 2007).
TPS-f member CaTPS39 catalyses both the formation of (E)-nerolidol and (E,E)-geranyl linalool, and its transcripts were induced by both spider mites and JA. Both (E)-nerolidol-derived DMNT and (E,E)-geranyl linalool-derived TMTT are detectable in the Capsicum spider-mite induced volatile blend. CaTPS39 expression correlates well with the expression of a putative CYP82 (Capana04G000466), which is localized in the proximity of CaTPS39 on chromosome IV, suggesting that this cytochrome P450 could be responsible for the conversion of geranyl linalool to TMTT. A CYP82 member was shown to convert geranyl linalool into DMNT and TMTT, in Zea mays and Arabidopsis (Richter et al. 2016; Lee et al. 2010). Expression of a (E)-nerolidol/(E,E)-geranyl linalool synthase in Arabidopsis resulted in enhanced emission of (E)-nerolidol, DMNT and TMTT, and improved attraction of parasitoid wasps, Diadegma semiclausum (Houshyani et al. 2013) as well as predatory mites Phytoseiulus persimilis (Kappers et al. 2005). The presence of TMTT in the headspace of Capsicum upon spider-mite feeding is likely due to the concerted expression of TPS39 and CYP82 gene candidates. TPS-g clade member, CaTPS76 encodes a monoterpene synthase catalysing the conversion of GPP to acyclic linalool and also accepts FPP as substrate to produce acyclic (E)-nerolidol. CaTPS76 has high sequence similarity with SlTPS37 and SlTPS39 from tomato with similarities of 89.0% and 86.5% respectively, catalysing the formation of linalool and (E)-nerolidol from GPP and (E,E)-FPP, respectively (Falara et al. 2011). Multi-substrate accepting enzymes capable to catalyse both the formation of monoterpenes and sesquiterpenes have been reported in multiple species (Degenhardt et al. 2009) and linalool/nerolidol synthases have been identified in Arabidopsis (Chen et al. 2011), tomato (Falara et al. 2011), cucumber (He et al. 2022) and grape (Zhu et al. 2014). In Capsicum, the ratio between linalool and nerolidol in the volatile blend differed when leaves were biotically challenged, which might be explained by the altered flux of nerolidol towards DMNT in response to herbivory. However, we did not detect any highly correlated genes for CaTPS76 possibly involved in the conversion of nerolidol to DMNT.
Genes encoding biosynthetic enzymes for specialized metabolites are often located physically close on chromosomes, forming clusters in the plant genome, such as the biosynthetic genes responsible for the formation of triterpenoid limonoids in citrus and neem (De La PeÑa et al. 2023), petirucalla-7,24-dien-3b-ol in Arabidopsis (Boutanaev et al. 2015), cucurbitacin C in Cucumis sativus (Shang et al. 2014) and diterpenoid casbene in Euphorbia peplus (King et al. 2014). Combinations of terpenoid synthases with cytochromes P450s are found clustered together in plant genomes far more commonly than would be expected by chance and this gene pairing likely has evolved from a common ancestral pairing in eudicots (Boutanaev et al. 2015). Many metabolites formed as result of genes within such plant metabolic gene clusters appear to provide protection against pathogens and herbivores. Clustering genes and coinheritance of optimized gene combinations that confer selective advantage is probably beneficial for plants (Yeaman and Whitlock 2011; Takos and Rook 2012). Besides, co-localization of metabolic pathway genes increases the likelihood of co-regulating gene expression and prevents the accumulation of auto-toxic intermediates (Field et al. 2011; Xu et al. 2012).
In Capsicum, multiple genes including cytochromes P450s, prenyl transferases, and MVA and MEP pathway genes were detected to localise physically close to TPS genes, suggesting specialized gene-clusters putatively under the control of a shared regulatory mechanism as many of them are co-expressed upon biotic stress. Especially members of the TPS-a subfamily tend to cluster with cytochrome P450 genes. The clustering of TPSs with glycosyl transferases and with cytochrome P450s matches with the findings that multiple monoterpene glycosides, oxygenated sesquiterpenes, diterpene glycosides and triterpene glycosides increased upon spider-mite feeding and/or JA treatment.
