
Abstract
REM (reproductive meristem) transcription factors have been proposed as regulators of plant reproductive development mainly based on their specific expression patterns in reproductive structures, but their roles are still largely unknown probably because of their redundancy. We selected three REM genes (REM13, REM34 and REM46) for functional analysis, based on their genome position and/or co-expression data.
Our results suggest that these genes have a role in flowering time regulation and may modulate cell cycle progression. In addition, protein interaction experiments revealed that REM34 and REM46 interact with each other, suggesting that they might work cooperatively to regulate cell division during inflorescence meristem commitment.
Previous attempts of using co-expression data as a guide for functional analysis of REMs were limited by the transcriptomic data available at the time. Our results uncover previously unknown functions of three members of the REM family of Arabidopsis thaliana and open the door to more comprehensive studies of the REM family, where the combination of co-expression analysis followed by functional studies might contribute to uncovering the biological roles of these proteins and the relationship among them.
Key message
REM13, REM34 and REM46 were selected, through co-expression analysis of the whole REM family, as involved in flowering time regulation and show alteration in the cell division rate during this process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
REM (Reproductive Meristem) proteins belong to the plant-specific B3 superfamily of transcription factors (Franco-Zorrilla et al. 1999). REM genes appear consistently in transcriptomic studies focused on reproductive development (Mantegazza et al. 2014; Wynn et al. 2011), but their roles are still largely unknown. Currently, no phenotypes have been observed for several single or higher-order mutants, even if the REM genes under study were strongly expressed during specific phases and/or tissues during reproductive development (Mantegazza et al. 2014; Romanel et al. 2011; Wynn et al. 2011). Functional redundancy might contribute to this, as all investigated plant genomes contain dozens of REM genes often located in tandem in the genome, suggesting they arose by fragment duplications (Ahmad et al. 2019; Romanel et al. 2009; Ruan et al. 2021; Swaminathan et al. 2008; Verma and Bhatia 2019; Wang et al. 2012).
REM transcription factors are characterized by the presence of multiple B3 domains, a plant-specific DNA binding domain (Swaminathan et al. 2008), but differently to other families in the B3 superfamily, like ARFs, the mechanism for DNA binding of REM proteins is unclear, as different modes of binding have been described. While VRN1 was reported to bind DNA in a non-sequence specific mode (Levy et al. 2002) and, more recently, to undergo liquid-liquid phase separation with DNA (Zhou et al. 2019), REM1/REM34 binds DNA in a sequence-specific mode (Franco-Zorrilla et al. 2014). In addition, many of the REM proteins characterized so far can homo- and heterodimerize (Caselli et al. 2019; Mendes et al. 2016), suggesting that dimerization might be a common mechanism of action in this family.
So far, only a handful of REM genes have known roles in reproductive development. A few REMs have assigned roles in ovule development. Namely, in Arabidopsis, VERDANDI (VDD, REM20) (Matias-Hernandez et al. 2010), VALKYRIE (VAL, REM11) (Mendes et al. 2016), REM34 and REM35 (Caselli et al. 2019) have been linked to gametophyte development, whereas REM22 contributes to ovule primordia formation (Gomez et al. 2018). In rice, OsREM20 was recently shown to regulate grain number per panicle (Wu et al. 2021).
The other few REM genes functionally characterized includeVERNALIZATION1 (VRN1/REM5) (Levy 2002), RELATED TO VERNALIZATION 1 (RTV1/REM4) (Heo et al. 2012), TARGET OF FLC AND SVP 1 (TFS1/REM17) (Richter et al. 2019), and REM16 (Yu et al. 2020), all involved in flowering time regulation.
Floral transition is considered one of the most important developmental switches undertaken by the shoot apical meristem (SAM) and it affects the number of leaves, flowers and fruits produced by a plant. After the vegetative to reproductive transition, the SAM remodels into an inflorescence meristem (IM) which has the ability to initiate floral meristems (FMs) that will give rise to flowers (Fornara et al. 2010; Kwiatkowska 2008). This transition has to be precisely coordinated through internal and external signals, to ensure the reproductive success of the plant. A broad range of studies done in Arabidopsis thaliana and expanded to crops and other species suggest substantial conservation of this process (Blümel et al. 2015). Several signals such as developmental, hormonal and environmental cues are precisely perceived and processed from six main genetic pathways, which converge into a few genes called “floral integrators” (Simpson and Dean 2002). Control of flowering in response to seasonal changes is mastered by the vernalization and photoperiod pathways, whereas changes in ambient temperature are perceived by the ambient temperature pathway. In Arabidopsis thaliana, flowering is promoted by long-day photoperiod (LD, when night length falls below a certain threshold), but this is not a mandatory requirement. Therefore, Arabidopsis plants grown in short-day photoperiod (SD) will eventually flower as well (Wang et al. 2019). The age, autonomous, and gibberellin pathways, act mainly independently of environmental stimuli (Srikanth and Schmid 2011). The synergetic action of these pathways ultimately converges into floral integrator genes which modulate flowering time and activate floral meristem identity genes (Srikanth and Schmid 2011). FLOWERING LOCUS T (FT), which belongs to the PEBP (phosphatidylethanolamine-binding protein) family (Kobayashi et al. 1999), and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1/AGL20) (Lee et al. 2000), a MADS-box transcription factor, are two floral integrators; FT interacts with FLOWERING LOCUS D (FD) in the SAM to activate the floral promoter SOC1 and, later on, the floral meristem identity genes APETALA 1 (AP1) and the AP1 paralog CAULIFLOWER (CAL) (Abe et al. 2005). SOC1 also activates floral meristem identity through LEAFY (LFY). Together, LFY, AP1, CAL, AGL24 and SVP orchestrate the differentiation of a group of cells at the flank of IM into FMs (Gregis et al. 2008; Liu et al. 2008).
During the vegetative to reproductive transition, an increase in cell division occurs in the SAM (Jacqmard et al. 2003; Kwiatkowska 2008). This phenomenon was first described in the 1960s (Bernier 1969; Corson, 1969; Miksche and Brown, 1965) at the cytological level, and since then it has been described in a variety of plants suggesting that it is widespread among angiosperms (Kurokura et al. 2006; Lyndon and Battey 1985; Marc and Palmer, 1984). A more recent study in Arabidopsis thaliana (Jacqmard et al. 2003), showed that, upon transfer to LD, the increase of mitotic activity is one of the earliest events at the SAM in the vegetative to reproductive transition, preceding the enlargement and doming of the SAM and the bolting and initiation of the first floral meristem (Jacqmard et al. 2003). The authors found also that mitotic activity increases throughout the SAM, except in the organizing center, where stem cells reside (Jacqmard et al. 2003).
However, in all these pioneering works, molecular events were not characterized. In 2015, Klepikova et al. published a study based on RNA-seq, where they found that the increase in cell division observed during floral transition is supported by the upregulation of genes related to the cell cycle that occurs at a precise stage, where FLC expression has decreased and LFY expression has not started to increase yet. Based on the expression patterns of cell cycle-related genes, the authors proposed that the increase in cell division was caused by a shortening of G1 and G2 phases, which also causes a partial synchronization of the cell cycle. This hypothesis is also in line with most of the previous observations by cytological means. More recently, Kinoshita et al. (2020) showed that the increase in cell division is, at least, partially controlled by the photoperiodic and the gibberellin flowering pathways.
In this work, we selected three REM genes (REM13, REM34 and REM46) for functional analysis based on their genome position or co-expression data. Our results indicate that these genes have a role in flowering time regulation and may modulate cell cycle progression. In addition, protein interaction experiments revealed that REM34 and REM46 interact with each other, suggesting that they might work cooperatively. Overall, our findings add pivotal information about the biological role of three members of the REM family of Arabidopsis thaliana and the co-expression patterns of the REM family, which might contribute to uncovering the biological roles of these genes and the functional relationship among them.
