
Abstract
The wide and not yet fully uncovered potential of plant secondary metabolites make plants a rich source of drug leads. Metabolomics enables the study of the metabolic perturbations taking place in bacterial cells under the influence of plant-based bioactive molecules. It reveals the changes in metabolic pathways within bacteria, reflecting the reprogramming of the biochemical networks. From this point of view, metabolomics is valuable in understanding the alteration of cell functions when bacteria are subjected to metabolic stress caused by treatment with secondary metabolites, that inhibit their growth. In this review the application of metabolomics in revealing bacteria response to plant-derived secondary metabolites is presented. Metabolomics may be a way to select antibacterial plant-based bioactive secondary metabolites and to understand their mode of action. Therefore, herein the usefulness of metabolomic approach in screening for antimicrobials from plants, as well as in exploring the target points in bacteria metabolism and in uncovering the mechanisms of bacteria adaptation and resistance to natural antimicrobials are discussed. Basic chemometrics and molecular networking are successfully applied for the identification of antimicrobial molecules in complex plant mixtures. Determination of antibacterial modes of action is done through classification strategy, pathway analysis and integration of transcriptomics, genomics and metabolomics, whereas, comparative metabolomics and integrative approach is useful in revealing the bacterial mechanisms of resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Food and medicinal plants have always been used by human as a natural remedy for bacterial infections and associated diseases. The impact of plant-based foods and their bioactive secondary metabolites on a human health and wellbeing in undoubtful. The wide and still not fully uncovered potential of plant secondary metabolites make plants a rich source of drug leads (Anand.et al. 2019). What is more, the antibacterial action of many food-derived secondary metabolites is well documented, like it was done for cinnamaldehyde from cinnamon, thymol and carvacrol from thyme, betulinic acid from ber tree (Ziziphus jujuba), allicin from garlic, curcumin from curcuma, piperine from piper (Gorlenko et al. 2020) or proanthocyanidins from cranberries (Wan et al. 2016), just to mention a few. Some of plant antimicrobial compounds found an application as food preservatives (Hintz et al. 2015). However more importantly, plant-based foods have been recently reported to modulate the composition of human gut microbiota, showing the possibility to supress pathogenic species and support symbiotic ones (Rajoka et al. 2017; Yang et al. 2020). This fact increases the importanceof plant secondary metabolites in fighting external but also internal bacterial infections.
The high antibacterial potential of medicinal plants and plant-based foods is based on the enormous diversity of their constituents (Gorlenko et al. 2020). This diversity is nowadays being successfully studied with the application of metabolomics (Nagana Gowda and Raftery 2017). Metabolomics enables the comprehensive characterization of the set of low-molecular-weight compounds as starting, intermediate, or end products of metabolic transformations in the living organisms (Markley et al. 2017). Since metabolites are directly related to biochemical, physiological, and pathophysiological processes, they reflect the activity of the genes at a particular time, in a given environment. Hence metabolomics is complementary to the other global omics platforms like genomics, transcriptomics or proteomics (Mirsaeidi et al. 2015). The metabolomic analysis captures the metabolome in the single time point while its subsection—fluxomics further tracks metabolites containing stable isotope-labelled elements and depicts the metabolic transformations in full (Mirsaeidi et al. 2015). Metabolomics supported by chemometrics and bioinformatics provide the insight into changes occurring the living organism under given conditions.
The most commonly applied analytical techniques used in metabolomics are: mass spectrometry (LC–MS and GC–MS) and nuclear magnetic resonance (NMR). Regardless of the technique used, metabolomic workflow requires spectral processing with features detection, normalization and deconvolution and consequent multivariate data analysis (Alonso et al. 2015). Bioinformatic calculations include statistical analysis, but also pathway and network analysis, which depicts more complicated correlations within metabolite sets (Gardinassi et al. 2017). All these bioinformatic manipulations on metabolomic data and possible integration with other OMICS data will be discussed in details in this review.