In conclusion, we identified the TPS gene family in the Capsicum genome and analysed their phylogeny, gene structure and expression profile upon spider-mite feeding and JA treatment. Upstream biosynthetic genes from the MEP and MVA pathway and downstream modification genes including cytochrome P450s, UDP-glycosyl transferases and transcription factors that under herbivory display co-expression with CaTPS genes were identified. Finally, recombinant proteins of selected TPSs were functionally characterized. Together, our work demonstrates the importance of the terpenoid biosynthetic network for the formation of volatile and non-volatile defence-related terpenoids in Capsicum annuum.
Data availability
RNA-sequencing data are available at Dryad Data Repository: https://doi.org/10.5061/dryad.n34h180. Metabolomics data files (LC–MS, GC–MS) will be deposited in Dryad upon acceptance of the manuscript.
Code availability
Not applicable.
References
Aubourg S, Lecharny A, Bohlmann J (2002) Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol Genet Genomics 267:730–745
Bathe U, Tissier A (2019) Cytochrome P450 enzymes: a driving force of plant diterpene diversity. Phytochemistry 161:149–162
Bohlmann J, Meyer-Gauen G, Croteau R (1998) Plant terpenoid synthases: molecular biology and phylogenetic analysis. Proc Natl Acad Sci USA 95:4126–4133
Bohlmann J, Stauber EJ, Krock B, Oldham NJ, Gershenzon J, Baldwin IT (2002) Gene expression of 5-epi-aristolochene synthase and formation of capsidiol in roots of Nicotiana attenuata and N. sylvestris. Phytochemistry 60:109–116
Boutanaev AM, Moses T, Zi J, Nelson DR, Mugford ST, Peters RJ, Osbourn A (2015) Investigation of terpene diversification across multiple sequenced plant genomes. Proc Natl Acad Sci 112:E81–E88
Carrizog C (2013) Wild Capsicums: identification and in situ analysis of Brazilian species. In: Breakthroughs in the genetics and breeding of capsicum and eggplant; Proceedings of the XV EUCARPIA meeting, pp 205–213
Chen F, Tholl D, Bohlmann J, Pichersky E (2011) The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J 66:212–229
Davis EM, Croteau R (2000) Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes. Biosynthesis 209:53–95
de La Peña R, Hodgson H, Chun-Ting LIUJ, Stephenson MJ, Martin AC, Owen C, Harkess A, Leebens-Mack J, Le J, Osbourn A, Sattely ES (2023) Complex scaffold remodeling in plant triterpene biosynthesis. Science 379:361–368
de Vos RC, Moco S, Lommen A, Keurentjes JJ, Bino RJ, Hall RD (2007) Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat Protoc 2:778–791
Degenhardt J, Köllner TG, Gershenzon J (2009) Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70:1621–1637
Dornelas MC, Mazzafera P (2007) A genomic approach to characterization of the citrus terpene synthase gene family. Genet Mol Biol 30:832–840
Ellermeier TDH, Craig D (2012) Extra cytoplasmic function σ factor activation. Curr Opin Microbiol 15:182–188
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Falara V, Akhtar TA, Nguyen TT, Spyropoulou EA, Bleeker PM, Schauvinhold I, Matsuba Y, Bonini ME, Schilmiller AL, Last RL, Schuurink RC, Pichersky E (2011) The tomato terpene synthase gene family. Plant Physiol 157:770–789
Ferrieri AP, Arce CCM, Machado RAR, Meza-Canales ID, Lima E, Baldwin IT, Erb M (2015) A Nicotiana attenuata cell wall invertase inhibitor (NaCWII) reduces growth and increases secondary metabolite biosynthesis in herbivore-attacked plants. New Phytol 208:519–530
Field B, Fiston-Lavier AS, Kemen A, Geisler K, Quesneville H, Osbourn AE (2011) Formation of plant metabolic gene clusters within dynamic chromosomal regions. Proc Natl Acad Sci USA 108:16116–16121