Materials and methods
Plant material and growth conditions
Mutant lines for the different REMs were obtained from the Nottingham Arabidopsis Stock Center (NASC): SALK_022885 (rem13-1/rem13_oe), SALK_050242 (rem13-2/rem13_kd), FLAG_566H04 (rem34-1), and SALK_151966 (rem46-1). Primers for the genotyping of mutants are listed in Supplementary Table 1. For flowering time assays plants were grown in a growth chamber under long-day conditions (LD: 16 h light/8 h darkness) at 22/24 °C. For expression analysis and cell cycle analysis, plants were grown in a growth chamber under short-day conditions (SD: 8 h light/16 h darkness) for 21 days. Then, plants were transferred to long-day conditions to induce flowering.
Co-expression and GO term enrichment analysis
For co-expression analysis, the “co-expression neighborhood” (CEN, list of the 20 most closely co-expressed genes with a particular gene) of each REM gene was retrieved from the athrna database (Zhang et al. 2020) and the number of co-expressed genes shared between a particular REM gene with every other REM gene was used for the clustering analysis. Clustering analysis and heatmaps were obtained with ClustVis, using Euclidean distances and average linkage method. CENs for each REM gene can be found in Supplementary File 1 (downloaded on the 6th of September 2021 from athrna database). GO term enrichment analysis was performed using Panther (Mi et al. 2019a, b).
Flowering time measures
Flowering time was measured in terms of the number of rosette leaves at the time of bolting and of days from sowing to bolting. Plants were considered bolted and rosette leaves were counted when the inflorescence was 1 cm long. A minimum of 10 plants per genotype and replicate were used.
Gene expression analysis
RNA was extracted using the LiCl method (Verwoerd et al. 1989). For each sample, 500 ng of RNA were retro-transcribed using iScript kit (BioRad) following the manufacturer’s instructions. qRT-PCR assay was performed using iTaq Universal SYBR Green supermix (BioRad) in a Bio-Rad iCycler iQ Optical System (software version 3.0a). Three biological replicates, with three technical replicates for each sample, were analyzed. Relative transcript enrichment of genes of interest was calculated by normalizing the amount of mRNA against EIF4 (Yamaguchi et al. 2009). Expression of genes was calculated using the 2−ΔΔCt method, using the Wild Type or Wild Type T0 as normalizer. Statistical significance was calculated on ΔCt values with a t-test. The primers used for this analysis are listed in Supplementary Table 1.
In situ hybridization analysis
SAMs were dissected by removing fully developed leaves, fixed in FAA (50% ethanol, 5% acetic acid, 3.7% formaldehyde) under vacuum for 15 min, dehydrated in ethanol and bioclear (Bioptica) and embedded in Paraplast Plus (Sigma-Aldrich). In situ hybridization was performed as previously described (Coen et al. 1990) with slight modifications. Digoxigenin-labelled antisense probes were synthesized with T7 RNA polymerase (Promega). For REM13 detection we used the probe described in Villarino et al. 2016, for REM34 we used the probe described in Mantegazza et al. 2014; for REM46 we designed a new specific probe and for H4 we employed the probe described in Petrella et al. 2020. Primers for probe amplification are listed in Supplementary Table 1. As the REM46 probe was designed downstream of the rem46-1 T-DNA insertion, specificity of the REM46 probe was tested on the rem46-1 mutant where, in contrast to wild type tissue, no signal was detected (Supplementary Fig. 5). The specificity of the signal given by all the other in situ hybridization probes employed in this study was already assessed in previous studies (Mantegazza et al. 2014; Robert et al. 1994; Villarino et al. 2016).
Flow cytometry analysis of cell cycle
Plants were grown in a growth chamber under SD (non-inductive) conditions for 20 days. Then, the photoperiod was shifted to LD and samples were collected at three time points: before the light shift (T0), 24 h (T1) and 72 h (T3) after the transfer to inductive conditions. Each sample consisted of 3 meristems, with three technical replicates per time point and genotype. Samples for flow cytometry were prepared as described by Yang et al. 2019. Flow cytometry was performed using a BD FACS Canto II (BD Biosciences) equipped with FACSDiva Software v6.1.3. 10,000 events per sample were analyzed. Stages of the cell cycle in the nuclei population were determined using FlowJo® 10.8.1 (BD Life Sciences) using the Watson model.
Plasmid construction
The coding sequences of REM13, REM17, REM34, REM36 and REM46 were amplified by PCR from cDNA, cloned in pDONR207 and subsequently transferred to pGADT7 and pGBKT7 (Clontech Laboratories, Inc) for yeast-two-hybrid assays; and to pYFN43 and pYFC43 (Belda-Palazón et al. 2012) for BiFC assays, by Gateway cloning (Invitrogen). Primers used for cloning are listed in Supplementary Table 1.
Yeast-two-hybrid assay
The yeast-two-hybrid experiments were performed in the AH109 strain. The bait (pGBKT7, Clontech Laboratories, Inc) and prey (pGADT7, Clontech Laboratories, Inc) vectors were co-transformed in yeast as described by de Folter and Immink (de Folter and Immink 2011). The protein-protein interaction assays were performed on selective yeast synthetic dropout medium lacking leucine, tryptophan and histidine supplemented with different concentrations of 3-aminotriazole (1, 2.5, or 5 mM of 3‐AT). Plates were grown for 5 days at 28 °C. The already published REM34-REM34, REM34-REM35 and REM35-REM35 interactions were employed as negative and positive controls (Supplementary Fig. 4 and Caselli et al. 2019).
Bimolecular Fluorescence Complementation (BiFC) assays
Nicotiana benthamiana leaves were infiltrated with Agrobacterium tumefaciens GV3103 previously transformed with the vectors of interest and the viral suppressor p19. Three days after inoculation, the abaxial surface of the leaves was imaged employing a Laser Scanning Confocal Microscope Nikon A1. As for the yeast-two hybrid experiment, the already published REM34-REM34 and REM35-REM35 interactions were employed as negative and positive controls, respectively (Supplementary Fig. 4 and Caselli et al. 2019).
Results
REM13 is a positive regulator of floral transition
REM13 (AT3G46770) is located on the long arm of chromosome 3. It is one of the few family members that is not part of a cluster of REM genes (Mantegazza et al. 2014) (Supplementary Fig. 1b), making it a good candidate for genetic analysis, as it could be potentially less redundant than other REMs that have undergone tandem duplications.
Unfortunately, lines with insertions or mutations in exons were not available in public collections. We thus analyzed two independent lines: SALK_022885 (rem13-1) and SALK_050242 (rem13-2), which contain insertions on the promoter region of REM13. The insertions were confirmed by sequencing to be at positions − 481 and − 342 respectively (Fig. 1a). The expression of REM13 in these lines was analyzed by qRT-PCR and, interestingly, we found that, while rem13-1 overexpressed REM13 (henceforth called rem13_oe for “overexpressor”), rem13-2 showed a moderate reduction (20%) of REM13 expression level (henceforth called rem13_kd for “knock-down”) (Fig. 1b).

Characterization of REM13 mutants. a Position of rem13-1 (rem13_oe) and rem13-2 (rem13_kd) T-DNA insertions in REM13 (At3g46770) genomic region, light blue boxes: UTR, dark blue boxes: exons, lines: introns. b Expression of REM13 in rem13_oe and rem13_kd mutants. Graph shows 2−ΔΔCt average of three biological replicates, significance was calculated on ΔCt using t-test (* p-value < 0.05, ** < 0.01) c Flowering time of rem13_oe and rem13_kd mutants, measured as the number of rosette leaves at bolting. Significance was calculated using a t-test coupled with Bonferroni correction for multiple testing (*** p-value < 0.001). d REM13 expression profile, detected by in situ hybridization in wild type SAM, showing REM13 ubiquitously expression in the meristem and the adaxial side of developing leaves primordia. Scale bar 100 μm e Expression level of REM13 in wild type SAM-enriched tissue in non-inductive SD conditions (T0) and 24 h (T1), 72 h (T3) after the switch to LD conditions. Graph shows 2−ΔΔCt average of three biological replicates, normalized on T0
While growing the plants for propagation, we readily noticed that rem13_oe seemed to flower earlier than wild type plants. Therefore, we decided to analyze the flowering time of both mutant lines in inductive LD conditions (16 h light, 8 h dark) to verify the involvement of REM13 in this character. While wild type plants produced on average 12.44 leaves at bolting, rem13_oe plants showed a reduction of flowering time, with plants bolting with an average of 8.39 leaves (Fig. 1c). Despite the moderate reduction in the expression of REM13 displayed by rem13_kd, this line showed a delay in flowering time, with plants bolting with an average of 18.25 leaves (Fig. 1c). To better contextualize the putative role of REM13 during the floral transition, the expression profile of this gene was investigated. In situ hybridization, carried out on wild type SAM, highlighted that REM13 is expressed both in the meristematic dome and in the developing leaf primordia (Fig. 1d). Furthermore, we performed a time course qRT-PCR experiment, prepared from hand-dissected apices (SAM-enriched tissue) of wild type plants grown for three weeks in non-inductive SD conditions and transferred to LD to induce flowering. Samples were collected at T0 (SD conditions), T1 (24 h LD) and T3 (72 h LD), to assess the changes in the expression of the gene of interest during floral transition. This analysis showed that REM13 mRNA level is stable during the switch between SD and LD (T0 and T1), and quickly rises 72 h after exposure to inductive conditions, further suggesting a putative role of REM13 in the control of vegetative to reproductive transition (Fig. 1e). Overall, these results suggest that REM13 promotes floral transition, since an increase in REM13 expression leads to a reduction of flowering time, while its downregulation delays floral transition.