Metabolomics covers a number of topics in a basic and applied research. It is used in the food production, for the evaluation of plant response to environmental conditions and stress factors or for profiling of crop varieties. Afterwards, it is utilized to monitor food processing, quality, safety and microbiology. It also provides biomarkers of nutraceutical and functional effects of food. Metabolomics is suitable to explore food, especially a plant-based food, for the presence of pharmacologically active secondary metabolites (Adebo et al. 2017; Silva et al. 2019). When combined with bioactivity studies, particularly with bioactivity-guided isolation, it enables fast tracking and identification of molecules with desired biological potential (Sebak et al. 2019).
Plant metabolome serves as a countless source of phytochemicals providing various benefits for humans, however the ability of plant secondary metabolites to inhibit the growth of microorganisms is evolutionary strategy (Baldwin 2010). Plants interact with microbes using different molecules, which can promote the growth of synergistic microorganisms and inhibit the opportunistic ones (Berg et al. 2017). This natural mechanism makes plants a source of antimicrobial compounds, which influence the bacterial biochemical processes in different manners. Bacteria are susceptible to external stimuli and have to respond to a wide range of environmental factors to survive. By sensing these changes and initiating metabolic regulation they are able to maintain the cell system homeostasis (Chauhan.et al. 2016). The metabolic regulation usually starts at transcriptional level, involving interactions between transcriptional factors and their target genes (Drapal et al. 2014; Shimizu 2013). The subsequent up/down-regulation of activity of enzymes results in adjusted production of corresponding molecules, which are the final products reflecting the metabolic state of the cell (Markley et al. 2017). In comparison to genomics, transcriptomics and proteomics, metabolomics gives more complete insight into the direct, indirect, and secondary responses of bacteria to different stress factors challenges (Mack et al. 2018). Metabolomics enables to study the metabolic perturbations taking place in bacterial cells under the influence of plant secondary metabolites. It reveals the changes in metabolic pathways reflecting the reprogramming of the biochemical networks within bacteria (Zampieri et al. 2017a). From this point of view, metabolomics is valuable in understanding altered cell functions when bacteria are subjected to metabolic stress caused by secondary metabolites inhibiting their growth. To date, metabolomic studies have explored at length the bacterial response to changing environmental conditions (carbon source, pH, starvation) (Drapal et al. 2014), the mechanisms of action of known antibiotics (Hoerr et al. 2016; Koen et al. 2018; Schelli et al. 2017; Vincent et al. 2016; Zampieri 2017a) or bacterial resistance mechanisms (Mack et al. 2018; Nandakumar et al. 2014; Zampieri et al. 2017b), however the application of metabolomics in revealing bacteria response to plant derived natural products still needs more attention and detailed research.
Therefore, herein we attempt to focus on metabolomics as a way to select antimicrobial plant-based bioactive secondary metabolites and to understand their mode of action. This involves the usefulness of metabolomics in I) screening for antimicrobials from plants, II) understanding the mode of action of antimicrobial plant-based bioactive secondary metabolites, and III) revealing the mechanisms of bacterial adaptation and resistance.
Screening for antimicrobials—Identification of antimicrobial molecules in complex plant mixtures
Basic chemometrics for the identification of antimicrobial molecules
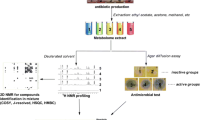
One of the significant advantages of metabolomics is the possibility to screen mixtures of plant secondary metabolites, like crude extracts or essential oils, for the presence of active molecules/biomarkers without fractionation step(s) (Bittencourt et al. 2015; Ebrahimabadi et al. 2016; dos Santosa et al. 2018; Maree et al. 2014; de Oliveira Dembogurski et al. 2018). This can be achieved through the combination of metabolomic analysis (normally non-targeted at this stage) and multivariate data analysis (Fig. 1). For this kind of experiments two data sets are required: set of metabolites present in the studied samples and the results of activity assay. The correlation of these data sets provides the putative biomarkers, eventually responsible for the activity. In such case, chemometrics enables the elucidation of patterns in complicated chemical matrices. It shows possible similarities or dissimilarities among samples, which can result in the presence of clusters. Such a characteristics of explored data sets helps to discover relevant variables and discard not significant features (Biancolillo and Marini 2018). The exploratory data analysis aims to reduce the data dimensionality via projection of multidimensional space (whose axes are the variables) to low-dimensional representation of the data. For this purpose, the principal components analysis (PCA) is commonly used. It provides the information about the distribution of the samples into PCA subspace based on the principal components and allows to evaluate the relative contribution of the experimental variable to the definition of the principal components (Kumar et al. 2014). Multivariate data analysis applied in metabolomic studies includes PCA, but also several methods derived from PCA, like partial least squares discriminant analysis (PLS-DA), orthogonal projections to latent structures discriminant analysis (OPLS-DA), or principal component regression (PCR). All these techniques aim to predict a set of response variables based on the relationship between a descriptor matrix X and a response matrix Y (Worley and Powers 2016). In the comparative studies, when more than one factor can influence the evaluated biomarkers PCR may be not sufficient to build a useful statistical model. Hence in that case a supervised PLS-DA should be applied, because it allows to determine the most discriminant metabolites and to predict group membership. Although PLS-DA is considered to be superior over PCA and PCR, it’s application is limited in complex mixtures when data is very variable and the correlation with the membership to the test group is not always clear. As a solution in that case, OPLS-DA model was proposed, because it enables to eliminate the variation from matrix X that does not correspond to Y (dos Santos et al. 2018; Worley and Powers 2016).

