
Abstract
Key message
Degradation of nitrogen-rich purines is tightly and oppositely regulated under drought and low nitrogen supply in bread wheat. Allantoin is a key target metabolite for improving nitrogen homeostasis under stress.
Abstract
The metabolite allantoin is an intermediate of the catabolism of purines (components of nucleotides) and is known for its housekeeping role in nitrogen (N) recycling and also for its function in N transport and storage in nodulated legumes. Allantoin was also shown to differentially accumulate upon abiotic stress in a range of plant species but little is known about its role in cereals. To address this, purine catabolic pathway genes were identified in hexaploid bread wheat and their chromosomal location was experimentally validated. A comparative study of two Australian bread wheat genotypes revealed a highly significant increase of allantoin (up to 29-fold) under drought. In contrast, allantoin significantly decreased (up to 22-fold) in response to N deficiency. The observed changes were accompanied by transcriptional adjustment of key purine catabolic genes, suggesting that the recycling of purine-derived N is tightly regulated under stress. We propose opposite fates of allantoin in plants under stress: the accumulation of allantoin under drought circumvents its degradation to ammonium (NH4+) thereby preventing N losses. On the other hand, under N deficiency, increasing the NH4+ liberated via allantoin catabolism contributes towards the maintenance of N homeostasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nitrogen (N) is a macronutrient required in large quantities for plant development and growth with N deficiency causing chlorosis in older leaves and significant yield losses. Under N deficiency and natural senescence plants translocate available N from source tissues to sink tissues, such as young leaves (Masclaux-Daubresse et al. 2010) and developing grains, accounting for 60–92% of total grain N (Barbottin et al. 2005). The glutamine synthetase–glutamate synthase (GS-GOGAT) cycle plays an important role in this process since it recycles N liberated from the catabolism of N-rich macromolecules, such as protein and nucleic acids, into low-molecular-weight organic compounds for long-distance N transport (Lea and Miflin 2010).
Purines are the most abundant N heterocyclic compounds in nature and are found in nucleic acids (DNA, RNA) and many other cellular components, such as ATP, GTP or NADH (Werner and Witte 2011). Plants undergo the complete breakdown of the purine ring via a catabolic pathway enabling the recycling of both carbon (C) and N (Fig. S2). Overall, the oxidation of one molecule of xanthine to one molecule of glyoxylate liberates three molecules of CO2 and four molecules of ammonium (NH4+), which are likely to be reassimilated by the GS-GOGAT cycle into amino acids. The pathway starts with the conversion of xanthine to urate catalysed by xanthine dehydrogenase (XDH) (Triplett et al. 1982; Werner and Witte 2011). Urate is further processed by urate oxidase (UOX) producing 5-hydroxyisourate (5-HIU), 5-HIU is then converted to allantoin via the 2-oxo-4-hydroxy-4-carboxy-5-ureido-imidazoline (OHCU) intermediate by allantoin synthase (AS) (Hanks et al. 1981; Ramazzina et al. 2006; Kim et al. 2007; Lamberto et al. 2010; Pessoa et al. 2010). Allantoin is metabolised to allantoate by allantoinase (ALN) and then to ureidoglycine by allantoate amidohydrolase (AAH) (Yang and Han 2004; Todd and Polacco 2006; Werner et al. 2008). The last two enzymatic steps are catalysed by ureidoglycine aminohydrolase (UGAH), which converts ureidoglycine to ureidoglycolate (Serventi et al. 2010). Ureidoglycolate amidohydrolase (UAH) converts ureidoglycolate to hydroxyglycine and, lastly, hydroxyglycine decays to glyoxylate by a non-enzymatic reaction (Werner et al. 2010).
In addition to its housekeeping role in N recycling, the purine catabolic pathway has an important function in certain dinitrogen (N2)-fixing legumes (Schubert 1986; Sinclair and Serraj 1995; Alamillo et al. 2010; Coleto et al. 2014). Allantoin and allantoate (also known as ureides) are the main products of atmospheric N2 fixation in root nodules, which are then translocated to the shoot (Herridge et al. 1978; Pate et al. 1980). In recent years, the ureide allantoin has gained attention by the scientific community as several metabolomics studies reported this metabolite to accumulate in a broad range of plant species under drought (Bowne et al. 2011; Oliver et al. 2011; Silvente et al. 2012; Degenkolbe et al. 2013; Yobi et al. 2013; Casartelli et al. 2018), high salt (Kanani et al. 2010; Wu et al. 2012; Nam et al. 2015; Wang et al. 2016), cold (Kaplan et al. 2004) and sulfate starvation (Nikiforova et al. 2005). In contrast, allantoin was found to be reduced under prolonged N deficiency in maize and rice (Amiour et al. 2012; Coneva et al. 2014). Further, the work of Watanabe et al. (2014) revealed that Arabidopsis AtALN mutants, which constitutively accumulated allantoin, were more tolerant to desiccation stress. It was demonstrated that allantoin mediates abscisic acid (ABA) signalling by stimulating the activity of genes and enzymes belonging to the ABA-producing pathways. In addition, allantoin was shown to accumulate in Arabidopsis leaves grown under drought and salt stress in coordination with transcriptional changes of a number of purine catabolic genes (Irani and Todd 2016; Lescano et al. 2016). To date, there are few studies on the regulation of the purine catabolic pathway under N deficiency, however, early reports showed that AtALN was strongly up-regulated when Arabidopsis seedlings were grown under N starvation (Yang and Han 2004).
Given the potentially important role of allantoin in N metabolism and stress tolerance but the limited information available in cereals, the aim of the present study was to characterise the purine catabolic pathway in bread wheat. For this, we selected two wheat genotypes (RAC875 and Mace) that are adapted to Australian environments. The gene loci were identified based on the reference genome of the cultivar Chinese Spring and their chromosomal location was experimentally verified. Quantification of allantoin in different tissues throughout plant development revealed accumulation of allantoin under drought and reduced allantoin levels under N limitation and this was associated with differential regulation of the genes in the purine catabolic pathway. Monitoring of N-metabolite pools in seeds revealed that allantoin levels progressively increase in developing grains and that genotypic differences exist for accumulation of allantoin and other important N-containing metabolites.
Materials and methods
Plant material
Two semi-dwarf South Australian wheat genotypes RAC875 and Mace were evaluated in this study. RAC875 (RAC655/3/Sr21/4*LANCE//4*BAYONET) is high-yielding in the drought and heat-prone South Australian environments (Izanloo et al. 2008; Bennett et al. 2012). Mace (WYALKATCHEM/STYLET//WYALKATCHEM) was bred and released by Australian Grain Technologies (AGT) in 2008 and preliminary studies suggest that Mace has high N-use efficiency across different South Australian environments (Mahjourimajd et al. 2016).
Plant growth
The experiment was conducted in a controlled environment with day/night cycle of 12 h/12 h at a flux density at canopy level of 300 µmol m−2 s−1, 20 °C/15 °C day/night temperature and 82% average humidity. Potting mixture was composed of river sand and coco-peat and prepared according to Melino et al. (2015). Granular urea was provided as basal N application with rates of 150 and 75 mg N kg−1 for high and low N treatments, respectively. At stem elongation (39 days after sowing) a third of the basal urea rates for each treatment were applied by soil drenching. A soil water retention curve was constructed by measuring the pre-dawn leaf water potential of 3-week old seedlings under progressive drought stress with a Scholander-type pressure chamber (Soil Moisture Equipment Corp., Santa Barbara, USA) (Fig. S1b). Overall, the experiment was comprised of two cultivars (RAC875 and Mace), three treatments (high nitrogen well-watered (HN-WW), low nitrogen well-watered (LN-WW) and high nitrogen drought (HN-D)), six sampling time points and six biological replicates for a total of 216 experimental units. Individual seeds were germinated in black square plastic pots (11 × 11 cm2 area, 14 cm height) containing 1.5 kg of soil mix arranged in a randomised complete block design. WW conditions were maintained by daily watering of the pots to 20% soil water content (SWC), calculated as SWC = [(mwet soil − mdry soil):mdry soil] × 100. Drought was induced by withholding water until signs of leaf rolling appeared (approximately 6.5% SWC; Fig. S1a). Samples collected during the reproductive stages were harvested according to the anthesis date of individual plants and water was first withheld 2 weeks after anthesis. Whole spikes were collected from the main stem of each plant and to minimise the differences in development of the grains along the spike, only the middle part of the spike was employed for further analyses. Rachis was removed and the vegetative part of the spike (hereafter referred as spikelet) was separated from the developing grains. Details on the sample collection throughout the experiment are given in Fig. S1a. Samples for molecular analysis were snap-frozen in liquid nitrogen and stored at − 80 °C until further use.
Identification of the purine catabolic genes in wheat and other grass genomes
The purine catabolic genes of Oryza sativa, Zea mays, Sorghum bicolor and Brachypodium distachyon were identified with a BLASTP search using Arabidopsis thaliana protein sequences as a query in Phytozome (https://phytozome.jgi.doe.gov). B. distachyon protein sequences were then used for a TBLASTN search on the barley WGS Morex Assembly version 3 (https://ics.hutton.ac.uk/morexGenes/blast_page.html) and the barley predicted protein sequences were used for a TBLASTN search against the Chinese Spring TGACv1 genome assembly (http://pre.plants.ensembl.org/Triticum_aestivum/Info/Index) (Clavijo et al. 2017). In cases where full- length sequences were not found in Chinese Spring TGACv1, we searched the Chinese Spring IWGSC chromosome survey sequence (css) version 3 (IWGSC 2014—http://www.wheatgenome.org) and the sequences were merged with TGACv1 sequences using Geneious version 10.0.2 (http://www.geneious.com) (see Table S1 for details). Each purine catabolic gene was assigned with the name of the Arabidopsis orthologous according to Watanabe et al. (2014). For wheat, the sub-genome localisation (e.g., Chr 1AL) was additionally included in the gene name to allow distinction of the three homeologous sequences (Table S1).