Gershenzon J, Dudareva N (2007) The function of terpene natural products in the natural world. Nat Chem Biol 3:408–414
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Hayashi KI, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H (2007) Corrigendum to “Identification and functional analysis of bifunctional ent -kaurene synthase from the moss Physcomitrella patens ” [FEBS Lett. 580 (2006) 6175–6181]. FEBS Lett 581:2748–2748
He J, Verstappen F, Jiao A, Dicke M, Bouwmeester HJ, Kappers IF (2022) Terpene synthases in cucumber (Cucumis sativus) and their contribution to herbivore-induced volatile terpenoid emission. New Phytol 233:862–877
Heidel-Fischer HM, Vogel H (2015) Molecular mechanisms of insect adaptation to plant secondary compounds. Curr Opin Insect Sci 8:8–14
Heiling S, Schuman MC, Schoettner M, Mukerjee P, Berger B, Schneider B, Jassbi AR, Baldwin IT (2010) Jasmonate and ppHsystemin regulate key malonylation steps in the biosynthesis of 17-Hydroxygeranyllinalool diterpene glycosides, an abundant and effective direct defense against herbivores in Nicotiana attenuata. Plant Cell 22:273–292
Heuston S, Begley M, Davey MS, Eberl M, Casey PG, Hill C, Gahan CGM (2012) HmgR, a key enzyme in the mevalonate pathway for isoprenoid biosynthesis, is essential for growth of Listeria monocytogenes EGDe. Microbiology 158:1684–1693
Hofberger JA, Ramirez AM, van den Bergh E, Zhu X, Bouwmeester HJ, Schuurink RC, Schranz ME (2015) Large-scale evolutionary analysis of genes and supergene clusters from terpenoid modular pathways provides insights into metabolic diversification in flowering plants. PLoS ONE 10:e0128808
Hong GJ, Xue XY, Mao YB, Wang LJ, Chen XY (2012) Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 24:2635–2648
Houshyani B, Assareh M, Busquets A, Ferrer A, Bouwmeester HJ, Kappers IF (2013) Three-step pathway engineering results in more incidence rate and higher emission of nerolidol and improved attraction of Diadegma semiclausum. Metab Eng 15:88–97
Hu B, Jin J, Guo A, Zhang H, Luo J, Gao G (2015) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31:1296–1297
Jiang S-Y, Jin J, Sarojam R, Ramachandran S (2019) A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol Evol 11:2078–2098
Kappers IF, Aharoni A, van Herpen T, Luckerhoff LLP, Dicke M, Bouwmeester HJ (2005) Genetic engineering of terpenoid metabolism attracts, bodyguards to Arabidopsis. Science 309:2070–2072
Kappers IF, Dicke M, Bouwmeester HJ (2008) Terpenoids in plant signaling, chemical ecology. Wiley Encyclopedia of Chemical Biology, New York
Keeling CI, Bohlmann J (2006) Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defence of conifers against insects and pathogens. New Phytol 170:657–675
Kim S, Park J, Yeom S-I, Kim Y-M, Seo E, Kim K-T, Kim M-S, Lee JM, Cheong K, Shin H-S, Kim S-B, Han K, Lee J, Park M, Lee H-A, Lee H-Y, Lee Y, Oh S, Lee JH, Choi E, Choi E, Lee SE, Jeon J, Kim H, Choi G, Song H, Lee J, Lee S-C, Kwon J-K, Lee H-Y, Koo N, Hong Y, Kim RW, Kang W-H, Huh JH, Kang B-C, Yang T-J, Lee Y-H, Bennetzen JL, Choi D (2017) Multiple reference genome sequences of hot pepper reveal the massive evolution of plant disease resistance genes by retroduplication. Genome Biol 18:210
King AJ, Brown GD, Gilday AD, Larson TR, Graham IA (2014) Production of bioactive diterpenoids in the euphorbiaceae depends on evolutionarily conserved gene clusters. Plant Cell 26:3286–3298
Kurze E, Wüst M, Liao J, Mcgraphery K, Hoffmann T, Song C, Schwab W (2022) Structure–function relationship of terpenoid glycosyltransferases from plants. Nat Prod Rep 39:389–409