Selection of antibacterial plant secondary metabolites
All three subtypes of PCA analysis are used in screening of crude plant extracts, essential oils and food-related products for the presence of antimicrobial secondary metabolites (Bittencourt et al. 2015; Ebrahimabadi et al. 2016; dos Santosa et al. 2018; Maree et al. 2014; de Oliveira Dembogurski et al. 2018). The examples discussed below refer to essential oils. Ebrahimabadi et al. (2016) showed that PCR, PLS-DA and OPLS-DA methods result in approximately similar plots indicating their equal abilities for data analysis and communication between predictor matrixes and response vectors, however OPLS-DA appeared to be the best choice because of its facility, repeatability and lower time-consuming. Nevertheless, the data pre-treatment method has an influence on the results and level scaling is supposedly better for active principles identification (dos Santos et al. 2018).
The chemometrics was most frequently combined with GC–MS in order to reveal the antimicrobial constituents within essential oils, obtained from aromatic plants. The application of PCA performed to identify and evaluate groupings, followed by OPLS-DA analysis enabled to identify eugenol to be responsible for the high antimicrobial activity against Bacillus cereus, Staphylococcus aureus and Enterococcus faecalis (Gram-positive), Escherichia coli and Pseudomonas aeruginosa (Gram-negative) as well as Candida albicans. Eugenol was detected in all samples, demonstrating minimal inhibitory concentration (MIC) value below 2 mg/mL. On the contrary, high concentration of α-pinene, limonene and sabinene in the studied commercial essential oils appeared negatively correlated with antimicrobial activity, hence demonstrating its poor effect against B. cereus, S. aureus, E. coli and E. faecalis (Maree et al. 2014). Similarly, the principal component regression (PCR), PLS and OPLS were adopted in order to identify the active antimicrobial constituents in Myrtus communis essential oil obtained from different locations. The regression coefficients for studied microorganisms obtained by PCR, PLS and OPLS plotted with the GC chromatograms of myrtle samples revealed that α-pinene and 1,8-cineol which consisted more than 75% of the essential oil samples, contributed the most to the activity against C. albicans, Shigella dysanteriae and Klebsiella pneumonia. The two additional compounds strongly correlated with activity were β-pinene and limonene. Although, these two consisted only nearly 2% of the essential oil samples their regression coefficient plots negative peaks obtained by PCR, PLS and OPLS suggest their considerable contribution in antimicrobial activity of essential oil samples. What is more, the better correlation was obtained for α-pinene peaks in case of C. albicans and S. dysanteriae compared to K. pneumonia, suggesting its higher antimicrobial activities against these microbial strains (Ebrahimabadi et al. 2016). Interestingly, the activity of α-pinene and limonene against C. albicans was described as strong (Ebrahimabadi et al. 2016) or poor (Maree et al. 2014) indicating that any synergistic effect reflecting the superior activity of compounds acting together should not be unnoticed. Therefore, besides the identification of active principles, metabolomics combined with i.e., decision tree analysis reveal the possible interactions between different constituents in the mixture. dos Santos et al. (2018), for instance, observed such interactions among palmitic acid, bornyl acetate and 4-terpineol in the samples of essential oils tested against Streptococcus mutans. Essential oils were described as very active if they contained palmitic acid at higher concentration, moderately active when bornyl acetate without palmitic acid was part of the composition and further inactive when 4-terpineol was present. Hence, the applied decision tree classifier suggested possibility of synergism or antagonism between different constituents. In that sense, metabolomics combined with chemometrics efficiently allows to observe the mutual interactions among compounds in the mixture. The weak positive correlation between chemical composition and antimicrobial activity may surprisingly suggest nonlinear concentration-dependent inhibition of the microorganism, which could be explained by the ability of the identified bioactive secondary metabolites to act synergistically (Bittencourt et al. 2015).