Synteny analysis
Synteny analysis were conducted based on available genome sequences of Sorghum bicolor, Oryza sativa and Brachypodium distachyon using the Phytozome 11 ‘Ancestry’ tool (phytozome.jgi.doe.gov). Generally, five genes upstream and downstream of the respective purine catabolic pathway gene were included in the orthology analysis. These genes in bread wheat were identified by TBLASTN searches of Brachypodium genes against the TGACv1 wheat genome assembly (http://pre.plants.ensembl.org/Triticum_aestivum/Info/Index). Genes were considered syntenic if they were present on the same TGACv1 scaffold where the wheat purine catabolic genes were annotated.
Wheat nulli-tetrasomic lines
To verify the chromosomal location of the wheat purine catabolic gene Chinese Spring nulli-tetrasomic (NT) lines were used in which individual wheat chromosomes were replaced (nullisomic) by an extra pair (tetrasomic) of their homeologs (Sears 1954). PCR was performed using 150 ng of genomic DNA of the NT lines as template using standard reaction conditions of ThermoPol (BioLabs, USA) using the primers provided in Table S1.
Allantoin quantification
Metabolite extraction
Metabolites were extracted from 10 mg of homogenised, freeze-dried tissue with 500 µl of 100% (v/v) methanol containing 12.5 µM 13C15N-allantoin (internal standard), except for mature grains samples for which 25 µM 13C15N-allantoin was used. Samples were vortexed and incubated in an Eppendorf Thermomixer at 1400 rpm and 30 °C for 15 min followed by a 15 min centrifugation at 13,000 rpm (4 °C). The supernatant was transferred to a new tube. 500 µl of milli-Q water was added to the remaining pellet, vortexed and centrifuged for 15 min at 15,000 rpm. The supernatant was combined with the previous one, vortexed for 30 s and centrifuged for 15 min at 15,000 rpm. The resulting supernatant was transferred to a new tube and 300 µl of 100% (v/v) chloroform was added, vortexed and centrifuged for 5 min at 15,000 rpm. 800 µl of the top (polar) phase was transferred into a new tube for allantoin analysis.
Sample derivatization
200 µl of the upper polar phase was aliquoted in a glass insert and dried under vacuum (RVC 2-33 CD plus, John Morris Scientific Australia) set at ambient temperature. All samples were re-constituted in 10 µl of methoxyamine hydrochloride (30 mg ml−1 pyridine) and derivatised at 45 °C for 60 min at 500 rpm before adding 20 µl of N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA) with 1% (w/v) trimethyl chlorosilane (TMCS) and incubated at 45 °C for 45 min.
GC–MS instrument conditions
1 µl of derivatised sample was injected into a GC-QqQ-MS system comprised of a Gerstel 2.5.2 Autosampler, a 7890A Agilent gas chromatograph and a 7000 Agilent triple-quadrupole MS (Agilent, Santa Clara, USA) with an electron impact (EI) ion source. The GC was operated in constant flow mode with helium as carrier gas. The MS was adjusted according to the manufacturer’s recommendations using tris-(perfluorobutyl)-amine (CF43). A J&W Scientific VF-5MS column (30 m long with 10 m guard column, 0.25 mm inner diameter, 0.25 µm film thickness) was used. The injection temperature was set at 250 °C, the MS transfer line at 290 °C, the ion source was adjusted to 230 °C and the quadrupole at 150 °C. Helium was used as carrier gas at flow rate of 1 ml min−1. Nitrogen (UHP 5.0) was used as collision cell gas at flow rate of 1.5 ml min−1. Helium (UHP 5.0) was used as quenching gas at a flow rate of 2.25 ml min−1. Gain factor for the triple axis detector was set at 2. Derivatised sample was injected into the column at 100 °C followed by 1 min hold, followed by a ramp of 25 °C min−1 to 325 °C.
Method optimization
Allantoin and 13C15N-allantoin standards were purchased from Sigma Aldrich (Australia). Retention time, a corresponding unique precursor ion and product ions were identified on the GC-QqQ-MS instrument for each standard. Collision energy was optimised between 0 and 20 V for the identified precursor to product ion transitions (Multiple Reaction Monitoring; MRM). A product ion was selected as the Target ion (T) and the other subsequent MRM transition was set as the Qualifier ion (Q) (see Table S2 for details). Linearity of the method was tested using serial dilutions of the calibration standard and showed a linear calibration range between 0.98 µM to 250 µM 13C15N-allantoin. Correlation coefficient (R2) of the calibration curve was 0.99 for the target allantoin MRM (398 → 171). The limit of detection for the allantoin MRM, was 0.294 µM based on a signal to noise ratio of 3.
Data was processed using Agilent MassHunter QQQ Quantitative Analysis software (B.07.00). Allantoin was quantified by single point calibration based on the relative response (the response area of allantoin (MRM 398 → 171) divided by the response area of 13C15N-allantoin (MRM 400 → 173)) and 13C15N-allantoin concentration. Sample dry weight and extraction volume were taken into consideration when calculating the final allantoin concentration.
Metabolome-wide analysis
Metabolites were extracted from 60 mg dry weight (DW) of freeze-dried wheat spikelet and developing grain samples using 100% methanol containing 13C-sorbitol as internal standard. Chloroform followed by water were added to the mixture. Polar phase aliquots were freeze-dried using a speed vacuum. Aliquots for GC-time of flight (TOF)-MS analysis were reconstituted in N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) and methoxyamine hydrochloride as derivatizing agents. Samples were analysed as described previously by Lisec et al. (2006) and Erban et al. (2007). Aliquots for ion chromatography (IC) were reconstituted in milli-Q water and analysed according to Moschen et al. (2016). Aliquots for free spermidine analysis were reconstituted in 0.2 N perchloric acid solution, dansylated and quantified by HPLC according to Do et al. (2013).
Amino acid analysis
Amino acids were extracted from 50 mg DW of freeze-dried wheat samples with 1 ml of 10 mM sodium acetate containing 250 nmol ml−1 Norvaline (internal standard). Amino acids were quantified on a Waters Acquity™ UPLC system using the Waters AccQ-Tag Ultra Chemistry Kit following the manufacturer’s instructions (Waters Corp., USA). Chromatograms were analysed with Empower® 3 Software (Waters Corp., USA).
Total N analysis
Total N concentration was determined using the Elementar rapid N exceed® (Elementar Analysensysteme GmbHanalyser, Germany) using 100 mg DW of homogenised wheat grain samples. Aspartic acid (250 mg) was used as standard for calibration: the theoretical aspartic acid N% (10.52) was divided by the N% measured by the instrument generating a N factor. The N factor was then used to correct the N% measured for each sample.
RNA-Seq reads mapping for the TaXDH2 paralogous genes
Publicly available bread wheat (cv. Chinese Spring) RNA-Seq data was used for generating the per-base expression values. This data set covers 15 duplicated samples corresponding to five organs (root, leaf, stem, spike, grain), each at three developmental stages (http://urgi.versailles.inra.fr/files/RNASeqWheat/) (Choulet et al. 2014). The RNA-Seq reads were quality-, adapter- and length-trimmed using Trimmomatic (Bolger et al. 2014), version 0.30 with a custom list of adapter sequences and the following settings: ‘ILLUMINACLIP:adapters.fa:1:6:6 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:6 MINLEN:60’. The reads were aligned to the scaffolds (version 3) from the Chinese Spring whole genome assembly (version 0.4) using STAR (version 2.5.1b) (Dobin et al. 2013), with the following settings: --outFilterMultimapScoreRange 0; --outFilterMultimapNmax 5; --outFilterMismatchNoverLmax 0 --outFilterMatchNminOverLread 1; --outSJfilterOverhangMin 35,20,20,20; --outSJfilterCountTotalMin 10,3,3,3; --outSJfilterCountUniqueMin 5,1,1,1 --alignEndsType EndToEnd; --alignSoftClipAtReferenceEnds No; --alignIntronMax 10,000; --alignMatesGapMax 10,000. The remaining settings were left at their defaults. The resulting BAM files were merged using samtools merge (version 1.2) (Li et al. 2009). The manually annotated coordinates of the genes define the regions for which the depth of the aligned reads was computed from the aligned BAM file using the depth module of samtools.
RNA preparation
cDNA libraries were prepared from snap-frozen samples of the youngest fully emerged leaf (YFEL), flag leaf, stem, spikelet and developing grain under control (HN-WW) and treatment (HN-D and LN-WW) conditions. Samples were ground to a fine powder using a 2010 Geno/Grinder® (SPEX SamplePrep, USA) and total RNA was extracted from frozen samples with a phenol and guanidine thiocyanate buffer according to Chomczynski (1993). To extract RNA from developing grains which have a high polysaccharide content, an extraction buffer (1% (w/v) sarcosyl, 150 mM NaCl, pH 9) and a guanidine hydrochloride-based buffer for purification according to Singh et al. (2003) was employed. Genomic DNA was removed using the TURBO DNA-free™ Kit (Ambion®, Thermo Fisher Scientific, USA) following the manufacturers’ instructions. High RNA quality was confirmed in a 2% (w/v) agarose gel visualized under UV light and RNA concentrations were quantified using a ND-1000 spectrophotometer (NanoDrop Technologies, USA). 1 µg of RNA was used for cDNA synthesis using the SuperScript® III kit (Thermo Fisher Scientific, USA) as per manufacturers’ instructions. cDNA quality was verified by PCR amplification of the actin gene (Table S3).
Primer design and quantitative reverse transcription PCR (qRT-PCR) analysis
Quantitative real-time PCR was performed with KAPA SYBR® Fast qPCR kit Master Mix, and amplification was real-time monitored on a QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems, USA). Gene-specific primers targeted to amplify all three homeologs simultaneously were designed with AlleleID® software (Premiere Biosoft) (Table S3) and the specificity of each pair was verified by melting curve analysis and sequencing of the products. Change in gene expression were calculated using qBASE + software and reported as calibrated normalised relative quantities (CNRQ) that represents the relative gene expression level between different samples for a given target gene:

NRQ is then divided by a calibration factor (CF) (Hellemans et al. 2007). Four reference genes (Table S3) were quantified by qRT-PCR: TaActin, TaGAPdH, TaCyclophilin and TaEFA2. CNRQ values were calculated using the most stable genes within a specific tissue (selected by qBASE + software).