Clustering analysis of REM genes supports independent roles in flowering time control
To better understand the redundancy relationship between the several REMs of Arabidopsis, and the role of REM13 in the context of the whole REM family, we performed a co-expression analysis. First, we obtained a comprehensive list of all the REM genes that have been predicted in Arabidopsis thaliana (Romanel et al. 2009; Wang et al. 2012; Swaminathan et al. 2008). We checked the overlap between the lists and found that 44 REM genes were predicted by all the works, 10 by 2 works, and 22 were only predicted by Swamithanan and collaborators (Supplementary Fig. 1a). One of the predicted genes (AT2G21920) (Swaminathan et al. 2008) is presently annotated as an F-box protein, so we removed it from the list. We generated a non-redundant list containing the 75 predicted REM genes (Supplementary Table 2) and depicted their position in the chromosomes of A. thaliana (Supplementary Fig. 1b). Romanel and collaborators named the 45 REM genes they described as REM1-45 (Romanel et al. 2009). REM46 was named in a separate work (Villarino et al. 2016), while the remaining 29 REM genes were not named, so we kept this nomenclature in this work.
Recently, Zhang and collaborators published a database (athrna database) based on more than 20,000 publicly available RNA-seq (Zhang et al. 2020). athrna database can generate a list of the 20-top co-expressed genes with a particular gene (from here on its “co-expression neighborhood”, “CEN”). Therefore, to gain further insight into the function of REM13, we performed a co-expression analysis based on the CENs provided by athrna database (Supplementary Table 3) and we produced a distance matrix comparing 75 REM genes using ClustVis (Metsalu and Vilo 2015) (Fig. 2). This analysis revealed that several groups of REMs cluster together, as their CENs share numerous genes.

Co-expression matrix for REM genes. Co-expression data was extracted from athrna database. Columns and rows were hierarchically clustered using Euclidean distance and the average linkage method using ClustVis (biit.cs.ut.ee/clustvis/) (Metsalu and Vilo 2015). REM13 cluster is highlighted in purple. REM17/TFS1 (light blue), REM16 (pink), REM4/RTV1 (orange) and REM5/VRN1 (green) and their respective clusters are also highlighted, as they have also been linked with flowering time regulation
REM13 forms a small cluster (highlighted in purple in Fig. 2) with REM11/VAL, REM20/VDD, REM21 and AT1G10455. None of these genes have been linked to flowering time regulation. REM20/VDD and REM11/VAL modulate synergid degeneration upon pollen tube arrival (Matias-Hernandez et al. 2010; Mendes et al. 2016), while REM21 and AT1G10455 do not have a known function yet.
We also investigated the REM genes already known to be involved in flowering time. TFS1/REM17, an activator of floral transition and target of FLC and SVP (Richter et al. 2019), is located in a big cluster containing 22 REMs in total (highlighted in light blue in Fig. 2). In this cluster, besides TFS1, the only genes with known biological roles are REM34, REM35, and REM36 which have been related to gametophyte development (Caselli et al. 2019).
REM16, a promoter of flowering that regulates SOC1 and FT (Yu et al. 2020), forms a small group of 6 genes with REM15, REM19, REM22, REM24 and REM25 (highlighted in pink in Fig. 2). Among these, REM15 was found to be a target of AGAMOUS (Gómez-Mena et al. 2005) expressed in the megaspore mother cell (Wynn et al. 2011). Instead, REM22 and REM24 were upregulated in the gibberellin-insensitive mutant gai-1, which is involved in the regulation of ovule number (Gomez et al. 2018).
Finally, REM5/VRN1 and REM4/RTV1, which regulate flowering time through the vernalization pathway (Heo et al. 2012; Levy et al. 2002), do not cluster among them or with any other REMs.
In summary, co-expression analysis based on athrna data showed that REM13 does not cluster with any of the REM genes previously linked with floral transition, suggesting that it might regulate flowering time through an independent pathway. In addition, some of the genes that cluster with REM13, like VDD/REM20 or VAL/REM11, are involved in other processes along the reproductive phase. Interestingly, this same pattern was observed for the rest of the REMs with a known role in floral transition (TFS1/REM17, REM16, VRN1 and RTV1), as all of them clustered separately and the respective clusters contained REMs with functions in other reproductive phases. Overall, this observation suggests two conclusions. First, that all the REMs with a known role in floral transition act independently of each other, and second, that either REMs have recursive roles along the reproductive process, or that different REMs might regulate different processes through the control of similar sets of genes.
REM34 and REM46 are negative regulators of floral transition
The co-expression analysis revealed that TFS1/REM17, known to be involved in flowering time regulation (Richter el al., 2019), belongs to a cluster containing 22 REM genes (Fig. 2). Interestingly, some of the genes of the cluster are linked to other processes throughout Arabidopsis life cycle, like REM34 (Caselli et al. 2019), involved in gametophyte development, or REM46, which was found to be a marker gene for the carpel margin meristem (Villarino et al. 2016). This poses the question of whether each of these REM genes regulates more than one process along reproduction, or if each one acts in a specific phase, but they all exert their function through the control of similar sets of genes. To investigate this, we examined whether REM34 and REM46 also have a role in flowering time control.
First, we verified the expression pattern of these two genes in the SAM during floral transition. In situ hybridization revealed that REM34 and REM46 share a similar expression domain, being expressed mainly in the meristematic dome (Fig. 3a, control in supplementary Fig. 5). A qRT-PCR time-course analysis revealed that REM34 and REM46 mRNA level is steady during the shift between SD (T0) and LD (24 h LD) and it increases in T3 (72 h LD) (Fig. 3b).

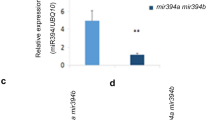
Characterization of rem34-1 and rem46-1. a in situ hybridization analysis showing the expression profiles of REM34 and REM46 within the SAM and the adaxial side of developing leaves primordia for REM34 and the whole developing leaves primordia for REM46. Scale bar: 100 μm (Control for the REM46 probe is presented in Supplementary Fig. 5) b RT-PCR showing the expression level of REM34 and REM46 in wild type SAM-enriched tissue in non-inductive SD conditions (T0) and 24 h (T1), 72 h (T3) after the switch to LD conditions. Graph shows 2−ΔΔCt average of three biological replicates, normalized on T0. c Position of rem34-1 and rem46-1 T-DNA insertion in the REM34 (AT4G31610) and REM46 (AT5G60142) genomic sequences. Light boxes: UTR, dark boxes: exons, line: introns. d Expression of REM46 in rem46-1, compared to the wild type, rem46-1 shows a reduction in the expression of around 70%. significance was calculated on ΔCt using t-test (***p-value < 0.001) d Flowering time of wild type, rem34-1 and rem46-1 plants measured as leaves at bolting. Significance was calculated using a t-test coupled with Bonferroni correction for multiple testing. (****p-value < 0.0001)
We then analyzed whether flowering time was altered in rem34-1 and rem46-1 mutant lines. For REM34, we employed the already characterized FLAG_566H04 line (rem34-1), which shows downregulation of the REM34 transcript (Mantegazza et al. 2014). For REM46, we chose the uncharacterized SALK_151966 line (rem46-1 from here on), which carries a T-DNA insertion in an exonic region in the four splice forms of REM46 (Fig. 3c). The insertion is located at the end of the third exon in the canonical splice isoform (AT5G60142.1) and causes a 70% reduction of REM46 transcript level (Fig. 3d).