Another advantage of metabolomics combined with chemometrics is the power to predict the resultant activity of the mixture. It can be obtained by prior analysis of range of samples of interest which are characterized by the presence of chemically similar secondary metabolites. When the level of activity is assigned to identified chemical biomarkers, statistical prediction can be used to forecast the antimicrobial activity of a new mixture that was not previously included in the model. By analysing the contribution of particular constituents to the new mixture, and correlating them with assigned level of activity, the probability of level of activity can be described. Such an approach was applied and validated by dos Santos et al. (2018), who used chemometric methods to predict the antimicrobial activity of Aldama arenaria essential oil. The model predicted 41% of probability for the essential oil to have very good activity, 30% of probability to be moderately active, 19% of chance to be weakly active and 10% of chance to have no activity. The experimental verification appeared in agreement with the statistical prediction with the MIC value in very active classification (dos Santos et al. 2018). This kind of studies undoubtedly contribute to the development of useful strategies leading to the identification of antimicrobial molecules in the complex mixtures obtained from plants.
Molecular networking for selection of antimicrobial targets and for the identification of antimicrobial molecules
The basic concept of molecular network includes the relationships between genes, proteins or metabolites expressed as nodes. This relationships are described as edges, which can be direct or indirect interactions, such as transcription regulation for genes, physical interactions for proteins, or chemical similarity for metabolites. More complex networks can be also generated in system biology, like gene-proteins or gene-metabolites relationships, which reflects the working machinery of the cell/organism (Grennan et al. 2014). The use of metabolic network analysis for selection of antimicrobial targets was conceptualized by Wang et al (2013). They suggested the cellular metabolite concentration as a criterion for selection of enzymatic targets in antibacterial discovery. The targets working with low substrate concentrations (< 0.5 mM) should be favourable drugs hits, because high concentration of antimicrobial can effectively compete with low amount of the substrate. In other words, the solubility of enzyme inhibitor should be is at least 100 times higher than the concentrations of the competed substrate (Wang et al. 2013). The molecular networking was proposed as a tool to predict cellular concentrations of metabolites. The evaluation of the chemical basis of metabolic network organization showed that bacterial metabolite concentrations correlated with metabolic network topology and metabolite chemical properties. Based on the observation that high-concentrated metabolites have higher degrees of connectivity authors hypothesized that using topological and chemical properties of metabolites it is possible to predict their cellular concentrations (Wang et al. 2013).
The other application of molecular networking is screening of antimicrobial constituents within crude extracts (Fig. 1). This approach allows for the identification of the variety of unknown natural products with potential antimicrobial activity even from plant species that have already been extensively studied (Quinn et al. 2017). Metabolite molecular networking is a tool based on a tandem mass spectrometry (MS/MS) analysis. It enables data organization according to the MS/MS fragmentation patterns of molecules, assuming that chemically related molecules are likely to display similar fragment ion spectra. In other words, the analysis can recognize sets of spectra from related molecules even when the spectra themselves are not matched to any known secondary metabolites (Allard et al. 2017; Raheem et al. 2019; Quinn et al. 2017; Wang et al. 2016). It results in detection and visualization of clusters of metabolites as a relational spectral network (Allard et al. 2017). Molecular networking functions within a Global Natural Products Social Molecular Networking (GNPS), which is a public online platform—the growing repository of MS/MS data enabling the annotation of compounds present in plant extracts or microbial cultures and bring about the generation of spectral connections among them (Quinn et al. 2017; Wang et al. 2016).