Data analysis
Statistical analyses were performed using GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla California USA. http://www.graphpad.com). All data are reported as mean ± SEM. Significant differences between means of two groups of data were tested by Student’s t-test. Significant differences between means of more than two groups were tested by two-way ANOVA. For metabolome-wide analysis, metabolites levels were log transformed to improve the normality of the data set and then scaled by subtracting the median metabolite value in each metabolite distribution. Hierarchical clustering by Pearson’s correlation distance and PCA analyses were performed with the support of ClustVis web tool (Metsalu and Vilo 2015). Transcriptional data was reported as log2 ratio of the fold-change between treatment (drought or low N) and control conditions.
Results
Identification of the wheat purine catabolic genes
Based on comparative sequence analyses, a total of 24 wheat genes that are homologous to the purine catabolic genes of Arabidopsis and rice were identified (Fig. 1a). Generally, for each rice gene three wheat homeologous sequences were identified, except for XDH that had paralogous copies on two different chromosome groups. Specifically, the two allelic variants were located on the long arm of chromosome group 1 (TaXDH1-1AL/TaXDH1-1BL/TaXDH1-1DL) and short arm of chromosome group 6 (TaXDH2-6AS/TaXDH2-6BS/TaXDH2-6DS), respectively. The TaXDH1 and TaXDH2 paralogs share 89.6–90.5% sequence identity within the predicted coding region (CDS) and the TaXDH1 homeologs showed a higher CDS identity (98.6–98.8%) than the TaXDH2 homeologs (93.5–95.3%) (Table S4). The TaALN genes (TaALN-2AL/TaALN-2BL/TaALN-2DL) and TaUGAH genes (TaUGAH-2AS/TaUGAH-2BS/TaUGAH-2DS) were localised on chromosome group 2, whilst the TaUOX genes were localised on chromosome group 3 (TaUOX-3AL/TaUOX-3BL/TaUOX-3DL). The TaAS genes were identified on chromosome group 4 and the TaUAH genes were localised on chromosome group 5 (TaUAH-5AS/TaUAH-5BS/TaUAH-5DS). The TaAAH genes were found located on chromosome group 7 (TaAAH-7AL/TaAAH-7BL/TaAAH-7DL) and were the least conserved among the analysed genes with 95–92.9% sequence identity within the predicted CDS (Table S4).

Distribution of the purine catabolic genes across the bread wheat allohexaploid genome (Triticum aestivum cv. Chinese Spring). a The 24 purine catabolic genes are represented by grey bars and their putative location on chromosomes was estimated based on the related Munich Information Centre for Protein Sequences (MIPS) gene annotation on EnsemblPlants (plants.ensembl.org). The figure was adapted from Gill et al. (2004). b Homoelog-specific primers were used for PCR amplification of the genes using DNA derived from nulli-tetrasomic lines of the cv. Chinese Spring. Labels shown in the agarose gel indicate the nullisomic chromosome, e.g. “N1A” indicates that chromosomal group 1A is absent (see Table S1 for details). The caption indicates the primer set employed to amplify the NT DNA (details provided in Table S1). For example, (i) XDH1-1AL,-1BL and − 1DL primer sets were used to amplify nullisomic lines for chromosome subgroup 1. The absence of an amplicon in a specific NT line (whilst amplicons are derived in the two remaining NT lines) indicates localisation of the gene on the respective absent chromosome
To experimentally validate the chromosomal localization of the wheat orthologues genes predicted by the TAGCv1 and IWGSC css assembly, nulli-tetrasomic (NT) lines of the wheat cultivar Chinese Spring were used (Fig. 1b; Sears 1954; see M&M for details) and homeolog-specific primers (Table S1) designed for PCR analysis of genomic DNA from the NT lines. The absence of an amplicon in the respective NT line confirms that the target gene is physically located on that nullisomic chromosome pair.
To further corroborate the orthology between the identified loci we analysed the synteny of the genomic regions between bread wheat, Brachypodium, rice and sorghum (for gene IDs see Supplementary Tables S1 and S5). The fragmented nature of the bread wheat TGACv1 assembly (Clavijo et al. 2017) may have reduced the resolution of the analysis, however, wheat showed a high degree of synteny with the three diploid genomes included in the analysis and the position of several purine catabolic genes and their neighbouring genes was highly conserved (Fig. 2). The only exception was XDH, for which we could identify only one syntenic gene (Bradi1g15910, depicted in blue) among all analysed genomes. This gene is located upstream of XDH in sorghum, downstream of XDH in rice and Brachypodium, and on chromosome group 1 in wheat. Interestingly, we identified two Brachypodium genes (Bradi1g15820 and Bradi1g15826, depicted in brown and grey, respectively) with orthologous sequences on wheat chromosome group 6, supporting the evidence that a duplication of the genomic region harbouring XDH may have occurred during wheat genome evolution. Analysis of the predicted open reading frames (ORFs) of the TaXDH genes revealed five premature stop codons in TaXDH2-6AS and several mutations in TaXDH2-6BS, both likely to result in non-functional proteins. In contrast, the TaXDH2-6DS ORF appeared to translate into a functional protein (data not shown). This was supported by in-silico analysis of publicly available Chinese Spring RNA-seq transcriptomics data which revealed that TaXDH2-6DS was the only homeolog expressed (Fig. S3a). Synteny analysis further revealed the presence of two copies of UAH in the Brachypodium genome of which only one gene (BdUAH1) showed a syntenic relationship with the other genomes.

Syntenic relations of the genomic regions harbouring purine catabolic genes in sorghum, rice, Brachypodium and wheat. The four species are shown in four distinct rows underneath each other and genes are represented as coloured shapes with abbreviations of the respective purine catabolic genes indicated. For each gene, the presence of the same coloured shape in different genomes denotes the presence of gene orthologs. Solid lines represent chromosomes and the chromosome number is indicated. This figure is not drawn to scale and therefore genome distances cannot be determined from the length of lines. Dashed lines indicate that genes are located on the same chromosome but not adjacent to the genes of interest (GOI). In the bread wheat genome, genes that were located on the same TGACv1 scaffold as the purine catabolic genes are represented on the same solid line. Genes that were identified on a different scaffold but assembled on the same chromosome are separated from the GOI by a dashed line
The effects of low N and drought on plant performance
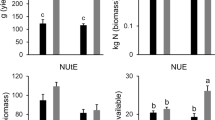
RAC875 and Mace plants were initially grown under well-watered (WW) conditions divided in two subsets supplied with either high N (HN) or low N (LN). During the course of the experiment, a subset of plants grown under HN were subjected to drought (D) at tillering stage and during grain filling, (Fig. S1a). Therefore, the experiment comprised of three treatments: control (high N, well-watered; HN-WW), low N (low N, well-watered; LN-WW) and drought (high N, drought; HN-D). Analysis of plants at maturity revealed that the low N treatment reduced above ground biomass by 20.8% and 23.3% in RAC875 and Mace, respectively, whilst grain yield was reduced by 24.4% and 25.2% in RAC875 and Mace as compared to the controls (HN-WW), respectively (Fig. 3). When plants were grown under drought conditions, above-ground biomass was reduced by approximately 15% in both genotypes as compared to their respective controls. Under drought conditions, Mace plants were also higher yielding (grain yield) than RAC875 plants (p < 0.02 by Student’s t-test); this corresponded to a 16.0% and 26.6% yield loss for Mace and RAC875, respectively, relative to control conditions (Fig. 3).

Analysis of agronomic traits of plants at maturity. a Above ground biomass (g DW plant−1) and b grain yield (g plant−1) were measured from the remaining tillers of wheat RAC875 and Mace plants used for molecular analyses. Plants were grown under control (HN-WW), low N (LN-WW) and drought (HN-D) conditions and each value represent the mean ± SEM of 16–18 biological replicates. Two-way ANOVA analysis was performed with Tukey’s correction and letters indicate significant differences between genotypes and treatments at p < 0.05. The analysis revealed a significant treatment effect (p < 0.0001) for both traits and a significant genotype × treatment interaction (p < 0.05) for grain yield. Asterisk denotes significant difference (p < 0.02) between grain yield of RAC875 and Mace under drought according to Student’s t-test
Allantoin accumulation under low N and drought stress
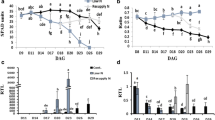
To assess whether N and water stress alter allantoin levels in the two wheat genotypes, allantoin concentration was quantified in the youngest fully emerged leaf (YFEL) during vegetative growth and in flag leaf, stem, spikelet and developing grain samples during reproductive growth in plants grown under HN-WW, LN-WW and HN-D (Fig. 4). Nitrogen deficiency significantly reduced allantoin concentration at different time points across different tissues and in both genotypes when compared to control conditions (Fig. 4a). In both genotypes grown under LN-WW conditions, allantoin levels were generally below 50 nmol g−1DW in YFEL, flag leaves and stems, but much higher (50 to 100 nmol g−1DW) in spikelets and developing grains. The highest reduction of allantoin in both genotypes was measured in the stem under LN-WW as compared to the HN-WW control, with a 22-fold and 10-fold reduction in RAC875 and Mace, respectively (Fig. 4a). Interestingly, under control conditions (HN-WW) Mace flag leaves significantly accumulated much higher levels of allantoin at 19 DAA (305 nmol g−1 DW) as compared to RAC875 (34 nmol g−1 DW), whilst a significant but relatively smaller difference was recorded under low N at 17 DAA (36 and 12 nmol g−1 DW for Mace and RAC875, respectively) (Fig. 4a). Similarly, allantoin concentration in Mace developing grains was significantly higher compared to RAC875 at 22DAA under both control (HN-WW) and low N (LN-WW) conditions.