We analyzed flowering time under LD conditions in rem34-1 and rem46-1 and we observed that both mutants showed an early flowering phenotype compared to wild type plants which, in our growing conditions, had on average 11.06 leaves at bolting. rem46-1 produced indeed an average of 9.82 leaves at the bolting stage, while rem34-1 showed a higher reduction, having 6.1 leaves at bolting (Fig. 3e). The negative effect on flowering time regulation by REM34 was confirmed by analyzing three REM_RNAi lines and two 35S:REM34-EAR fusion lines, previously described by Caselli et al. 2019 (Supplementary Fig. 2). In conclusion, our results suggest that REM34 and REM46 share a similar expression profile throughout the floral transition, and they act as negative regulators of this process.
Expression of floral transition markers in the early flowering REM mutants
To better understand the role of REM13, REM34 and REM46 during the reproductive switch, we measured the expression of three floral integrators in the rem mutant backgrounds of interest. To synchronize the plants in order to compare the different genotypes, the plants were grown under non-inductive SD conditions for 21 days and then switched to LD conditions to induce flowering. As described above, SAM-enriched tissue was collected at 0 h (T0), 24 h (T1) and 72 h (T3) after the transfer from SD to LD conditions.
The MADS-box gene SOC1 marks the floral transition, as it starts to be expressed at T1, as soon as the SAM acquires the competence to become IM (Yoo et al. 2005). In the early flowering mutants rem34-1 and rem46-1, as well as in rem13_oe, SOC1 expression at T0, before the photoperiodic induction, is higher than in the wild type. SOC1 upregulation was particularly dramatic in all of the analyzed time points in the rem34-1 background, which is the mutant showing the earliest flowering phenotype (Fig. 4a).

Expression analysis of SOC1 (a), AP1 (b) and LFY (c) in the wild type, rem13_oe, rem34-1 and rem46-1 genetic backgrounds. The expression levels are normalized on the EIF4 housekeeping gene and wild type at T0 was set to 1. Graph shows 2−ΔΔCt average of three biological replicates, significance was calculated on ΔCt using t-test (* p-value < 0.05, ** < 0.01, *** < 0.001)
As a second marker, we used the floral meristem identity gene AP1 (Liljegren et al. 1999). In wild type SAMs, AP1 expression starts to increase at T1 and reaches its highest level of expression 72 h after the switch to LD (T3). The AP1 transcript was highly upregulated already at T0 in rem34-1 and is continuously upregulated in the subsequent time points. AP1 was slightly upregulated also in rem46-1 in all three time points. In rem13_oe, AP1 transcript is upregulated at T1 and T3 compared to the wild type, suggesting that also in this line the floral meristem starts to be specified earlier (Fig. 4b).
Finally, we analyzed the expression of LFY (Liljegren et al. 1999). LFY was upregulated in rem34-1 throughout all the time points analyzed while in rem13_oe a slight upregulation was visible at T1. rem46-1 instead showed a pattern similar to the wild type (Fig. 4c).
These data fit with the phenotypical analysis, as in the early flowering mutants rem34-1, rem46-1, and rem13_oe, most or all the analyzed floral transition markers show higher and/or earlier expression than the wild type.
Moreover, in rem34-1, which has the most drastic reduction of the flowering time, SOC1, LFY and AP1 were strongly upregulated already at T0, before the switch to LD conditions. To verify whether this upregulation correlates with an early flowering time in non-inductive SD conditions, wild type and rem34-1 plants were grown in SD and bolting time was recorded. rem34-1 plants started to bolt 45 days after germination, while wild type plants remained in a vegetative state (Supplementary Fig. 3a).
Co-expression neighborhoods link REM clusters to specific biological processes
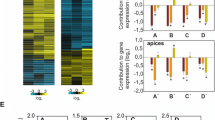
To gain further insight into how REM13, REM34 and REM46 regulate flowering time, we performed an enrichment analysis of Biological Process GO terms associated with the genes belonging to the CENs of the clusters containing REM13, REM34 and REM46. For that, we obtained non-redundant lists of all the genes in the CENs of every REM belonging to the clusters of interest and performed an enrichment analysis using Panther (Mi et al. 2019a, b) (Fig. 5, Supplementary Table 4).

GO term enrichment analysis of the 20-top co-expressed genes (“CEN”) with REM13 (a) and REM17/TFS1 (b) clusters. Enrichment analysis was performed using Panther (Mi et al. 2018). For simplicity, only tip-most GO terms are plotted
In the case of the REM13 cluster (Fig. 5a), the terms ‘synergid cell death’ and ‘plant ovule development’ were highly enriched, in accordance with the presence of VDD and VAL, but no terms that can be linked to flowering time were found.
REM34 and REM46 belong to the same cluster (Fig. 2). REM genes in this cluster are co-expressed with genes associated with cell cycle progression (Fig. 5b), as GO terms like ‘DNA integrity checkpoint signaling’, ‘cell cycle replication’, or ‘mitotic cell cycle process’ were enriched. Terms like ‘cell cycle DNA replication’, ‘microtubule-based movement’, ‘spindle organization’ or ‘nuclear division’ seem to suggest a role in the replication (S phase) and division (M phase) phases of the cycle. Moreover, these genes might be also involved in meiosis, as terms linked to meiosis like ‘DNA recombination’ or ‘meiotic cell cycle process’ are also enriched. Although no terms explicitly linked to flowering time control were found, the floral transition involves an increase in cell division at the SAM (Jacqmard et al. 2003; Kinoshita et al. 2020; Klepikova et al. 2015; Kwiatkowska 2008), suggesting that the role of REM genes located in this cluster might be linked to cell division.
REM13, REM34 and REM46 modulate cell cycle progression in the SAM during floral transition
During the transition from the vegetative SAM to the IM, there is an increase in cell division (Kinoshita et al. 2020; Klepikova et al. 2015; Marc and Palmer, 1984). GO term enrichment analysis suggested that REM34 and REM46 might be involved in the regulation of the cell cycle (Fig. 5b), which could be linked to the early flowering time phenotype of these mutants.
To check if REM13, REM34 and REM46 can influence the cell cycle, we measured cell cycle progression in the meristem of wild type and mutants during the floral transition, through the estimation of DNA content by flow cytometry staining nuclei with propidium iodide (PI) (Fig. 6). In particular, we measured the cell cycle in hand-dissected apices before transferring the plants from SD to LD (T0), and 24 (T1) and 72 (T3) hours after the switch. The results were analyzed using FlowJo 10.8.1 (BD Life Sciences).

Cell cycle progression analysis. a The histograms show the percentage of cells in the different cell cycle stages (G1, S, G2, >G2) at T0, T1 and T3 as the average of three biological replicates, each one consisting of three meristems. At T0, rem13_oe, rem34-1 and rem46-1 exhibit a significative increase in the percentage of cells in the S phase, consistent with an increase of cell division rate. Statistical significance was evaluated with a t-test (*p < 0.05, **<0.01). b shows an example of the nuclei distribution and their difference in DNA content at T0 for each genotype. c Expression of the S phase marker H4 was investigated via in situ hybridization, in meristems collected at T0 (SD conditions). In the wild type and rem13_kd H4 show a similar expression pattern, as the cells expressing this marker are mainly localized in the peripheral zone of the meristem where the new leaf primordia are differentiating. In rem13_oe, rem34-1 and rem46-1, however, the expression profile of H4 is broader and cells expressing this marker are localized also in the central zone of the meristem, confirming a general increase in cell cycle rate. Scale bar 100 μm
The wild type presented an increase in the percentage of cells in the S phase 24 h after the transfer from SD (T0) to LD conditions (T1) (Fig. 6a), which is symptomatic of an increased cell cycle rate associated with floral transition and is consistent with the increase in SOC1 expression observed previously (Fig. 4a). The higher cell division rate is maintained also 72 h (T3) after transfer to LD (Fig. 6a) and it indicates that the SAM is enlarging and changing its identity, becoming the reproductive IM.