The identification of new antimicrobial secondary metabolites within crude mixtures through molecular networking is complementary to the matching of the m/z peaks with available databases and dereplication of the data (Raheem.et al. 2019). In the classical approach, the list of known and unknown metabolites ranked according to their peak intensity is created and subjected to discriminative analysis, which performed on active versus inactive extracts allows to specify structures that are likely to mediate the bioactivity. In case of molecular networking the data can be further processed in two ways. First is to identify putative antimicrobial biomarkers from extracts and to use spectral data of these biomarkers for analysis at GNPS Molecular Networking website in order to find the similar fragmentation patterns with other molecules not necessarily present in the studied extracts, but which also might contribute to the activity (Lee et al. 2019). The second way is to create molecular network for all secondary metabolites present in the extracts and then statistically predict the putatively identified clusters for their bioactivity according to their presence in the active extracts and absence in the inactive extracts. This approach is not dependent on the concentration of the individual metabolites in the respective extracts and as a result even compounds present in very low concentration can be indicated as highly potent (Raheem et al. 2019).
The limitation of molecular networking is the possibility of designation of putatively active unidentified structures for which only precursor ions and mass spectra are known. In that case the molecular formula prediction is possible (Raheem et al. 2019), though only targeted isolation can result in undoubtful identification. However, because the analysis is not dependent on the concentration of the individual constituents in the samples, the isolation is still not always feasible, since some constituents are present in minor in the initial plant material.
Another limitation of basic chemometrics and molecular networking is the fact that the correlation of chemical composition of the crude mixture with its antimicrobial activity is not sufficient to explain the mechanism of action. For complete understanding of the mode of action of plant secondary metabolites the analysis of bacterial metabolome under the influence of these compounds is hence required.
Determination of antibacterial modes of action
Understanding the mechanism of action of natural (also applicable to synthetic) antimicrobial compounds is essential for their full consideration as antibacterial agents or future drug leads. The research on this field was previously done mainly through experimental bacteriology, involving whole-genome sequencing, transcriptome sequencing (RNA-seq), microarray analysis, two-dimensional protein gel electrophoresis, and gene knockout and overexpression studies (Hong et al. 2016). The drug targets were identified through tools enabling generation of knockout mutant strains not able to produce or producing inactive target protein (Tuyiringire et al. 2018). The mutations were usually induced by exposure to the increasing concentrations of antibacterial. In the next step, the overexpression of the mutation was proven to be responsible for the resistance (Awasthi et al. 2017). This “one target” oriented approach is, however, very laborious and usually based on the previous results of the cell assays, which give the premise that particular protein can be a hit. However, it is likely that this strategy misses the targets other than selected proteins or even other than proteins. The recent research already indicated that antibacterial natural products can influence bacterial cells on different metabolic and functional levels (Božik et al. 2018; Chen et al. 2020; Liu et al. 2020; Mack et al. 2018; Senizza et al. 2020). For that reason, untargeted observations of changes taking place in bacterial cells under the influence of compounds, inhibiting their growth give a better chance for identification of target points (Zampierii et al. 2018) (Fig. 2). Metabolomics perfectly applies in this case. It detects a large number of measurable analytes in a sample and reflects the complexity of metabolic changes caused by causative agent. The application of untargeted mode is preferred since measurements of selected predefined analytes (targeted strategy) give only very narrow view on the metabolomics perturbations as it is difficult to correlate limited sets of metabolites and specific physiological states (Senizza et al. 2020).