Allantoin concentration in bread wheat genotypes RAC875 and Mace under N deficiency and drought stress. Allantoin concentration (nmol g−1DW) was quantified by GC-QqQ-MS under a low N and b drought conditions with respect to control conditions in youngest fully emerged leaf (YFEL), flag leaf, stem, spikelet and developing grain. YFEL samples were collected during tillering stages at 23, 24 and 27 days after sowing, corresponding to 2, 3 and 6 days of drought stress (days after last watering), respectively. Flag leaf, stem, spikelet and developing grain samples were collected during grain filling stages at 17, 19 and 22 days after anthesis (DAA), corresponding to 3, 5 and 8 days of drought (days after last watering), respectively (see Fig. S1 for details). Data are mean ± SEM of four to six biological replicates and letters indicate significant differences between treatments and time points within a genotype (lower case: RAC875; upper case: Mace) by two-way ANOVA with Tukey’s correction at p < 0.05. Asterisks indicate significant allantoin differences between the genotypes RAC875 and Mace for a given treatment and time point by Student’s t-test, corrected for multiple comparison using the Bonferroni-Dunn method (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Student’s t-test)
In contrast to the reduced accumulation of allantoin under N deficiency, allantoin significantly positively accumulated under drought in all tissues assessed, with the exception of the stem where, despite a clear positive trend, allantoin accumulation was not significant (Fig. 4b). During the tillering stage, under severe drought stress (day 8; 6.3% SWC) allantoin significantly accumulated 3.6- and 2.5-fold in the YFEL of RAC875 and Mace, respectively. The higher magnitude of allantoin accumulation under drought in RAC875 were due to significantly higher levels of allantoin in the YFEL of RAC875 under drought compared to Mace (Fig. 4b). Similarly, during grain filling, allantoin significantly accumulated in Mace flag leaves at day 5 and 8 of the drought treatment (8.6% and 6.6% SWC, respectively), whilst it significantly accumulated in RAC875 flag leaves only at day 8 (Fig. 4b). In spikelet and developing grains, allantoin significantly increased only at day 8 in both genotypes, when drought stress was most severe (Fig. 4b). Interestingly, flag leaves of RAC875 progressively accumulated allantoin during the course of the drought treatment, increasing from 136 nmol g−1 DW at day 5 to 1197 nmol g−1 DW at day 8, representing an almost 30-fold increase relative to HN-WW. On the other hand, Mace accumulated 1614 nmol of allantoin per gram DW after only 5 days of drought treatment and levels remained constant until day 8. However, the magnitude of allantoin accumulation under HN-D in Mace was only 2.5-fold as compared to HN-WW at day 8 (Fig. 4b). The large difference in the magnitude of allantoin accumulation between RAC875 and Mace flag leaf can be associated with the higher allantoin levels in Mace already present under control conditions at day 5 (19DAA).
Transcription of purine catabolic genes under low N and drought stress
To assess stress responsiveness of wheat purine catabolic genes, transcript abundance was quantified in all of the RAC875 samples collected during the course of the experiment (Fig. 5) and on YFEL (Z22) Mace samples (Fig. S4). The data for each gene and treatment is presented in Fig. 5 as log2 ratio of the calibrated normalised relative quantities (CNRQ) (Hellemans et al. 2007) between treatment (LN-WW or HN-D) and control conditions (HN-WW). For this analysis qRT-PCR primers were designed to amplify all three homeologs of each purine catabolic gene. The only exception was the design of primers to specifically amplify TaXDH2-6DS, which is the only TaXDH2 homeolog that is expressed (Fig. S3a). This likely explains why the abundance of TaXDH2-6DS transcripts in RAC875 was approximately 20 times lower than in TaXDH1 (Fig. S3b).

qRT-PCR analysis of the purine catabolic genes in different tissues of the bread wheat genotype RAC875 under stress. RNA was extracted from the youngest fully emerged leaf (YFEL), flag leaf, stem, spikelet and developing grain of plants grown under control (HN-WW), low N (LN) and drought (D) conditions collected at different time points (see Fig. S1a for details). Transcript abundance of the genes of interest were determined by quantitative real-time PCR (qRT-PCR). Calibrated normalised relative quantity (CNRQ) was calculated using the most stable reference genes across tissues: TaActin and TaGAPdH for YFEL and spikelet; TaCyclophilin and TaGAPdH for flag leaf and stem; TaCyclophilin and TaEFA2 for developing grain. Data is expressed as log2 calibrated normalised relative quantity (CNRQ) of gene transcription under N deficiency or drought divided by CNRQ under control. Colour denotes significant differences between treatment and the respective control conditions (low N/high N; well-watered/drought) as determined by Student’s t-test using the Two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5% within a given time point. Red indicates up-regulation and blue down-regulation with respect to the control. Time points highlighted in green indicate that significant changes in allantoin concentration were detected between treatment and control conditions according to two-way ANOVA with Tukey’s test (p < 0.05)
Analysis of the transcriptional regulation of purine catabolism under LN-WW revealed that purine catabolic genes were up-regulated in RAC875 in most tissues and developmental stages analysed (Fig. 5). In particular, the genes TaALN and TaAAH, putatively coding for the key enzymes involved in allantoin and allantoate degradation, respectively, were highly responsive to the LN-WW treatment. The differential expression of purine catabolic genes in RAC875 flag leaves, stem and spikelets at 17DAA corresponded to a significant reduction of allantoin measured in the same tissues as presented in Fig. 4a (time point highlighted in green in Fig. 5). At 19DAA, all purine catabolic genes were significantly up-regulated in flag leaves and, with a lower magnitude, also in stems and spikelets (Fig. 5).
In contrast to the transcriptional response under low N conditions, RAC875 droughted plants displayed a differential regulation of specific sets of purine catabolic genes (Fig. 5). Generally, TaALN was down-regulated in all analysed tissues of droughted plants (HN-D) relative to plants grown under HN-WW, except in the flag leaves and spikelets where no significant changes in transcription of TaALN were detected. A decrease in transcript abundance of TaALN in the YFEL occurred already after exposure to mild drought conditions (day 3), suggesting that this gene is particularly drought responsive in this tissue (Fig. 5). TaALN down-regulation at day 6 in YFEL corresponded with accumulation of allantoin as presented in Fig. 4b (time point highlighted in green in Fig. 5). The TaXDH1 and TaUOX genes, coding for the enzymes that synthesise allantoin, were up-regulated under HN-D across the analysed tissues, except in developing grain (Fig. 5). Interestingly, TaXDH2 showed the opposite transcriptional regulation to its paralog, TaXDH1. In fact, TaXDH2 was down-regulated under HN-D in the YFEL, the flag leaf and the stem (Fig. 5). TaAAH and TaUGAH presented different drought responses depending on the tissue analysed (e.g. were down-regulated in stem and up-regulated in the spikelet).
Analysis of the transcriptional profile of purine catabolic genes in the YFEL of Mace are largely in agreement with those in RAC875. N deficiency increased transcription of TaXDH1, TaALN and TaAAH in Mace YFEL; whilst TaALN was down-regulated andTaXDH1 up-regulated under drought (Fig. S4).
Metabolite signature of spikelet and developing grain under drought and low N conditions
To assess the involvement of allantoin in N and C metabolism in wheat under drought and N limitation during grain filling, the metabolic signatures from the spikelet and the developing grain of RAC875 and Mace were assessed and visualised by hierarchical clustering (Figs. 6, 7) and PCA (Supplementary Fig. S5 and S6).

Metabolite response of RAC875 and Mace spikelet and developing grain at day 8 of the drought treatment. Metabolite levels measured with GC-MS, ion chromatography and HPLC were log transformed and scaled by subtracting the median metabolite value in each metabolite distribution. Hierarchical clustering and heatmap of metabolite levels of RAC875 and Mace spikelet a and developing grain b at day 8 of the drought treatment was performed with the support of ClustVis web tool (Metsalu and Vilo 2015). Pearson’s correlation distance of scaled data was used for the hierarchical clustering. Each column represents a biological replicate. Representative cluster containing allantoin (measured with GC-QqQ method) is noted with a green line, other representative clusters are noted with a blue line

Metabolite response of RAC875 and Mace spikelet and developing grain under High N and Low N treatments during grain filling. Metabolite levels measured with GC–MS, ion chromatography and HPLC were log transformed and scaled by subtracting the median metabolite value in each metabolite distribution. Hierarchical clustering and heatmap of metabolite levels of RAC875 and Mace spikelet a and developing grain b at 17, 19 and 22 days after anthesis (DAA) was performed with the support of ClustVis web tool (Metsalu and Vilo 2015). Pearson’s correlation distance of scaled data was used for the hierarchical clustering. Dashed lines represent different time points within a given genotype and treatment. Each column represents a biological replicate. Representative cluster containing allantoin (measured with GC-QqQ method) is noted with a green line, other representative clusters are noted with a blue line
This analysis revealed that allantoin accumulation under drought conditions (HN-D) at day 8 showed the same pattern with additional metabolites forming a distinct cluster in both the spikelet (D.S.ii; Fig. 6a) and the developing grain (D.Dg.ii; Fig. 6b) of RAC875 and Mace. In both tissues, these clusters contained mostly amino acids including the well-known drought responsive amino acids proline and 4-amino-butanoic acid (GABA) and, in the developing grain, the polyamine putrescine. Generally, metabolites belonging to these clusters appeared to accumulate with higher magnitude in RAC875 compared to Mace. A genotype-specific response was also apparent in two additional clusters in developing grain with metabolites specifically accumulated (D.Dg.i) and reduced (D.Dg.iv) in RAC875 under drought, respectively (Fig. 6b).
Clusters containing metabolites with an apparent reduction under drought in both genotypes were also identified, in both the spikelet (D.S.i; Fig. 6a) and the developing grain (D.Dg.iii; Fig. 6b).These clusters contained sucrose, myo-inositol and quinic acid. In addition, in spikelets, this cluster contained other organic acids (e.g., isocitric acid, 3-phosphate-glyceric acid) as well as the tri-saccharides raffinose and 1-kestose. The cluster in developing grain further contained glucose-6-phosphate and fructose-6-phosphate, as well as tryptophan and glutamate.
Analysis of the metabolite signature under low N condition revealed contrasting patterns compared to the drought treatment. Generally, the drought treatment at day 8 clearly separated metabolites between the two treatments (HN-WW and HN-D; Fig. S5). However, the low N treatment appeared to influence the metabolite levels to a lower extent as supported by PCA analysis (Fig. S6). In particular, in spikelet the major dividing component of the data points was PC2 that separated according to the genotype (Fig. S6a). On the other hand, PCA of developing grain revealed no major differences between genotypes and treatment, although a weak separation according to the time point, especially for 22 DAA, was detected along PC1 (Fig. S6b).