At T0, rem34-1 and rem46-1 showed already a 10% increase in the number of cells in the S phase compared with the wild type, suggesting that in the SAM of these mutants, cells divide faster than those of the wild type (Fig. 6ab). As the plants are shifted to LD (T1-T3), the cell cycling rate increases also in the wild type and the percentage of cells in the S phase reaches a similar level to rem34-1 and rem46-1.
The two rem13 mutant alleles, rem13_kd and rem13_oe, exhibit opposite behavior, in accordance with their different effects on flowering time regulation. In rem13_oe, indeed, which is characterized by a shorter flowering time, we measured a higher percentage of cells in S phase than in the wild type at T0, suggesting a possible increase in cell cycle also in this mutant. On the other hand, rem13_kd plants, characterized by a delayed flowering time, showed a similar trend of cell cycling and division of the wild type (Fig. 5 ab).
Overall, these results suggest that REM13, REM34 and REM46 are involved in the control of flowering time and show alterations in the progression of the cell cycle during floral transition. Furthermore, while REM34 and REM46 appear to act as negative regulators of cell cycle and division rate, REM13 seems to promote these processes.
To further confirm the GO term enrichment and the flow cytometer analyses, which strongly suggested a link between the role of the REM genes under analysis in flowering time regulation and the modulation of cell division, the expression pattern of the Histone4 (H4) mRNA was investigated by in situ hybridization (Fig. 6c). The H4 is indeed considered a marker of cell cycle activity, being expressed specifically during the S phase (Geier et al. 2008; Wang and Liu 2006; Xu et al. 2008). The analysis was performed on tissues collected at T0, grown under short-day conditions, to allow the comparison of the results with the flow cytometer data. In the wild type SAM, the majority of the H4-expressing cells are located in the developing primordia, on the flanks of the meristematic area, where the cells are dividing and differentiating into leaves. In rem13_kd the H4 expression pattern is similar to the one of the wild type, confirming that the number of cells in the S phase is similar in these two genetic backgrounds as suggested by the flow cytometer analysis. In rem13_oe, rem34-1 and rem46-1, where a significative increase in the percentage of cells in the S phase was recorded, the expression profile of H4 is wider than what was observed in the wild type. In particular, the cells expressing this cycling marker are not confined in the peripheral zone of the meristem but are spread into all three layers of the central zone, suggesting that the increase in the cell cycle rate observed in rem13_oe, rem34-1 and rem46-1 is due to an increase of dividing cells in the meristematic tissue. Furthermore, this analysis revealed a dramatic increase in the dimensions of the SAM of rem34-1 compared to the wild type. The meristem, in this genetic background, appears to be both wider and higher and has a more pronounced dome than the one of the wild type, a shape that is usually observed in meristems that already underwent floral transition (Supplementary Fig. 3 bc). As the meristems employed for this analysis were collected under SD non-inductive conditions, this observation, coupled with the early flowering time phenotype observed in SD for rem34-1, strongly suggests that REM34 might have a role in the light-dependent flowering time regulation.
Interaction analysis of REM13, REM17, REM34 and REM46.
Several REMs are able to homo/heterodimerize (Caselli et al. 2019; Mendes et al. 2016). Therefore, we decided to test whether REM13, REM34 and REM46 can interact by yeast-two-hybrid (Y2H). Caselli and co-workers (Caselli et al. 2019) showed that REM34 heterodimerizes with REM35 but is unable to homodimerize, so these pairs were used as positive and negative controls respectively (Supplementary Fig. 4ab).
Y2H assays showed that REM46 can form heterodimers with REM34, suggesting that these proteins might cooperate (Fig. 7b). TFS1 was able to homodimerize but did not show any positive interaction with REM34 and REM46, even though they belong to the same co-expression cluster (Fig. 7a). This observation was in accordance with their opposite role in the regulation of flowering time (Richter et al. 2019).

Protein-protein interactions. a Yeast-two-Hybrid assay showing that TFS1 is able to homodimerize but does not interact with REM34 and REM46, b REM46 can interact with REM34, c the empty pGADT7 and PGBKT7 were used as controls. For each tested interaction four independent colonies were tested on selective media lacking leucine, tryptophan and histidine, supplemented with different concentrations of 3-aminotriazole (1, 2.5, or 5 mM of 3‐AT) d BiCF assay, confirming the TFS1-TFS1 homodimerization and the REM34-REM46 heterodimers formation Bars = 50 µM
Finally, in line with the co-expression data (Fig. 2), no interactions were found between REM13 and any of the other REMs tested (Supplementary Fig. 4b).
All the positive interactions found in the yeast-two hybrid analysis were further confirmed by Bimolecular Fluorescence Complementation (BiFC) (Fig. 7d). REM34-REM34 and REM34-REM35 interaction were used as negative and positive control respectively (Supplementary Fig. 4c).
The interaction between REM34 and REM46 indicates that these proteins might cooperate, as also suggested by the similar negative effect that these genes have on floral transition. Instead, in line with the lack of co-expression of REM13 with REM34 and REM46, REM13 did not heterodimerize with any of those, suggesting an independent role.
Discussion
Here, we report roles in flowering time regulation for REM13, REM34 and REM46 potentially through the modulation of cell division. Based on the phenotype of the mutants, co-expression and protein interaction data, we propose the existence of independent networks involving different sets of REM proteins that influence flowering time in Arabidopsis.
All investigated plant genomes contain dozens of REM genes, many located in tandem, suggesting that they arose through gene duplication events, (Ahmad et al. 2019; Romanel et al. 2009; Swaminathan et al. 2008; Verma and Bhatia 2019; Wang et al. 2012) so redundancy might be hiding the role of most of them.
For this reason, we initially focused our attention on REM13, a ‘solitary’ REM gene located in chromosome 3, as it might be less redundant than REMs located in tandem. We characterized two rem13 mutant alleles, one of which showed an increase in the expression of REM13 (rem13-1 or rem13_oe), and the other a slight reduction (rem13-2 or rem13_kd) and they were early and late flowering, respectively (Fig. 1).
To better understand the role of REM13 and to investigate if it cooperates with other REMs in flowering time regulation or other traits, we performed a clustering analysis of the Arabidopsis REM family based on the co-expression data present in athrna database (Zhang et al. 2020) (Fig. 2). Most REMs grouped in clusters, suggesting that they might cooperate or be functionally redundant since they are co-expressed with similar sets of genes. REM13 belonged to a small cluster containing VDD and VAL, involved in synergid identity and degeneration (Matias-Hernández et al. 2010; Mendes et al. 2016).
We also observed that although genes located closely in the genome tend to be co-expressed, on some occasions, they can be co-expressed with REMs at other genomic locations (i.e., REM34 and REM46) (Supplementary Fig. 1), and conversely, genes like VRN1 or RTV1 are not co-expressed with any other REMs even if they are located in tandem with other REM genes (Supplementary Fig. 1). Overall, this suggests that the genomic position and the presence of other REMs close by are not entirely predictive of the potential redundancy of a particular REM and that the co-expression analysis proposed here might be a successful strategy to identify redundancy relationships in this highly redundant family.
Another interesting finding of the co-expression analysis is that none of the REMs previously described as regulators of flowering time (VRN1, RTV1, REM16 and TFS1) clustered together, suggesting that they might control floral transition independently. This observation led us to wonder whether genes clustering together, such as TFS1, REM34 and REM46, which have been linked to different biological processes, participate in more than one process throughout reproductive development, or if each gene is specialized in a specific stage but they regulate similar sets of genes. To explore this hypothesis, we checked if REM34 and REM46 have also a role in flowering time.
rem34-1 and rem46-1 mutants showed reduced flowering time in LD (Fig. 3), which correlated with the higher expression at early time points of the floral integrators SOC1 and AP1 compared to the wild type. This confirms that REM34 and REM46 have multiple roles in reproduction and suggests that this situation could be extended to other REM genes.