Understanding of the mode of action of plant secondary metabolites
Bacterial metabolome consists of numerous and diverse metabolites. Majority of these metabolites are placed inside the cells and when characterized gives a fingerprint. The smaller number of metabolites is transported outside the cell membrane and results in a footprint (Rácz et al. 2018; Pinu and Villas-Boas 2017). Fingerprint reflects the biochemical state of the cell in a given moment and can be related to all metabolic processes ongoing within bacteria and needed to adapt and survive (Rácz et al. 2018). Extracellular metabolites play important role in the formation of cell wall, biofilm and quorum sensing (Pinu and Villas-Boas 2017). Metabolomic intracellular fingerprinting and extracellular footprinting of the treated bacteria can provide complementary and classifying information about the mechanisms of action of antibacterial molecules. Both sets of metabolites are affected in a distinct manner when bacteria are under the influence of plant secondary metabolites, having intracellular or extracellular targets such as protein synthesis, DNA and cell wall (Hoerr et al. 2016). Treatment with antibiotics disturbing intracellular targets results in modes of action specific fingerprints. The footprints could be differentiated from controls, but with much smaller magnitude and significance of the separation than that for the cell wall affecting compounds. The opposite situation takes place when bacteria are influenced with antibiotics targeting the call wall, the fingerprints are similar to those of control cultures however footprints are significantly different (Hoerr et al. 2016). For that reason, comprehensive metabolomic determination of mechanism of action of plant secondary metabolites should include the analysis of both, intracellular and extracellular metabolites.
The classification strategy
NMR-, GC–MS- and LC–MS-based metabolomics were already successfully applied for the characterization of antibacterial mechanisms of action of various compounds—known and new antibiotics (Hoerr et al. 2016; Koena et al. 2018; Vincent et al. 2016, Zampieri et al. 2017a), uncharacterized antimicrobial compounds (Zampieri et al. 2018), human-targeted pharmacologically diverse drugs (Campos and Zampieri 2019), and also plant secondary metabolites in mixtures or as individual molecules (Božik et al. 2018; Chen et al. 2020; Hossain et al. 2013; Liu et al. 2020; Senizza et al. 2020; Zhao et al. 2020; Zhi et al. 2008). In 2016 Dos Santos and co-workers published a review about the application of omics technologies for evaluation of antibacterial mechanisms of action of plant-derived products. At that time the authors stated that the topic is still largely unexplored with Staphylococcus aureus being the most studied organisms (Dos Santos et al. 2016). Since that review many papers appeared describing the mechanism of antibacterial action of natural products not only against S. aureus, but also against Escherichia coli (Božik et al. 2018), Bifidobacterium breve (Senizza et al. 2020), Listeria monocytogenes (Zhao et al. 2020) or Mycobacterium tuberculosis (Sieniawska et al. 2020).
The simplest approach, enabling the prediction of mechanism of action based on the antibiotic class membership is the comparison of mode of action of unknown molecule to mode of action of well characterized antibiotics or chemotherapeutics (Dos Santos et al. 2016). This strategy can be performed with the application of multivariate data analyses (usually PLS-DA), which groups the molecules in clusters according to their molecular targets. The statistical model is then built to predict the bacterial response to compounds that had not been included in the original training set. This methodology was validated by Hoerr et al. (2016), who correctly predicted the classification of streptomycin, tetracycline and carbenicillin against Escherichia coli K12 BL21 by their fingerprints. First, they described that molecules with intracellular targets influence the levels of alanine, glutamate acetamide as well as of energy-related metabolites such as ethanol, citrate, formate and isobutyrate, whereas antibiotics affecting the cell wall results in decreased levels of tricarboxylic acid energy-related metabolites and increased levels of metabolites derived from anaerobic energy pathways such as formate, acetate, and acetone. Later Hoerr et al. (2016) calculated that with 89% prediction the metabolomic profile of bacteria treated with streptomycin is similar to that of kanamycin. Tetracycline showed 76.4% probability of being in the same cluster that doxycycline and that carbenicillin may target cell wall with probability of 62.1% (Hoerr et al. 2016). The same approach was recently extended to the evaluation of the mode of action of plant-based antibacterial natural products. The cluster and correlation analyses performed by Božik et al. (2018) revealed that essential oils constituents affect the levels of small proteins in E. coli in a more complicated manner than antibiotics, chlorine and peroxide. Differences in protein expression were characteristic for treatments with various volatiles, however some of the changes were shared among different groups of constituents. The guaiacol fingerprint appeared partially similar to that of antibiotic tetracycline. The increased levels of UPF0434 protein involved in biofilm formation and 23S rRNA methylase leader peptide involved in the regulation of the synthesis of the erythromycin resistance protein were detected at very high levels only in the guaiacol-treated sample. The other volatiles: citral, eugenol, geraniol, thymol, carvacrol, cinnamaldehyde, carvomenthenol, carvone and careen induced the expression of DNA-binding protein HU-alpha which stabilizes DNA by wrapping to prevent its denaturation in stress conditions. The characteristic fingerprints provided the evidence that essential oils constituents not only impact bacterial membranes and the cell but also influence biofilm formation, resistance to antibiotics, and ribosomal functionality (Božik et al. 2018). The PCA was also sufficient to compare the antibacterial mode of action of extract of Chinese plant Hemsleya pengxianensis and its main constituent—dihydrocucurbitacin F-25-O-acetate with the known modes of action of nine antibacterial substances. The metabolic profiles of Staphylococcus aureus occurred to be different for antibiotics targeting protein synthesis, RNA polymerase, gyrase and topoisomerase IV, as well as cell wall peptidoglycan. The fingerprint of dihydrocucurbitacin F-25-O-acetate was close to that of vancomycin, which inhibit peptidoglycan formation. What is more, results of PCA analysis indicated that dihydrocucurbitacin F-25-O-acetate play the main antimicrobial role against S. aureus in the studied extract (Zhi et al. 2008).