Hierarchical clustering of the spikelet data under different N conditions placed allantoin into a representative cluster (Ln.S.i) which contained a large number of metabolites, mostly carbon-rich compounds (Fig. 7a). Specifically, this allantoin cluster contained eleven organic acids (including citric acid, malic acid, succinic acid, glyceric acid and shikimic acid) and ten sugars (including fructose, sucrose, myo-inositol, raffinose, 1-kestose). These metabolites appeared to accumulate to higher levels in spikelet of Mace as compared to RAC875, which showed a small or no treatment effect (Fig. 7a).
Similarly, in the developing grain allantoin grouped in a representative cluster (Ln.Dg.iii) which was composed of allantoin and three carbon-rich metabolites, including raffinose, galactinol and 1-kestose. This is in contrast to the metabolite profile in the drought treatment, where allantoin grouped with amino acids. These amino acids, under low N conditions, are scattered between two representative clusters in spikelets (Ln.S.ii and Ln.S.iii; Fig. 7a) or, in developing grain, largely not assigned to any representative cluster (Fig. 7b). Although, in the developing grain, this large area clustered according to the time points (DAA).
However, the accumulation of allantoin, galactinol and raffinose in developing grain under N starvation appears to be developmental rather than representing a specific response to N starvation as similar metabolic changes occurred also under control conditions at 22DAA (Fig. 8b). In agreement with the quantitative data on allantoin shown in Fig. 4a, Mace appeared to accumulate higher levels of allantoin and the other metabolites in cluster Ln.Dg.iii suggesting genotypic differences within wheat.

Total N and N-containing metabolites in mature grain of bread wheat genotypes RAC875 and Mace. Comparison of a N concentration (%) and b allantoin concentration (nmol g−1 DW) in mature grain harvested from the tallest remaining tiller of RAC875 and Mace plants grown under control conditions (HN-WW), drought (HN-D) and low N (LN-WW) conditions. Letters indicate significant differences between genotypes and treatments by two-way ANOVA with Tukey’s test (p < 0.05); c free amino acids levels in mature grain of RAC875 and Mace grown under control conditions (HN-WW). Amino acid concentration is reported in black, whilst N atoms present in each amino acid in grey, values are expressed as nmol g−1 DW. Allantoin and corresponding N concentration are reported in lighter colours to allow comparison. Asterisks denote significant differences between RAC875 (R) and Mace (M) by Student’s t-test (*p < 0.05; **p < 0.01; ****p < 0.0001). All data are mean ± SEM of 16–18 biological replicates
In summary, the metabolite profiling showed that, under drought, allantoin clustered predominantly with N-rich compounds (amino acids), whereas under N-starvation allantoin clustered with C-rich compounds (sugars and organic acids) and therefore allantoin represented the only N-rich compound.
Allantoin accumulation in the grain
To assess the contribution of allantoin to the overall N pools in the grain under control (HN-WW) and stress conditions (HN-D and LN-WW), the plants minus the sampled tiller (Supplementary Fig. S1) were grown until maturity, corresponding overall to 16–18 plants per treatment. Total N, allantoin and free amino acid concentrations were then determined in grain harvested from the tallest remaining tiller (Fig. 8). Analysis of grain N% highlighted significant differences between treatments (two-way ANOVA, p < 0.0001), in particular, it showed a 20% reduction in grain N% of plants grown under LN-WW with no evident genotypic differences between RAC875 and Mace (Fig. 8a). Allantoin concentration was significantly responsive to the applied treatments and genotypes (two-way ANOVA, Genotype × Treatment, p = 0.0028) (Fig. 8b). Particularly, allantoin was reduced under LN-WW by 44% and 49% in RAC875 and Mace plants, respectively. In contrast, under HN-D, allantoin in RAC875 grains increased by 39% relatively to HN-WW. Interestingly, the largest difference in allantoin concentration between genotypes was observed under HN-WW, as previously observed for the flag leaf (Fig. 4). In fact, Mace grains accumulated 66% more allantoin than RAC875, whilst there were no significant differences between the genotypes under LN-WW and HN-D, as mentioned above (Fig. 8b).
Analysis of the free amino acids content of grain from plants grown under HN-WW revealed that allantoin accumulated to levels comparable to other amino acids, such as glutamine, aspartate, asparagine and arginine (Fig. 8c). When considering that each allantoin molecule contains four N atoms (1:1 nitrogen to carbon ratio, N:C), the overall N stored in allantoin in grain was even larger than the N present in glutamate and aspartate, which were the most abundant free amino acids identified but have only one N atom (1:4 N:C) (Fig. 8c). However, the N-rich amino acid arginine (2:3 N:C) retained approximately double the N content of allantoin in both RAC875 and Mace grains. The analysis also showed significant genotypic differences in certain amino acids between RAC875 and Mace. In particular, Mace had 70% and 31% higher concentration of arginine and alanine than RAC875, whilst RAC875 had 179%, 43% and 35% higher concentration of tryptophan, aspartate and serine than Mace, respectively (Fig. 8c).
Discussion
The aim of this study was to assess the role of the purine intermediate allantoin and the purine catabolic genes under water and nutrient stress in bread wheat. For this, we selected two Australian genotypes, specifically RAC875, a breeding line that has been characterised as tolerant to drought (Izanloo et al. 2008; Bennett et al. 2012; Bonneau et al. 2013) and the locally adapted variety Mace, which is suggested to have a higher N-use efficiency based on preliminary studies (Mahjourimajd et al. 2016).
The wheat purine catabolic genes show high synteny but also differences compared with other grasses
The genes of the purine catabolic pathway, identified in several grass genomes including hexaploid bread wheat, showed a high degree of synteny among three grass genomes, suggesting that the identified wheat loci are true gene orthologues (Fig. 2). However, the poor synteny displayed by XDH and its adjacent genes even in diploid genomes (Brachypodium, rice and sorghum) suggests that XDH is located in an unstable genomic region prone to rearrangements. The majority of the grass genomes had only one copy of the purine catabolic genes (Table S5), corresponding to three homeologs in wheat (Table S1), with the only exceptions of the fore-mentioned XDH, and UAH, for which there was no synteny for the Brachypodium orthologs BdUAH2.
For XDH we have identified a second copy on chromosome 6 of which, based on in-silico expression analysis (Fig. S3a), only the homeolog on chromosome 6, TaXDH2-6DS is a functional gene explaining its low expression level compared with TaXDH1 (Fig. S3b; Fig. 5). Interestingly, TaXDH2 shows an opposite transcriptional response to drought (reduced transcript level) compared with TaXDH1 suggesting different roles and/or functional divergence. A differential response to drought and other treatments (salt, cold, ABA) has also been shown in Arabidopsis, which carries a tandem duplication of AtXDH1 and AtXDH2 on a single chromosome (Hesberg et al. 2004). It will therefore be interesting to investigate the role and enzymatic function of TaXDH2 in more detail in relation to drought and other stresses.
Wheat highly accumulates allantoin under drought stress
There was a significant accumulation of allantoin in all analysed tissues and genotypes of wheat plants exposed to drought (Fig. 4b). This was accompanied by a significant reduction in transcript levels of TaALN, i.e., the first step in allantoin degradation (Fig. 5).
Allantoin was previously reported to accumulate in wheat and rice in response to abiotic stresses, including drought (Bowne et al. 2011; Degenkolbe et al. 2013; Casartelli et al. 2018), however, these studies did not investigate the regulation of the purine catabolic genes under those conditions.
Analysis of the expression of purine catabolic genes in Arabidopsis (Irani and Todd 2016) recently showed that leaf accumulation of allantoin upon drought stress was associated with transcriptional up-regulation of the purine catabolic genes leading to allantoin synthesis (AtXDH1, AtXDH2, AtUOX, AtAS) whilst expression of the allantoin-degrading gene AtALN was only marginally increased. Similarly, Yesbergenova et al. (2005) showed that, in tomato, LeXDH1 and LeXDH2 were up-regulated under drought in leaf and root tissues. In Arabidopsis, the allantoin pathway was also implicated with salt stress, showing reduced AtALN and AtAAH expression and increased AtXDH1 expression (Irani and Todd 2016; Hesberg et al. 2004), as well as an accumulation of allantoin accompanied by AtUOX and AtALNS up-regulation and AtALN down-regulatoin (Lescano et al. 2016).
Our data agree with the above studies confirming that allantoin significantly accumulates under drought (Fig. 4b) and that this is paralleled by the up-regulation of genes putatively encoding enzymes for allantoin synthesis (TaXDH1 and TaUOX) and/or down-regulation of TaALN (Fig. 5). Although we have not quantified xanthine, the gene expression data suggest that under drought, an increased amount of xanthine is likely to feed into this pathway which, in combination with a reduced allantoin degradation, results in the accumulation of allantoin under drought which was observed in our study.
In contrast, studies in common bean (Phaseolus vulgaris) showed up-regulation of both allantoin-synthetising genes (PvUOX) and allantoin-degrading genes (PvALN and PvAAH) in leaves under drought and this corresponded to increased levels of allantoin and allantoate (Alamillo et al. 2010; Coleto et al. 2014). Interestingly, Coleto et al. (2014) reported that these ureides were more concentrated in the tissues of drought-sensitive genotypes. This is in contrast to our data and other studies showing that allantoin specifically accumulates in drought tolerant genotypes of wheat, rice and resurrection plants (Bowne et al. 2011; Oliver et al. 2011; Degenkolbe et al. 2013; Yobi et al. 2013; Casartelli et al. 2018). This suggests that purine catabolism is regulated differently under drought stress in ureidic legumes.
Evidence of the functional role of purine catabolism under abiotic stress were revealed using reverse genetics approaches. Arabidopsis XDH mutants (xdh) showed that disrupting the first step of purine catabolism caused hypersensitivity to water stress (Watanabe et al. 2010) and impaired recovery from extended dark exposure (Brychkova et al. 2008), which in both cases led to excessive reactive oxygen species (ROS) accumulation compared to WT plants. In contrast, the constitutive accumulation of the intermediate allantoin in Arabidopsis ALN knock-out mutants (aln) led to enhanced performance under dehydration, drought and salt stress, which also corresponded to reduced ROS levels (Watanabe et al. 2014; Irani and Todd 2016). These studies further showed that increasing allantoin concentration, either constitutively (aln mutants) or by exogenous supplementation, stimulated ABA and jasmonic acid (JA) metabolism, key components of abiotic stress responses. However, a previous study suggested that allantoin does not possess any in-vitro antioxidant activity (Wang et al. 2012), prompting to speculate that allantoin and purine catabolism play a role in stress sensing and regulation rather than a direct ROS scavenging function.