In line with its strong early flowering phenotype, rem34-1 showed the highest upregulation of SOC1, LFY and AP1, which were strongly upregulated already before the transfer to LD (Fig. 4). In the case of rem13_oe and rem46-1 at T0 only SOC1 and AP1 were upregulated, while LFY showed a pattern similar to the wild type.
SOC1 and AP1 are directly regulated by FT (Fornara et al. 2010), which was recently found to be a direct target of REM16 (Yu et al. 2020). The upregulation of SOC1 and AP1 in rem34-1 and rem46-1 might suggest these genes directly or indirectly regulate FT. The fact that LFY is not upregulated in rem46-1 and rem13_oe at T0, despite being a target of SOC1 and AP1, could indicate that REM13 and REM46 regulate these genes through an independent pathway.
Then, we used GO term enrichment analysis of the combined CENs of the clusters of the REMs under study (Fig. 5) to gather new insight on the molecular function of the clusters of interest. In the case of REM34 and REM46, the analysis suggested that the genes in this cluster might be associated with cell division (Fig. 5b). Interestingly, we recently showed that REM34 modulates the expression of Kip-related protein 6 (KRP6), a cyclin-dependent kinase inhibitor (CKI), (Caselli et al. 2019) and TFS1 was also suggested to regulate cell division based on its pattern of expression in the meristem (Richter et al. 2019).
We thus analyzed whether our mutants present differences in cell division during the floral transition (Fig. 6). Although enrichment analysis did not give clues regarding the molecular role of REM13, we also tested if it controls cell division because REM13 is co-expressed with only 4 REMs so, maybe, the role for REM13 in cell division was not revealed due to the lower power of the enrichment analysis. Indeed, rem13_oe, as well as rem34-1 and rem46-1 showed an increased percentage of cells in the S phase at T0, suggesting that the rate of cell division in the SAM was higher than in the wild type (Fig. 6). As the increase of cell division at the SAM is one of the earliest events observed during the floral transition (Jacqmard et al. 2003; Klepikova et al. 2015) the presence of a higher percentage of cells in the S phase at T0 might explain the early flowering phenotype observed (Fig. 3). As so, our work constitutes an interesting starting point to explore the link between the higher cell division rate observed in these mutants and its effect on flowering time.
As some REM were reported to be able to homo- and/or heterodimerize (Caselli et al. 2019; Mendes et al. 2016), we checked the interaction of the REM proteins under study, as it could give further information regarding their possible cooperation. Protein interaction assays showed that REM34 heterodimerizes with REM46, suggesting that these two proteins could cooperate in the regulation of flowering time. Nevertheless, they are not completely redundant as both the mutants show an acceleration in the floral transition. The stronger phenotype of rem34-1 and the fact it is the only mutant in which the expression of all the floral integrators analyzed (SOC1, LFY and AP1) is altered, suggests that REM34 might participate in several regulatory networks. This could imply that REM34 and REM46 might modulate cell cycle progression together and that REM34 might participate in additional pathways not related to the control of the cell cycle, possibly partnering with other REMs.
We also investigated the possible interactions between TFS1 and REM34 and REM46, as they are co-expressed (Fig. 2) and all show phenotypes related to flowering time (Richter et al. 2019). TFS1 homodimerized but it did not interact with REM34 or REM46. This is coherent with their different roles in flowering time as TFS1 promotes floral transition (Richter et al. 2019) and REM34 and REM46 repress it. Finally, REM13 does not interact with itself or with any of the REMs tested in this work. This observation is not entirely surprising given the lack of co-expression with them (Fig. 2). Overall, the newly found interactions reported here, together with the interactions already reported for VDD, VAL, REM34 and REM35 suggest that dimerization could be a frequent event for the REM family.
It is remarkable that we have revealed the roles in the flowering time of REM13/34/46 using alleles that lead to changes in their expression level (Figs. 1 and 3), rather than a loss of function. Changes in the expression level of genes have led to interesting phenotypes on several occasions during plant domestication (Manrique et al. 2019). For example, the acquisition of photoperiod-insensitivity in maize that allowed its growth at high latitudes was caused by the downregulation of ZmCCT9 gene provoked by the insertion of a transposon in its regulatory region (Huang et al. 2017). Moreover, it was recently published that a mutation in the promoter of OsREM20 during rice domestication led to an increase in the expression of OsREM20 producing an increment in the number of grains per panicle (Wu et al. 2021). Similarly, our results show that the modulation of the level of expression of REM13/34/46 leads to changes in flowering time. This could be interesting for breeding programs, as it further suggests that the modulation of the expression of REM genes can affect important agronomical traits.
In summary, on one hand, our strategy of relying on the lack of other REMs located in tandem with REM13 has been successful in finding a biological role for it. On the other hand, using co-expression data as a starting point for planning functional analysis, we have uncovered roles in flowering time regulation and changes in cell division rate for REM34 and REM46. Our attempt of using co-expression data as a starting point for functional analysis of REMs has been more successful than previous ones (Mantegazza et al. 2014), probably due to the increase in the amount and quality of transcriptomic data available. Moreover, it has revealed interesting features regarding the redundancy patterns of REM genes that open the door to better-focused studies of the REM family where the combination of co-expression and interaction data might help to uncover more biological roles for these cryptic transcription factors.
References
Abe M, Kobayashi Y, Yamamoto S, Daimon Y, Yamaguchi A, Ikeda Y, Ichinoki H, Notaguchi M, Goto K, Araki T (2005) FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309(5737):1052–1056. https://doi.org/10.1126/SCIENCE.1115983/SUPPL_FILE/ABE.SOM.PDF
Ahmad B, Zhang S, Yao J, Ur Rahman M, Hanif M, Zhu Y, Wang X (2019) Genomic Organization of the B3-Domain transcription factor family in Grapevine (Vitis vinifera L.) and expression during seed development in Seedless and seeded Cultivars. Int J Mol Sci 2019 20(18):4553. https://doi.org/10.3390/IJMS20184553. 20
Belda-Palazón B, Ruiz L, Martí E, Tárraga S, Tiburcio AF, Culiáñez F, Farràs R, Carrasco P, Ferrando A (2012) Aminopropyltransferases involved in Polyamine Biosynthesis localize preferentially in the Nucleus of Plant cells. PLoS ONE 7(10):e46907. https://doi.org/10.1371/JOURNAL.PONE.0046907
Bernier G (1969) The induction of flowering.Some Case Histories. Macmillan. https://scholar.google.com/scholar_lookup?hl=en&publication_year=1969&pages=305-327&author=G.+Bernier&title=The+Induction+of+Flowering
Blümel M, Dally N, Jung C (2015) Flowering time regulation in crops—what did we learn from Arabidopsis? Curr Opin Biotechnol 32:121–129. https://doi.org/10.1016/J.COPBIO.2014.11.023