The classification strategy enables the accurate ranking of tested antimicrobial secondary metabolites to groups with known modes of action, however uncommon or unknown mechanisms of action could not be elucidated in this way.
Pathway analysis
More functional strategy, aiming to reveal new mechanisms of action of plant-based antibacterial secondary metabolites, which does not require comparison with drugs of a known class is pathway analysis. It can be used to determine the differences in the expression of genes, proteins and metabolites, and gives a possibility to reduce the complexity of data and hence provides a system-level view of changes in cellular activity in response to treatments (Ma et al. 2016). In metabolomics, pathway analysis is derived from Metabolite Set Enrichment Analysis, which identify biologically meaningful patterns containing significantly enriched molecules (García-Campos et al. 2015). Metabolites, which expression is significantly changed, are grouped in clusters and related to key cellular signaling and metabolic networks (Xia et al. 2015). The simplest analysis is to group the lists of individual metabolites into smaller sets of related molecules. Such sets can describe biological processes in which individual metabolites are known to be involved in and how they interact with each other (Khatri et al. 2012). However, metabolites can take part in different pathways at the same time, and their roles in the pathways may be different, depending on the cellular context. Therefore, the same pathways can be further related to build the pathway networks composed of metabolites and their connections representing the interactions between molecules (binding, activation or inhibition) (García-Campos et al. 2015). Identifying active pathways that differ between two conditions can be performed through functional enrichment or over-representation analysis (ORA), which identifies over-expressed molecules in relation to the control set. In other words, the analysis lists differentially active pathway members however does not provide any view on the interactions between them. The interactions between biomolecules in the pathway networks can be computed based on the curated biological databases (Ma et al. 2016). This type of analysis, called pathway topology, reveals information about connections between components, which can affect each other in direct/indirect manner. Direct connections result in activation or inhibition of reactions and describes the redirections of the metabolism (García-Campos et al. 2015).