Lee S, Badieyan S, Bevan DR, Herde M, Gatz C, Tholl D (2010) Herbivore-induced and floral homoterpene volatiles are biosynthesized by a single P450 enzyme (CYP82G1) in Arabidopsis. Proc Natl Acad Sci USA 107:21205–21210
Lee H-A, Kim S, Kim S, Choi D (2017) Expansion of sesquiterpene biosynthetic gene clusters in pepper confers nonhost resistance to the Irish potato famine pathogen. New Phytol 215:1132–1143
Lemfack MC, Brandt W, Krüger K, Gurowietz A, Djifack J, Jung J-P, Hopf M, Noack H, Junker B, von Reuß S, Piechulla B (2021) Reaction mechanism of the farnesyl pyrophosphate C-methyltransferase towards the biosynthesis of pre-sodorifen pyrophosphate by Serratia plymuthica 4Rx13. Sci Rep 11:3182. https://doi.org/10.1038/s41598-021-82521-9
Li G, Köllner TG, Yin Y, Jiang YL, Chen H, Xu Y, Gershenzon J, Pichersky E, Chen F (2012) Nonseed plant Selaginella moellendorffi [corrected] has both seed plant and microbial types of terpene synthases. Proc Natl Acad Sci USA 109:14711–147115. https://doi.org/10.1073/pnas.1204300109
Lommen A (2009) MetAlign: interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal Chem 81:3079–3086
Macel M, Visschers IGS, Peters JL, Kappers IF, de Vos RCH, van Dam NM (2019) Metabolomics of thrips resistance in pepper (Capsicum spp.) reveals monomer and dimer acyclic diterpene glycosides as potential chemical defenses. J Chem Ecol 45:490–501
Machado RAR, Baldwin IT, Erb M (2017) Herbivory-induced jasmonates constrain plant sugar accumulation and growth by antagonizing gibberellin signaling and not by promoting secondary metabolite production. New Phytol 215:803–812
Martin DM, Aubourg S, Schouwey MB, Daviet L, Schalk M, Toub O, Lund ST, Bohlmann J (2010) Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol 10:226. https://doi.org/10.1186/1471-2229-10-226
Matsuba Y, Nguyen TT, Wiegert K, Falara V, Gonzales-Vigil E, Leong B, Schäfer P, Kudrna D, Wing RA, Bolger AM, Usadel B, Tissier A, Fernie AR, Barry CS, Pichersky E (2013) Evolution of a complex locus for terpene biosynthesis in solanum. Plant Cell 25:2022–2036
Mitra S, Estrada-Tejedor R, Volke DC, Philips MA, Gershenzon J, Wright LP (2021) Negative regulation of plastidial isoprenoid pathway by herbivore-induced β-cyclocitral in Arabidopsis thaliana. Proc Natl Acad Sci USA 118:e2008747118. https://doi.org/10.1073/pnas.2008747118
Nieuwenhuizen NJ, Green SA, Chen X, Bailleul EJD, Matich AJ, Wang MY, Atkinson RG (2013) Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in Apple. Plant Physiol 161:787–804
Ninkuu V, Zhang L, Yan J, Fu Z, Yang T, Zeng H (2021) Biochemistry of terpenes and recent advances in plant protection. Int J Mol Sci 22:5710. https://doi.org/10.3390/ijms22115710
Parker MT, Zhong Y, Dai X, Wang S, Zhao P (2014) Comparative genomic and transcriptomic analysis of terpene synthases in Arabidopsis and Medicago. IET Syst Biol 8:146–153. https://doi.org/10.1049/IET-SYB.2013.0032
Peters RJ, Ravn MM, Coates RM, Croteau RB (2001) Bifunctional abietadiene synthase: free diffusive transfer of the (+)-copalyl diphosphate intermediate between two distinct active sites. J Am Chem Soc 123:8974–8978
Pichersky E, Gang DR (2000) Genetics and biochemistry of secondary metabolites in plants: an evolutionary perspective. Trends Plant Sci 5:439–445
Pichersky E, Raguso RA (2018) Why do plants produce so many terpenoid compounds? New Phytol 220:692–702
Piechulla B, Magnus N, Lemfack MC, von Reuss S (2020) Terpenoid cyclization by SAM-dependent C-Methyl transferase. Trends Chem 2:585–586