Caselli F, Beretta VM, Mantegazza O, Petrella R, Leo G, Guazzotti A, Herrera-Ubaldo H, de Folter S, Mendes MA, Kater MM, Gregis V (2019a) REM34 and REM35 control female and male Gametophyte Development in Arabidopsis thaliana. Front Plant Sci 10:1351. https://doi.org/10.3389/fpls.2019.01351
Coen ES, Romero JM, Doyle S, Elliott R, Murphy G, Carpenter R (1990) floricaula: a homeotic gene required for flower development in antirrhinum majus. Cell 63(6):1311–1322. https://doi.org/10.1016/0092-8674(90)90426-F
Corson GE, Cell division studies of the shoot apex of datura stramonium during transition to flowering (1969) Am J Bot 56(10):1127–1134. https://doi.org/10.1002/J.1537-2197.1969.TB09767.X
de Folter S, Immink RGH (2011) Yeast protein-protein interaction assays and screens. Methods Mor Biol (Clifton N J) 754:145–165. https://doi.org/10.1007/978-1-61779-154-3_8
Fornara F, de Montaigu A, Coupland G (2010) SnapShot: control of flowering in Arabidopsis. Cell. https://doi.org/10.1016/J.CELL.2010.04.024
Franco-Zorrilla J, Manuel, Fernández-Calvín B, Madueño F, Cruzalvarez M, Salinas J, Martínez-Zapater JM (1999) Identification of genes specifically expressed in cauliflower reproductive meristems. Molecular characterization of BoREM1. Plant Mol Biol 39(3):427–436. https://doi.org/10.1023/A:1006130629100
Franco-Zorrilla JM, López-Vidriero I, Carrasco JL, Godoy M, Vera P, Solano R (2014) DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc Natl Acad Sci USA 111(6):2367–2372. https://doi.org/10.1073/PNAS.1316278111/SUPPL_FILE/SAPP.PDF
Geier F, Lohmann JU, Gerstung M, Maier AT, Timmer J, Fleck C (2008) A quantitative and dynamic model for Plant Stem Cell Regulation. PLoS ONE 3(10):e3553. https://doi.org/10.1371/JOURNAL.PONE.0003553
Gomez MD, Barro-Trastoy D, Escoms E, Saura-Sańchez M, Sańchez I, Briones-Moreno A, Vera-Sirera F, Carrera E, Ripoll JJ, Yanofsky MF, Lopez-Diaz I, Alonso JM, Perez-Amador MA (2018) Gibberellins negatively modulate ovule number in plants. Dev. https://doi.org/10.1242/DEV.163865/264728
Gómez-Mena C, de Folter S, Costa MMR, Angenent GC, Sablowski R (2005) Transcriptional program controlled by the floral homeotic gene AGAMOUS during early organogenesis. Development 132(3):429–438. https://doi.org/10.1242/DEV.01600
Gregis V, Sessa A, Colombo L, Kater MM (2008) AGAMOUS-LIKE24 and SHORT VEGETATIVE PHASE determine floral meristem identity in Arabidopsis. Plant J 56(6):891–902. https://doi.org/10.1111/j.1365-313X.2008.03648.x
Heo JB, Sung S, Assmann SM (2012) Ca2+-dependent GTPase, extra-large G protein 2 (XLG2), promotes activation of DNA-binding protein related to vernalization 1 (RTV1), leading to activation of floral integrator genes and early flowering in Arabidopsis. J Biol Chem 287(11):8242–8253. https://doi.org/10.1074/JBC.M111.317412
Huang C, Sun H, Xu D, Chen Q, Liang Y, Wang X, Xu G, Tian J, Wang C, Li D, Wu L, Yang X, Jin W, Doebley JF, Tian F (2017) ZmCCT9 enhances maize adaptation to higher latitudes. Proc Natl Acad Sci USA 115(2):E334–E341. https://doi.org/10.1073/PNAS.1718058115/SUPPL_FILE/PNAS.1718058115.SAPP.PDF
Jacqmard A, Gadisseur I, Bernier G (2003) Cell Division and morphological changes in the shoot apex of Arabidopsis thaliana during Floral Transition. Ann Botany 91(5):571–576. https://doi.org/10.1093/AOB/MCG053
Kinoshita A, Vayssières A, Richter R, Sang Q, Roggen A, Van Driel AD, Smith RS, Coupland G (2020) Regulation of shoot meristem shape by photoperiodic signaling and phytohormones during floral induction of arabidopsis. ELife 9:1–29. https://doi.org/10.7554/ELIFE.60661
Klepikova AV, Logacheva MD, Dmitriev SE, Penin AA (2015) RNA-seq analysis of an apical meristem time series reveals a critical point in Arabidopsis thaliana flower initiation. BMC Genomics 16(1):1–16. https://doi.org/10.1186/S12864-015-1688-9/FIGURES/5
Kobayashi Y, Kaya H, Goto K, Iwabuchi M, Araki T (1999) A pair of related genes with antagonistic roles in mediating flowering signals. Science 286(5446):1960–1962. https://doi.org/10.1126/SCIENCE.286.5446.1960/SUPPL_FILE/1044707S2_THUMB.GIF
Kurokura T, Inaba Y, Sugiyama N (2006) Histone H4 gene expression and morphological changes on shoot apices of strawberry (Fragaria × ananassa Duch.) During floral induction. Sci Hort 110(2):192–197. https://doi.org/10.1016/J.SCIENTA.2006.07.013
Kwiatkowska D (2008) Flowering and apical meristem growth dynamics. J Exp Bot 59(2):187–201. https://doi.org/10.1093/jxb/erm290
Lee H, Suh SS, Park E, Cho E, Ahn JH, Kim SG, Lee JS, Kwon YM, Lee I (2000) The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev 14(18):2366–2376. https://doi.org/10.1101/GAD.813600
Levy YY (2002) Multiple roles of Arabidopsis VRN1 in vernalization and flowering Time Control. Science 297(5579):243–246. https://doi.org/10.1126/science.1072147
Levy YY, Mesnage S, Mylne JS, Gendall AR, Dean C (2002) Multiple roles of Arabidopsis VRN1 in vernalization and flowering Time Control. Science 297(5579):243–246. https://doi.org/10.1126/science.1072147
Liljegren SJ, Gustafson-Brown C, Pinyopich A, Ditta GS, Yanofsky MF (1999) Interactions among APETALA1, LEAFY, and TERMINAL FLOWER1 Specify Meristem Fate. Plant Cell 11(6):1007–1018. https://doi.org/10.1105/TPC.11.6.1007
Liu C, Chen H, Er HL, Soo HM, Kumar PP, Han JH, Liou YC, Yu H (2008) Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development 135(8):1481–1491. https://doi.org/10.1242/DEV.020255
Lyndon RF, Battey NH (1985) The growth of the shoot apical meristem during flower initiation. Biol Plant 1985 27(4):339–349. https://doi.org/10.1007/BF02879875. 27
Manrique S, Friel J, Gramazio P, Hasing T, Ezquer I, Bombarely A (2019) Genetic insights into the modification of the pre-fertilization mechanisms during plant domestication. J Exp Bot 70(11):3007–3019. https://doi.org/10.1093/JXB/ERZ231
Mantegazza O, Gregis V, Mendes MA, Morandini P, Alves-Ferreira M, Patreze CM, Nardeli SM, Kater MM, Colombo L (2014) Analysis of the arabidopsis REM gene family predicts functions during flower development. Ann Botany 114(7):1507–1515. https://doi.org/10.1093/aob/mcu124
Marc J, Palmer JH (1984a) Variation in cell-cycle time and nuclear DNA content in the apical meristem of Helianthus annuus L. during the transition to flowering. Am J Bot 71(4):588–595. https://doi.org/10.2307/2443334
Marc J, Palmer JH, Variation in cell-cycle time and nuclear DNA content in the apical meristem of helianthus annuus l. During the transition to flowering (1984) Am J Bot 71(4):588–595. https://doi.org/10.1002/J.1537-2197.1984.TB12544.X
Matias-Hernandez L, Battaglia R, Galbiati F, Rubes M, Eichenberger C, Grossniklaus U, Kater MM, Colombo L (2010) VERDANDI is a direct target of the MADS domain ovule identity complex and affects embryo sac differentiation in Arabidopsis. Plant Cell 22(6):1702–1715. https://doi.org/10.1105/tpc.109.068627
Mendes MA, Guerra RF, Castelnovo B, Silva-Velazquez Y, Morandini P, Manrique S, Baumann N, Groß-Hardt R, Dickinson H, Colombo L (2016) Live and let die: a REM complex promotes fertilization through synergid cell death in Arabidopsis. Development 143(15):2780–2790
Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43(W1):W566–W570. https://doi.org/10.1093/NAR/GKV468
Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD (2019a) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47(D1):D419–D426. https://doi.org/10.1093/NAR/GKY1038
Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD (2019b) Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protocols 2019b 14:3(3):703–721. https://doi.org/10.1038/s41596-019-0128-8. 14
Miksche JP, Brown JAM (1965) Development of vegetative and floral meristems of Arabidopsis Thaliana. Am J Bot 52(6):533–537. https://doi.org/10.1002/J.1537-2197.1965.TB06818.X
Petrella R, Caselli F, Roig-Villanova I, Vignati V, Chiara M, Ezquer I, Tadini L, Kater MM, Gregis V (2020) BPC transcription factors and a polycomb group protein confine the expression of the ovule identity gene SEEDSTICK in Arabidopsis. Plant J 102(3):582–599. https://doi.org/10.1111/TPJ.14673
Richter R, Kinoshita A, Vincent C, Martinez-Gallegos R, Gao H, van Driel AD, Hyun Y, Mateos JL, Coupland G (2019) Floral regulators FLC and SOC1 directly regulate expression of the B3-type transcription factor TARGET OF FLC AND SVP 1 at the Arabidopsis shoot apex via antagonistic chromatin modifications. PLoS Genet 15(4):e1008065. https://doi.org/10.1371/journal.pgen.1008065
Robert PR, Coen ES, Murphy GJP, Doonan JH (1994) Patterns of cell division revealed by transcriptional regulation of genes during the cell cycle in plants. EMBO J 13(3):616–624. https://doi.org/10.1002/J.1460-2075.1994.TB06299.X
Romanel E, Schrago CG, Couñago RM, Russo CAM, Alves-Ferreira M (2009) Evolution of the B3 DNA binding superfamily: new insights into REM family gene diversification. PLoS ONE. https://doi.org/10.1371/journal.pone.0005791
Romanel E, Das P, Amasino RM, Traas J, Meyerowitz E, Alves-Ferreira M (2011) Reproductive Meristem22 is a unique marker for the early stages of stamen development. Int J Dev Biol 55(6):657–664. https://doi.org/10.1387/IJDB.113340ER
Ruan CC, Chen Z, Hu FC, Fan W, Wang XH, Guo LJ, Fan HY, Luo ZW, Zhang ZL (2021) Genome-wide characterization and expression profiling of B3 superfamily during ethylene-induced flowering in pineapple (Ananas comosus L). BMC Genomics 22(1):1–12. https://doi.org/10.1186/S12864-021-07854-1/FIGURES/7
Simpson GG, Dean C (2002) Arabidopsis, the Rosetta stone of flowering time? Sci (New York N Y) 296(5566):285–289. https://doi.org/10.1126/SCIENCE.296.5566.285
Srikanth A, Schmid M (2011) Regulation of flowering time: all roads lead to Rome. Cell Mol Life Sci 68(12):2013–2037. https://doi.org/10.1007/S00018-011-0673-Y/FIGURES/3
Swaminathan K, Peterson K, Jack T (2008) The plant B3 superfamily. Trends Plant Sci 13(12):647–655. https://doi.org/10.1016/j.tplants.2008.09.006
Verma S, Bhatia S (2019) A comprehensive analysis of the B3 superfamily identifies tissue-specific and stress-responsive genes in chickpea (Cicer arietinum L). 3 Biotech 9(9):1–17. https://doi.org/10.1007/S13205-019-1875-5/FIGURES/7
Verwoerd TC, Dekker BMM, Hoekema A (1989) A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Res 17(6):2362–2362. https://doi.org/10.1093/NAR/17.6.2362
Villarino GH, Hu Q, Manrique S, Flores-Vergara M, Sehra B, Robles L, Brumos J, Stepanova AN, Colombo L, Sundberg E, Heber S, Franks RG (2016) Transcriptomic signature of the SHATTERPROOF2 expression domain reveals the Meristematic Nature of Arabidopsis Gynoecial Medial Domain. Plant Physiol 171(1):42–61. https://doi.org/10.1104/PP.15.01845
Wang C, Liu Z (2006) Arabidopsis Ribonucleotide Reductases are critical for cell cycle progression, DNA damage repair, and Plant Development. Plant Cell 18(2):350–365. https://doi.org/10.1105/TPC.105.037044
Wang Y, Deng D, Zhang R, Wang S, Bian Y, Yin Z (2012) Systematic analysis of plant-specific B3 domain-containing proteins based on the genome resources of 11 sequenced species. Mol Biol Rep 39(5):6267–6282. https://doi.org/10.1007/S11033-012-1448-8/FIGURES/4
Wang X, Liu Q, He W, Lin C, Wang Q (2019) Characterization of flowering time mutants. Methods Mol Biol. https://doi.org/10.1007/978-1-4939-9612-4_16/FIGURES/1
Wu X, Liang Y, Gao H, Wang J, Zhao Y, Hua L, Yuan Y, Wang A, Zhang X, Liu J, Zhou J, Meng X, Zhang D, Lin S, Huang X, Han B, Li J, Wang Y (2021) Enhancing rice grain production by manipulating the naturally evolved cis-regulatory element-containing inverted repeat sequence of OsREM20. Mol Plant 14(6):997–1011. https://doi.org/10.1016/J.MOLP.2021.03.016
Wynn AN, Rueschhoff EE, Franks RG (2011) Transcriptomic characterization of a Synergistic Genetic Interaction during Carpel Margin Meristem Development in Arabidopsis thaliana. PLoS ONE 6(10):e26231. https://doi.org/10.1371/JOURNAL.PONE.0026231
Xu B, Li Z, Zhu Y, Wang H, Ma H, Dong A, Huang H (2008) Arabidopsis genes AS1, AS2, and JAG negatively regulate boundary-specifying genes to promote Sepal and Petal Development. Plant Physiol 146(2):323–324. https://doi.org/10.1104/PP.107.113787
Yamaguchi A, Wu MF, Yang L, Wu G, Poethig RS, Wagner D (2009) The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev Cell 17(2):268–278. https://doi.org/10.1016/J.DEVCEL.2009.06.007
Yang L, Wang Z, Hua J (2019) Measuring cell ploidy level in arabidopsis thaliana by flow cytometry. Methods Mol Biol. https://doi.org/10.1007/978-1-4939-9458-8_11
Yoo SK, Chung KS, Kim J, Lee JH, Hong SM, Yoo SJ, Yoo SY, Lee JS, Ahn JH (2005) CONSTANS activates SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 through FLOWERING LOCUS T to promote flowering in Arabidopsis. Plant Physiol 139(2):770–778. https://doi.org/10.1104/PP.105.066928
Yu Y, Qiao L, Chen J, Rong Y, Zhao Y, Cui X, Xu J, Hou X, Dong CH (2020) Arabidopsis REM16 acts as a B3 domain transcription factor to promote flowering time via directly binding to the promoters of SOC1 and FT. Plant J. https://doi.org/10.1111/tpj.14807
Zhang H, Zhang F, Yu Y, Feng L, Jia J, Liu B, Li B, Guo H, Zhai J (2020) A Comprehensive Online database for exploring ∼20,000 public Arabidopsis RNA-Seq libraries. Mol Plant 13(9):1231–1233. https://doi.org/10.1016/J.MOLP.2020.08.001
Zhou H, Song Z, Zhong S, Zuo L, Qi Z, Qu LJ, Lai L (2019) Mechanism of DNA-Induced phase separation for transcriptional repressor VRN1. Angew Chem Int Ed 58(15):4858–4862. https://doi.org/10.1002/ANIE.201810373
Acknowledgements
We thank Claudia Bazzini (Unimi) and Chiara Cavallini (Univr) for technical assistance with flow cytometry experiments, as well as Valerio Parravicini and Mario Beretta for the technical assistance at the University of Milan Botanical Garden. Part of this work was carried out at NOLIMITS, an advanced imaging facility established by the Università degli Studi di Milano. We thank Marta Adelina Mendes for her helpful suggestions and valuable discussions.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. This work was supported by MUR, MADSMEC, Proposal number RBSI14BTZR. SM received post-doctoral fellowships from Fundación Alfonso Martín Escudero (Madrid, Spain) and the University of Milan and her research visit to RGF laboratory was funded by a RISE project (SEXSEED, Grant ID: 690946). The post-doctoral fellowship for FC is funded by the University of Milan.
Author information
Authors and Affiliations
Contributions
Conceptualization, SM, FC, LMH, RF, LC and VG: investigation, SM, FC and VG: writing—original draft preparation, SM, FC and VG: writing—review and editing, SM, FC, LMH, RGF, LC, and VG. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Manrique, S., Caselli, F., Matías-Hernández, L. et al. Assessing the role of REM13, REM34 and REM46 during the transition to the reproductive phase in Arabidopsis thaliana. Plant Mol Biol 112, 179–193 (2023). https://doi.org/10.1007/s11103-023-01357-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-023-01357-1