Metabolomic profiling of the spikelet (defined here as vegetative part of the spike) and the developing grain after 8 days of drought stress showed that allantoin clustered with a set of highly drought-responsive metabolites (Fig. 6a, b). These clusters were composed mainly of amino acids, with alanine, valine, leucine, and proline common to the allantoin clusters in spike samples (D.S.ii) and developing grain (D.Dg.ii). Proline is amongst the best characterized drought-related amino acids and has been shown to have a range of functions under stress, e.g., osmolyte, regulator of redox potential, molecular chaperone, ROS scavenger and signalling molecule (Yoshiba et al. 1995; Hare and Cress 1997; Verbruggen and Hermans 2008; Szabados and Savoure 2010; Mohanty and Matysik 2001; Khedr et al. 2003). The specific accumulation of allantoin under drought in a wide range of different plant species and the suggested role in stimulating the ABA and JA pathway (see above) justifies further investigations into the importance of allantoin in the mitigation or tolerance to drought.
Preventing N losses under drought and allantoin as a source of N under nutrient deprivation
Under drought, RNA and DNA as well as protein degradation and nutrient remobilisation caused by premature leaf senescence (Munné-Bosch and Alegre 2004) and increased photorespiration (Mattsson et al. 1997; Wingler et al. 1999; Kumagai et al. 2011b) are sources of high tissue NH4+ and related emission of volatile ammonia (Mattsson and Schjoerring 2002). In principle, free NH4+ can be recycled by the GS-GOGAT cycle (Fig. S2), however, this pathway is negatively responsive to drought (Nagy et al. 2013; Singh and Gosh 2013) and might therefore not be sufficiently effective in capturing NH4+. Increased ammonia emission due to inhibition of GS with MSO has been demonstrated (Mattsson and Schjoerring 1996) and has been directly linked with enhanced photorespiration under high light and high O2 stress (Kumagai et al. 2011b), as well as with senescence (Parton et al. 1988) and heat stress (Mattson et al. 1997).
Ammonia emission can be considered an efficient, though very wasteful mechanism, to prevent the build-up of high tissue concentrations, which is toxic to plants (for a review see Britto and Kronzucker 2002). The accumulation of allantoin (e.g. 30-fold accumulation in RAC875 flag leaves, Fig. 4b) would therefore be beneficial to plants because it prevents accumulation of NH4+ to toxic levels and also, to some extent, to retain organic N in the plant that could be lost to the atmosphere in the form of volatile ammonia. Although allantoin represents just a fraction of the total organic N in a wheat plant, allantoin accumulation under drought appears to occur simultaneously with the accumulation of several other small metabolites containing N, notably amino acids such as proline (Fig. 6). This response seems to be shared by other plant species (see Introduction for references), in fact we previously reported high accumulation of allantoin and several amino acids when rice plants were subjected to drought stress (Casartelli et al. 2018). Therefore, this could underlie a global strategy that plants adopt to improve N balance under stress, when GS-GOGAT enzymatic activity is reduced. Differences in the regulation of GS (Singh and Gosh 2013) and ammonia emission (Kumagai et al. 2011) between drought tolerant and intolerant genotypes have been shown in rice and genetic diversity and drought-tolerant specific GS responses have also been shown in tomato (Sanchez-Rodriguez et al. 2011) and wheat (Nagy et al. 2013).We suggest that the selection of high allantoin and maintenance of GS activity (reduced N losses) under drought stress could be a specific target for breeding (Fig. 9).

Schematic model for the dual-role of allantoin under stress in relation to N homeostasis. Under low N conditions, increased expression of ureide-degrading genes ALN and AAH indicate that NH4+ is liberated from purines providing an internal source of organic N that can be re-assimilated by the GS-GOGAT cycle. As a result, allantoin concentration in plant tissues is reduced. In contrast, during drought stress, allantoin accumulates due to down-regulation of the ALN gene preventing the liberation of NH4+ that could lead to cellular toxicity and part of it might be lost as volatile ammonia due to drought-impaired GS-GOGAT activity
The importance of allantoin and purine catabolism in N metabolism have long been established in ureidic legumes, which employ allantoin and allantoate as major forms for long-distance transport of N (Schubert 1986; Sinclair and Serraj 1995). However, recently an important role in maintaning N homeostasis in non-ureidic plant species is becoming evident. Soltabayeva et al. (2018) reported that ArabidopsisAtxdh1, Ataln and Ataah mutants displayed an early senescence phenotype when grown under low NO3− conditions and that the activity of nitrate reductase (NR) was increased in leaves of Atxdh1 mutants suggesting a higher demand for nitrate than wild type plants. Similarly, Nakagawa et al. (2007) reported an early onset of senescence in RNAi-induced xdh mutants which also displayed reduced chlorophyll content and increased cytosolic GS1 protein, which is known to be induced during senescence (Bernard et al. 2008).
In our study, the quantification of allantoin in the wheat plants grown under N-limited conditions revealed a significant reduction of allantoin in all tissues analysed (Fig. 4a) which is in contrast to the observed accumulation of allantoin under drought (Fig. 4b) as discussed above. A decrease of allantoin under N deprivation was also reported in other cereal species, specifically in maize and rice showing reduced allantoin in leaves and roots, respectively (Amiour et al. 2012; Coneva et al. 2014). In addition, high induction of OsALN expression under low N conditions in rice was recently shown (Lee et al. 2018), this was previously also observed in Arabidopsis and the leguminous tree Robinia pseudoacacia (Yang and Han 2004). In agreement with this, the reduction of allantoin under low N reported in this study (Fig. 4a) was accompanied by increased transcription of purine catabolic genes, and this was particularly evident in genes encoding for the ureide degrading enzymes TaALN and TaAAH (Fig. 5).
Downstream compounds of the purine catabolic pathway were recently analysed in more detail under N deprivation using the same wheat genotypes, RAC875 and Mace (Melino et al. 2018) showing, in addition to reduced levels of allantoin, a significant reduction of allantoate in leaves and an accumulation of glyoxylic acid, the end product of purine degradation. The assumption that the N (and C) remobilised from allantoin supports plant growth is supported by studies showing that Arabidopsis and rice seedlings could grow with ureides as a sole N source (Desimone et al. 2002; Brychkova et al. 2008; Lee et al. 2018), although growth was delayed in comparison to plants supplied with inorganic N. This is in contrast to wheat seedlings that, resupplied with allantoin and xanthine as a sole source of N after NO3− starvation, grew and photosynthesised as well as those re-supplied with NO3− (Melino et al. 2018).
Taken together, these findings suggest that the enhanced activity of the purine catabolic pathway provides an internal source of organic N used to maintain homeostasis under low N conditions whereas the accumulation of allantoin under drought releases pressure from the GS-GOGAT cycle thereby preventing accumulation of toxic levels of NH4+ and possibly N losses due to volatilization (Fig. 9).
Allantoin is a relevant component of grain nitrogen in wheat
In wheat, N translocated to the grain during plant senescence is an important determinant of grain quality and there is currently little information on the contribution of allantoin and/or other purine catabolites to grain N in non-ureide plants. The data reported here show progressive accumulation of allantoin during grain filling (Fig. 4) up to more than 1400 nmol g−1 DW at maturity in Mace (Fig. 8b). This was confirmed by metabolomics analyses of developing grains showing that allantoin levels correlated with other metabolites specifically accumulating at 22DAA, in contrast to the majority of metabolites that showed an opposite trend (Fig. 7).
Although we calculated that allantoin accounted for only about 0.3% of the total N measured in wheat grains, this represents a significant portion of the soluble N pool, comparable to the most concentrated and N-rich amino acids, such as asparagine and arginine (Fig. 8c). Previous studies have shown that allantoin stored in wheat grains is quickly utilised, from as early as 1 day after germination (Montalbini 1992), suggesting that it can be used as a readily available N substrate.
The data reported here further show that genotypic differences exist among bread wheat genotypes, in fact, Mace accumulated 66% more grain allantoin than RAC875, despite similar % grain N (Fig. 8a, b). This difference was also particularely evident in flag leaves of Mace, which had up to 10-fold higher allantoin content compared to RAC875 under both, control and low N conditions (Fig. 4a). Mace is widely grown in Australia because of its superior yield under drought and nitrogen-use efficiency (Mahjourimajd et al. 2016). High allantoin concentrations in the leaves and grain might indeed be a positive contributing factor to high NUE in Mace. This is in agreement with findings in rice where differences in allantoin levels in genotypes with contrasting drought tolerance were already apparent under control conditions (Casartelli et al. 2018). In addition to greater accumulation of allantoin, mature grains of Mace also accumulated 70% more arginine than RAC875 (Fig. 8c). Arginine is the proteinogenic amino acid with the highest N:C ratio (3:2), known to represent a major N form for storage and transport.
Our data thus indicate that the two analysed wheat genotypes prioritize different N compounds for transport and grain loading. Previous studies in Mace suggest that this cultivar has high N use efficiency (Mahjourimajd et al. 2016) and this study and Kastury et al. (2018) additionally demonstrated that Mace is higher yielding under drought than RAC875. The possibility that the superior performance of Mace may be related to the preferential use of metabolites with high N:C ratio, which are more energy effective forms for transporting and storing N, is an intriguing hypothesis that requires further validation. In addition, allantoin and arginine may also play a regulatory role in plant growth and development as they participate in ABA, JA, and nitric oxide (NO) signalling, respectively (Watanabe et al. 2014; Winter et al. 2015; Takagi et al. 2016).
Summary
In summary, our data are in support of the converging evidence that allantoin has a relevant role in non-ureide plants under water stress and in N homeostasis. In addition to the proposed role of allantoin in drought-induced ABA responses, accumulation of this N-rich molecule under drought would contribute to optimise N (and C) balance in the plant by preventing toxic build-up of NH4+ and possible N losses through volatile ammonia due to a reduced activity of the GS-GOGAT cycle (Fig. 9). Under low N conditions, induced expression of allantoin catabolic genes and decreased allantoin levels indicate that allantoin serves as an internal organic N source (Fig. 9). In wheat, allantoin is also a relevant component of grain N and comparable to that of other major amino acids.