Prasifka JR, Spring O, Conrad J, Cook LW, Palmquist DE, Foley ME (2015) Sesquiterpene lactone composition of wild and cultivated sunflowers and biological activity against an Insect Pest. J Agric Food Chem 63:4042–4049
Prisic S, Xu MM, Wilderman PR, Peters RJ (2004) Rice contains two disparate ent-copalyl diphosphate synthases with distinct metabolic functions. Plant Physiol 136:4228–4236
Qin C, Yu C, Shen Y et al (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 111:5135–5140. https://doi.org/10.1073/pnas.1400975111
Ralston L, Kwon ST, Schoenbeck M, Ralston J, Schenk DJ, Coates RM, Chappell J (2001) Cloning, heterologous expression, and functional characterization of 5-epi-Aristolochene-1,3-Dihydroxylase from Tobacco (Nicotiana tabacum). Arch Biochem Biophys 393:222–235
Rehrig EM, Appel HM, Jones AD, Schultz JC (2014) Roles for jasmonate- and ethylene-induced transcription factors in the ability of Arabidopsis to respond differentially to damage caused by two insect herbivores. Front Plant Sci 5:407. https://doi.org/10.3389/fpls.2014.00407
Richter A, Schaff C, Zhang Z, Lipka AE, Tian F, Köllner TG, Schnee C, Preiß S, Irmisch S, Jander G, Boland W, Gershenzon J, Buckler ES, Degenhardt J (2016) Characterization of biosynthetic pathways for the production of the volatile homoterpenes DMNT and TMTT in Zea mays. Plant Cell 28:2651–2665
Schnee C, Köllner TG, Held M, Turlings TCJ, Gershenzon J, Degenhardt J (2006) The products of a single maize sesquiterpene synthase form a volatile defense signal that attracts natural enemies of maize herbivores. Proc Natl Acad Sci USA 103:1129–1134
Shang Y, Ma YS, Zhou Y, Zhang HM, Duan LX, Chen HM, Zeng JG, Zhou Q, Wang SH, Gu WJ, Liu M, Ren JW, Gu XF, Zhang SP, Wang Y, Yasukawa K, Bouwmeester HJ, Qi X, Zhang Z, Lucas WJ, Huang SW (2014) Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346:1084–1088
Sharma E, Anand G, Kapoor R (2017) Terpenoids in plant and arbuscular mycorrhiza-reinforced defence against herbivorous insects. Ann Bot 119:791–801
Singh B, Sharma RA (2015) Plant terpenes: defense responses, phylogenetic analysis, regulation and clinical applications. Biotechnology 5:129–151
Sun M, Voorrips RE, van’t Westende W, van Kaauwen M, Visser RGF, Vosman B (2020) Aphid resistance in Capsicum maps to a locus containing LRR-RLK gene analogues. Theoret Appl Genet 133:227–237
Takahashi S, Koyama T (2006) Structure and function of cis-prenyl chain elongating enzymes. Chem Rec 6:194–205
Takos AM, Rook F (2012) Why biosynthetic genes for chemical defense compounds cluster. Trends Plant Sci 17:383–388. https://doi.org/10.1016/j.tplants.2012.04.004
Tamura K, Dudley J, Nei M, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027
Tholl D, Lee S (2011) Terpene specialized metabolism in Arabidopsis thaliana.. Arabidopsis Book/Am Soc Plant Biol 9:e0143
Tikunov YM, Laptenok S, Hall RD, Bovy A, de Vos RCH (2012) MSClust: a tool for unsupervised mass spectra extraction of chromatography-mass spectrometry ion-wise aligned data. Metabolomics 8:714–718
Trapp SC, Croteau RB (2001) Genomic organization of plant terpene synthases and molecular evolutionary implications. Genetics 158:811–832. https://doi.org/10.1093/genetics/158.2.811
Vaello T, Sarde SJ, Marcos-García MÁ, de Boer JG, Pineda A (2018) Modulation of plant-mediated interactions between herbivores of different feeding guilds: effects of parasitism and belowground interactions. Sci Rep 8:14424. https://doi.org/10.1038/s41598-018-32131-9
van Herpen TWJM, Cankar K, Nogueira M, Bosch D, Bouwmeester HJ, Beekwilder J (2010) Nicotiana benthamiana as a production platform for artemisinin precursors. PLoS ONE. https://doi.org/10.1371/JOURNAL.PONE.0014222