Sufficient evidence for genetic diversity for the purine catabolic pathway in relation to drought tolerance and enhanced NUE is now available and, in conjunction with reported genetic diversity for GS-GOGAT, provides an opportunity to explore this further for the development of crops with enhanced drought tolerance and NUE.
References
Alamillo JM, Diaz-Leal JL, Sanchez-Moran MV, Pineda M (2010) Molecular analysis of ureide accumulation under drought stress in Phaseolus vulgaris L. PlantCell. Environ 33:1828–1837
Amiour N, Imbaud S, Clément G et al (2012) The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. J Exp Bot 63:5017–5033
Barbottin A, Lecomte C, Bouchard C, Jeuffroy M-H (2005) Nitrogen remobilization during grain filling in wheat: genotypic and environmental effects. Crop Sci 45:1141
Bennett D, Izanloo A, Reynolds M, Kuchel H, Langridge P, Schnurbusch T (2012) Genetic dissection of grain yield and physical grain quality in bread wheat (Triticum aestivum L.) under water-limited environments. Theo Appl Genetics 125:255–271
Bernard SM, Møller ALB, Dionisio G et al (2008) Gene expression, cellular localisation and function of glutamine synthetase isozymes in wheat (Triticum aestivum L.). Plant Mol Biol 67:89. https://doi.org/10.1007/s11103-008-9303-y
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Bonneau J, Taylor J, Parent B, Bennett D, Reynolds M, Feuillet C, Langridge P, Mather D (2013) Multi-environment analysis and improved mapping of a yield-related QTL on chromosome 3B of wheat. Theo Appl Genetics 126:747–761
Bowne JB, Erwin TA, Juttner J, Schnurbusch T, Langridge P, Bacic A, Roessner U (2011) Drought responses of leaf tissues from wheat cultivars of differing drought tolerance at the metabolite level. Mol Plant 5:418–429
Britto DT, Kronzucker HJ (2002) NH4 + toxicity in higher plants: a critical review. Plant Physol 159:567–584
Brychkova G, Fluhr R, Sagi M (2008) Formation of xanthine and the use of purine metabolites as a nitrogen source in Arabidopsis plants. Plant Signal Behav 3:999–1001
Casartelli A, Riewe D, Hubberten HM, Altmann T, Hoefgen R, Heuer S (2018) Exploring traditional aus-type rice for metabolites conferring drought tolerance. Rice 11:9
Chomczynski P (1993) A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 15:532–534
Choulet F, Alberti A, Theil S, Glover N, Barbe V, Daron J, Pingault L, Sourdille P, Couloux A, Paux E (2014) Structural and functional partitioning of bread wheat chromosome 3B. Science 345:1249721
Clavijo BJ, Venturini L, Schudoma C et al (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res 27:885–896
Coleto I, Pineda M, Rodino AP, De Ron AM, Alamillo JM (2014) Comparison of inhibition of N2 fixation and ureide accumulation under water deficit in four common bean genotypes of contrasting drought tolerance. Ann Botany 113:1071–1082
Coneva V, Simopoulos C, Casaretto JA et al (2014) Metabolic and co-expression network-based analyses associated with nitrate response in rice. BMC Genom 15:1056
Degenkolbe T, Do PT, Kopka J, Zuther E, Hincha DK, Köhl KI (2013) Identification of drought tolerance markers in a diverse population of rice cultivars by expression and metabolite profiling. PLoS ONE 8:e63637
Desimone M, Catoni E, Ludewig U, Hilpert M, Kunze AS, Tegeder R, Frommer M, Schumacher WB (2002) A novel superfamily of transporters for allantoin and other oxo derivatives of nitrogen heterocyclic compounds in Arabidopsis. Plant Cell 14:847–856
Do PT, Degenkolbe T, Erban A, Heyer AG, Kopka J, Köhl KI, Hincha DK, Zuther E (2013) Dissecting rice polyamine metabolism under controlled long-term drought stress. PLoS ONE 8:e60325
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Erban A, Schauer N, Fernie AR, Kopka J (2007) Nonsupervised construction and application of mass spectral and retention time index libraries from time-of-flight gas chromatography–mass spectrometry metabolite profiles. Humana Press, New York pp 19–38
Gill BS, Appels R, Botha-Oberholster A-M et al (2004) A workshop report on wheat genome sequencing: international genome research on wheat consortium. Genetics 168:1087–1096
Hanks JF, Tolbert N, Schubert KR (1981) Localization of enzymes of ureide biosynthesis in peroxisomes and microsomes of nodules. Plant Physiol 68:65–69
Hare P, Cress W (1997) Metabolic implications of stress-induced proline accumulation in plants. Plant Growth Regul 21:79–102
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19
Herridge DF, Atkins CA, Pate JS, Rainbird RM (1978) Allantoin and Allantoic Acid in the Nitrogen Economy of the Cowpea (Vigna unguiculata [L.] Walp.). Plant Physiol 62:495–498
Hesberg C, Hänsch R, Mendel RR, Bittner F (2004) Tandem orientation of duplicated xanthine dehydrogenase genes from Arabidopsis thaliana: differential gene expression and enzyme activities. J Biol Chem 279:13547–13554
Irani S, Todd CD (2016) Ureide metabolism under abiotic stress in Arabidopsis thaliana. J Plant Physiol 199:87–95
Izanloo A, Condon AG, Langridge P, Tester M, Schnurbusch T (2008) Different mechanisms of adaptation to cyclic water stress in two South Australian bread wheat cultivars. J Exp Bot 59:3327–3346
Kanani H, Dutta B, Klapa MI (2010) Individual vs. combinatorial effect of elevated CO2 conditions and salinity stress on Arabidopsis thaliana liquid cultures: comparing the early molecular response using time-series transcriptomic and metabolomic analyses. BMC Syst Biol 4:177–177
Kaplan F, Kopka J, Haskell DW, Zhao W, Schiller KC, Gatzke N, Sung DY, Guy CL (2004) Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol 136:4159–4168
Kastury F, Rahimi Eichi V, Enju A, Okamoto M, Heuer S, Melino V (2018) Exploring the potential for top-dressing bread wheat with ammonium chloride to minimize grain yield losses under drought. Soil Sci Plant Nutr. https://doi.org/10.1080/00380768.2018.1493341
Khedr AHA, Abbas MA, Wahid AAA, Quick WP, Abogadallah GM (2003) Proline induces the expression of salt-stress-responsive proteins and may improve the adaptation of Pancratium maritimum L. to salt-stress. J Exp Bot 54:2553–2562
Kim K, Park J, Rhee S (2007) Structural and Functional basis for (S)-allantoin formation in the ureide pathway. J Biol Chem 282:23457–23464
Kumagai E, Araki T, Hamaoka N, Ueno O (2011) Ammonia emission from rice leaves in relation to photorespiration and genotypic differences in glutamine synthetase activity. Annals Bot 108:1381–1386
Lamberto I, Percudani R, Gatti R, Folli C, Petrucco S (2010) Conserved alternative splicing of arabidopsis transthyretin-like determines protein localization and S-allantoin synthesis in peroxisomes. Plant Cell 22:1564–1574
Lea PJ, Miflin BJ (2010) Nitrogen assimilation and its relevance to crop improvement. Ann Plant Rev 42:1–40
Lee D-K, Redillas MCFR, Jung H, Choi S, Kim YS, Kim J-K (2018) A nitrogen molecular sensing system, comprised of the ALLANTOINASE and UREIDE PERMEASE 1 genes, can be used to monitor N status in rice. Front Plant Sci 9:444
Lescano C, Martini C, González C, Desimone M (2016) Allantoin accumulation mediated by allantoinase downregulation and transport by Ureide Permease 5 confers salt stress tolerance to Arabidopsis plants. Plant Mol Biol 91:581–595
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR (2006) Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc 1:387
Mahjourimajd S, Kuchel H, Langridge P, Okamoto M (2016) Evaluation of Australian wheat genotypes for response to variable nitrogen application. Plant Soil 399:247–255
Masclaux-Daubresse C, Daniel-Vedele F, Dechorgnat J, Chardon F, Gaufichon L, Suzuki A (2010) Nitrogen uptake, assimilation and remobilization in plants: challenges for sustainable and productive agriculture. Ann Bot 105:1141–1157
Mattsson M, Schjoerring JK (1996) Ammonia emission from young barley plant: influence of N-source, light/dark cycles and inhibition of glutamine synthetase. J Exp Bot 47:477–484
Mattsson M, Schjoerring JK (2002) Dynamic and steady-state responses of inorganic nitrogen pools and NH3 exchange in leaves of Lolium perenne and Bromus erectus to changes in root nitrogen supply. Plant Physiol 128:742–750
Mattsson M, Häusler RE, Leegood RC, Lea PJ, Schjoerring JK (1997) Leaf-atmosphere NHâ exchange in barley mutants with reduced activities of glutamine synthetase. Plant Physiol 114:1307–1312
Melino VJ, Fiene G, Enju A, Cai J, Buchner P, Heuer S (2015) Genetic diversity for root plasticity and nitrogen uptake in wheat seedlings. Funct Plant Biol 42:942
Melino VJ, Casartelli A, George J, Rupasinghe T, Roessner U, Okamoto M, Heuer S (2018) RNA catabolites contribute to the nitrogen pool and support growth recovery of wheat. Front Plant Sci 9:1539
Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43:W566–W570
Mohanty P, Matysik J (2001) Effect of proline on the production of singlet oxygen. Amino Acids 21:195–200
Montalbini P (1992) Ureides and enzymes of ureide synthesis in wheat seeds and leaves and effect of allopurinol on Puccinia recondita f. sp. tritici infection. Plant Sci 87:225–231
Moschen S, Bengoa Luoni S, Di Rienzo JA, Caro MdP, Tohge T, Watanabe M, Hollmann J, González S, Rivarola M, García-García F (2016) Integrating transcriptomic and metabolomic analysis to understand natural leaf senescence in sunflower. Plant Biotechnol J 14:719–734