Verma V, Ravindran P, Kumar PP (2016) Plant hormone-mediated regulation of stress responses. BMC Plant Biol 16:86. https://doi.org/10.1186/s12870-016-0771-y
Vranová E, Coman D, Gruissem W (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol 64:665–700
Wang KC, Ohnuma S-I (2000) Isoprenyl diphosphate synthases. Biochim Biophys Acta 1529:33–48
Whitehead IM, Threlfall DR, Ewing DF (1989) 5-epi-aristolochene is a common precursor of the sesquiterpenoid phytoalexins capsidiol and debneyol. Phytochemistry 28:775–779
Xu MM, Wilderman PR, Morrone D, Xu JJ, Roy A, Margis-Pinheiro M, Upadhyaya NM, Coates RM, Peters RJ (2007) Functional characterization of the rice kaurene synthase-like gene family. Phytochemistry 68:312–326
Xu M, Galhano R, Wiemann P, Bueno E, Tiernan M, Wu W, Chung IM, Gershenzon J, Tudzynski B, Sesma A (2012) Genetic evidence for natural product-mediated plant-plant allelopathy in rice (Oryza sativa). New Phytol 193:570–575
Yeaman S, Whitlock MC (2011) The genetic architecture of adaptation under migration-selection balance. Evol Int J Org Evol 65:1897–1911
Zangerl AR, Hamilton JG, Miller TJ, Crofts AR, Oxborough K, Berenbaum MR, de Lucia EH (2002) Impact of folivory on photosynthesis is greater than the sum of its holes. Proc Natl Acad Sci USA 99:1088–1091
Zavalapáramo G, Chávezmoctezuma MP, García-Pineda E, Yin S, Chappell J, Lozoyagloria E (2000) Isolation of an elicitor-stimulated 5-epi-aristolochene synthase gene (gPEAS1) from chili pepper (Capsicum annuum). Physiol Plant 110:410–418
Zhang PJ, Zheng SJ, van Loon JJA, Boland W, David A, Mumm R, Dicke M (2009) Whiteflies interfere with indirect plant defense against spider mites in Lima bean. Proc Natl Acad Sci USA 106:21202–21207
Zhang Y, Bouwmeester HJ, Kappers IF (2020) Combined transcriptome and metabolome analysis identifies defence responses in spider mite-infested pepper (Capsicum annuum). J Exp Bot 71:330–343
Zhou F, Pichersky E (2020) The complete functional characterisation of the terpene synthase family in tomato. New Phytol 226:1341–1360
Zhu B-Q, Cai J, Wang Z-Q, Xu X-Q, Duan C-Q, Pan Q-H (2014) Identification of a plastid-localized bifunctional nerolidol/linalool synthase in relation to linalool biosynthesis in young grape berries. Int J Mol Sci 15:21992–22010
Acknowledgements
We thank Johannes Hofberger for the inventory of (putative) C. annuum terpenoid pathway genes, Francel Verstappen for volatile measurements, Bert Schipper for LC-MS measurements. The Dutch Genebank (CGN.wur.nl) is acknowledged for providing Capsicum seeds.
Funding
This work is part of the Perspective programme Green Defences Against Pests of the Netherlands Organisation for Scientific Research (NWO, TTW-grant no. 13551) with financial support by breeding companies ENZA seeds, RijkZwaan, and Syngenta. YZ acknowledges the China Scholarship Council (CSC) for a supporting grant.
Author information
Authors and Affiliations
Contributions
YZ and ABK contributed equally. Conceived funding and designed the experiments: HJB and IFK. Performed the experiments: YZ, ABK and SB under guidance of IFK. Analysed the data: YZ, ABK, TZ and IFK. Wrote the manuscript: YZ and IFK. All authors contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Kashkooli, A.B., Blom, S. et al. The Capsicum terpenoid biosynthetic module is affected by spider-mite herbivory. Plant Mol Biol 113, 303–321 (2023). https://doi.org/10.1007/s11103-023-01390-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-023-01390-0