Munné-Bosch S, Alegre L (2004) Die and let live: leaf senescence contributes to plant survival under drought stress. Funct Plant Biol 31:203–216
Nagy Z, Németh E, Guóth A, Bona L, Wodala B, Pécsváradi A (2013) Metabolic indicators of drought stress tolerance in wheat: glutamine synthetase isoenzymes and rubisco. Plant Physiol Biochem 67:48–54
Nakagawa A, Sakamoto S, Takahashi M, Morikawa H, Sakamoto A (2007) The RNAi-mediated silencing of xanthine dehydrogenase impairs growth and fertility and accelerates leaf senescence in transgenic Arabidopsis plants. Plant Cell Physiol 48:1484–1495
Nam M, Bang E, Kwon T, Kim Y, Kim E, Cho K, Park W, Kim B, Yoon I (2015) Metabolite profiling of diverse rice germplasm and identification of conserved metabolic markers of rice roots in response to long-term mild salinity stress. Int J Mol Sci 16:21959–21974
Nikiforova VJ, Kopka J, Tolstikov V, Fiehn O, Hopkins L, Hawkesford MJ, Hesse H, Hoefgen R (2005) Systems rebalancing of metabolism in response to sulfur deprivation, as revealed by metabolome analysis of Arabidopsis plants. Plant Physiol 138:304
Oliver MJ, Guo L, Alexander DC, Ryals JA, Wone BW, Cushman JC (2011) A sister group contrast using untargeted global metabolomic analysis delineates the biochemical regulation underlying desiccation tolerance in Sporobolus stapfianus. Plant Cell 23:1231–1248
Parton WJ, Morgan JA, Altenhofen JM, Harper LA (1988) Ammonia volatilization from spring wheat. Plants Agron J 80:419–425. https://doi.org/10.2134/agronj1988.00021962008000030008x
Pate JS, Atkins CA, White ST, Rainbird RM, Woo KC (1980) Nitrogen nutrition and xylem transport of nitrogen in ureide-producing grain legumes. Plant Physiol 65:961–965
Pessoa J, Sárkány Z, Ferreira-da-Silva F, Martins S, Almeida MR, Li J, Damas AM (2010) Functional characterization of Arabidopsis thaliana transthyretin-like protein. BMC Plant Biol 10:30
Ramazzina I, Folli C, Secchi A, Berni R, Percudani R (2006) Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat Cheml Biol 2:144–148
Sánchez-Rodríguez E et al (2011) Ammonia production and assimilation: Its importance as a tolerance mechanism during moderate water deficit in tomato plants. J Plant Physiol 168:816–823. https://doi.org/10.1016/j.jplph.2010.11.018
Schubert KR (1986) Products of biological nitrogen fixation in higher plants: synthesis, transport, and metabolism. Ann Rev Plant Physiol 539–574
Sears ER (1954) The aneuploids of common wheat. Missouri Agricult Exp Stat Annu Bull 572:1–59
Serventi F, Ramazzina I, Lamberto I, Puggioni V, Gatti R, Percudani R (2010) Chemical basis of nitrogen recovery through the ureide pathway: formation and hydrolysis of S-ureidoglycine in plants and bacteria. ACS Chem Biol 5:203
Silvente S, Sobolev AP, Lara M (2012) Metabolite adjustments in drought tolerant and sensitive soybean genotypes in response to water stress. PLoS ONE 7:e38554
Sinclair TR, Serraj R (1995) Legume nitrogen fixation and drought. Nature 378:344–344
Singh K, Ghosh S (2013) Regulation of glutamine synthetase isoforms in two differentially drought-tolerant rice (Oryza sativa L.) cultivars under water deficit conditions. Plant Cell Rep 32:183–193
Singh G, Kumar S, Singh P (2003) A quick method to isolate RNA from wheat and other carbohydrate-rich seeds. Plant Mol Biol Report 21:93–93
Soltabayeva A, Srivastava S, Kurmanbayeva A, Bekturova A, Fluhr R, Sagi M (2018) Early senescence in older leaves of low nitrate-grown Atxdh1 uncovers a role for purine catabolism in N supply. Plant Physiol. https://doi.org/10.1104/pp.18.00795
Szabados L, Savoure A (2010) Proline: a multifunctional amino acid. Trends Plant Sci 15:89–97
Takagi H, Ishiga Y, Watanabe S et al (2016) Allantoin, a stress-related purine metabolite, can activate jasmonate signaling in a MYC2-regulated and abscisic acid-dependent manner. J Exp Bot 67:2519
Todd CD, Polacco JC (2006) AtAAH encodes a protein with allantoate amidohydrolase activity from Arabidopsis thaliana. Planta 223:1108–1113
Triplett EW, Blevins DG, Randall DD (1982) Purification and properties of soybean nodule xanthine dehydrogenase. Arch Biochem Biophys 219:39–46
Verbruggen N, Hermans C (2008) Proline accumulation in plants: a review. Amino Acids 35:753–759
Wang P, Kong CH, Sun B, Xu XH (2012) Distribution and Function of Allantoin (5-Ureidohydantoin) in Rice Grains. J Agric Food Chem 60:2793–2798. https://doi.org/10.1021/jf2051043
Wang W-S, Zhao X-Q, Li M, Huang L-Y, Xu J-L, Zhang F, Cui Y-R, Fu B-Y, Li Z-K (2016) Complex molecular mechanisms underlying seedling salt tolerance in rice revealed by comparative transcriptome and metabolomic profiling. J Exp Bot 67:405–419
Watanabe S, Nakagawa A, Izumi S, Shimada H, Sakamoto A (2010) RNA interference-mediated suppression of xanthine dehydrogenase reveals the role of purine metabolism in drought tolerance in Arabidopsis. FEBS Lett 584:1181–1186
Watanabe S, Matsumoto M, Hakomori Y, Takagi H, Shimada H, Sakamoto A (2014) The purine metabolite allantoin enhances abiotic stress tolerance through synergistic activation of abscisic acid metabolism. Plant Cell Environ 37:1022–1036
Werner AK, Witte CP (2011) The biochemistry of nitrogen mobilization: purine ring catabolism. Trends Plant Sci 16:381–387
Werner AK, Sparkes IA, Romeis T, Witte CP (2008) Identification, biochemical characterization, and subcellular localization of allantoate amidohydrolases from Arabidopsis and soybean. Plant Physiol 146:418–430
Werner AK, Romeis T, Witte C-P (2010) Ureide catabolism in Arabidopsis thaliana and Escherichia coli. Nat Chem Biol 6:19–21
Wingler A, Quick WP, Bungard RA, Bailey KJ, Lea PJ, Leegood RC (1999) The role of photorespiration during drought stress: an analysis utilizing barley mutants with reduced activities of photorespiratory enzymes. Plant Cell Environ 22:361–373
Winter G, Todd CD, Trovato M, Forlani G, Funck D (2015) Physiological implications of arginine metabolism in plants. Front Plant Sci 6:534
Wu H, Liu X, You L, Zhang L, Zhou D, Feng J, Zhao J, Yu J (2012) Effects of salinity on metabolic profiles, gene expressions, and antioxidant enzymes in halophyte. J Plant Growth Reg 31:332–341
Yang J, Han KH (2004) Functional characterization of allantoinase genes from Arabidopsis and a nonureide-type legume black locust. Plant Physiol 134:1039–1049
Yesbergenova Z, Yang G, Oron E, Soffer D, Fluhr R, Sagi M (2005) The plant Mo-hydroxylases aldehyde oxidase and xanthine dehydrogenase have distinct reactive oxygen species signatures and are induced by drought and abscisic acid. Plant J 42:862–876
Yobi A, Wone BW, Xu W, Alexander DC, Guo L, Ryals JA, Oliver MJ, Cushman JC (2013) Metabolomic profiling in Selaginella lepidophylla at various hydration states provides new insights into the mechanistic basis of desiccation tolerance. Mol Plant 6:369–385
Yoshiba Y, Kiyosue T, Katagiri T, Ueda H, Mizoguchi T, Yamaguchi-Shinozaki K, Wada K, Harada Y, Shinozaki K (1995) Correlation between the induction of a gene for δ1-pyrroline-5-carboxylate synthetase and the accumulation of proline in Arabidopsis thaliana under osmotic stress. Plant J 7:751–760
Acknowledgements
We thank Akiko Enju, Pia Müller and Jessey George (University of Adelaide) for supporting with sample collection; Juan Carlos Sanchez-Ferrero (University of Adelaide) for support with bioinformatic analyses; Adam Lukaszewski (University of Riverside, California, US) for providing the Chinese Spring NT seeds stock and Margaret Pallotta (University of Adelaide) for providing DNA of the NT lines; Sanjiv Satija, Larissa Chirkova and Yuan Li (University of Adelaide) for technical support with molecular analyses; Julian Taylor (University of Adelaide) for support in experimental design; Siria Natera, Gina Barossa and Veronica Liu (Metabolomics Australia) for support with the allantoin quantification. We are grateful to Elmien Heyneke, Ines Fehrle and Astrid Basner (Max Plank Institute, Potsdam-Golm) for their support in performing the metabolomics analysis. This work was supported by an Australian Research Council linkage Grant LP1400100239 in partnership with and additional funding from DuPont-Pioneer (USA). Rothamsted Research receives strategic funding from the Biotechnological and Biological Sciences Research Council of the United Kingdom. We acknowledge support through the Designing Future Wheat (DFW) Strategic Programme (BB/P016855/1).
Author information
Authors and Affiliations
Contributions
SH was leading the project. MO, VJM and RH conceived the ideas, edited the manuscript and co-supervised A.C. who conducted the experiments as part of his PhD thesis. AC wrote the manuscript with inputs from SH, MO, VJM and RH. The quantitative allantoin method was developed by UR, NSJ and HM. The identification of the wheat genes and bioinformatics analyses was supported by UB and RS. Inputs on the quantitative gene expression analyses was provided by MR. Access to analytical pipelines, technical support and data analysis was provided by AE, MW, and EZ.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Casartelli, A., Melino, V.J., Baumann, U. et al. Opposite fates of the purine metabolite allantoin under water and nitrogen limitations in bread wheat. Plant Mol Biol 99, 477–497 (2019). https://doi.org/10.1007/s11103-019-00831-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-019-00831-z