The application of pathway analysis is advantageous to identify the modes of action of molecules which specifically target metabolic pathways (Nandakumar et al. 2014). However, the use of untargeted metabolomics and pathway analysis is helpful in determination of mechanisms of action of antimicrobial natural products in general, because they can indicate metabolic perturbations, which are effects of direct or indirect action of the studied secondary metabolites. Such metabolomic analysis was successfully applied to evaluate the antibacterial activity of the essential oil from the leaves of Cinnamomum camphora. Chen et al. (2020) studied the metabolic profile of methicillin-resistant Staphylococcus aureus (MRSA) under the essential oil treatment and found that seven pathways were enriched by shared differential metabolites. Metabolism of alanine, aspartate and glutamate, sulphur metabolism, arginine and proline metabolism, cysteine and methionine metabolism, aminoacyl-tRNA biosynthesis, cyanoamino acid metabolism and streptomycin biosynthesis were influenced and differed from the control group. The largest metabolic difference was observed for pathways related to cell metabolism, where most of metabolites enriched in the four pathways decreased indicating that essential oil act via disturbing the cell metabolism. Additionally, two upstream metabolites of tricarboxylic acid cycle (citrate and succinate) were downregulated, whereas two downstream metabolites (fumarate and malate) upregulated suggesting disruption of TCA cycle (Chen et al. 2020). The study provided the metabolomic evidence that antibacterial activity of C. camphora against MRSA is not only related to damage of cell membranes, but also disruption of amino acids metabolism. Another recently reported work elucidating the mode of antibacterial action of cinnamon essential oil by means of metabolomics evaluated the response of Mycobacterium tuberculosis H37Ra to Cinnamomum zeylanicum essential oil treatment. The significant alterations in metabolic pathways were observed for tetrahydrofolate biosynthesis, tRNA charging, factor 420 biosynthesis and biotin biosynthesis from 8‐amino‐7‐oxononanoate. The highest significant dysregulation was observed for tetrahydrofolate biosynthesis pathway, resulting in increased production of alpha- and keto-mycolic acids and consequently glucose monomycolates and phthiocerol dimycocerosates, meaning that mycobacteria reorganize their outer membrane as a physical barrier against stress. What is more, upregulated factor 420 biosynthesis pathway suggested that cinnamon essential oil may also contribute to disturbances in bacteria redox homeostasis and detoxification mechanisms (Sieniawska et al. 2020). Pathway analysis appeared also helpful to investigate the antimicrobial effect of nisin and grape seed extract against Listeria monocytogenes. Identified metabolomic markers were involved in total, 22 pathways, 8 of which were regarded as significantly contributing to the antimicrobial action. Disruption was described for alanine, aspartate and glutamate metabolism; aminoacyl-tRNA biosynthesis; valine, leucine and isoleucine biosynthesis; cysteine and methionine metabolism; glutathione metabolism; butanoate metabolism; TCA cycle and pyruvate metabolism leading to the conclusion that antibacterial action of combination of nisin and grape seed extract is mediated through blocking the TCA cycle, amino acid biosynthesis and energy-producing pathway (Zhao et al. 2020). Interestingly, the detected metabolic changes in bacteria exposed to food-derived natural products indicate the disruption of TCA cycle and redox homeostasis. Such observations were correlated with stress response activation and adaptive metabolic reaction, being an indirect effect leading to the bacteria growth inhibition (Mack et al. 2018). The better understanding of the stress/detoxification response was provided for Bifidobacterium breve challenging linoleic acid exposure. The untargeted metabolomics-based approach revealed that redox stress resulted in the downregulation of peroxide scavengers (low molecular-weight thiols, glutathione- and mycothiol-related compounds) and ascorbate precursors, together with the upregulation of oxidized forms of fatty acids suggesting that bacteria reprogrammed metabolism to counteract oxidative stress (Senizza et al. 2020).
As can be seen, the recent metabolomic studies, summarized above, add new knowledge to the modes of action of plant secondary metabolites against bacteria. They reveal that also other than known antibacterial-drugs targets may be involved when bacteria are treated with natural products, however applied methodology does not explore the regulation of bacteria response to stress factors. This can be obtained by more comprehensive gene-metabolite approach.
Integration of transcriptomics, genomics and metabolomics into systems biology
The combination of metabolomics with genomics or/and transcriptomics is a new powerful strategy giving insight into bacterial metabolism and consequently into antibacterial mechanisms of action of plant-based natural molecules (Wanichthanarak et al. 2015). Genomics enables the identification of genes related to different processes needed for bacteria survival and resistance. It indicates potential antibacterial drugs targets, which are checked then through screening platforms based on protein interactions and mutant libraries (Dos Santos et al. 2016). Transcriptomics provides the information about functional elements of the genome and reveals the global gene expression profiles in bacterial cells. It describes the association between genes, transcripts and the phenotype, which is depicted by metabolic state of the cells (Cruickshank-Quinn et al. 2018; Han et al. 2019). The integration of metabolomics and transcriptomics reveals the functional correlations between cellular metabolic perturbations and differentially expressed genes to identify metabolic pathways that are essential in cellular responses to antimicrobial compounds treatment (Han et al. 2019).
A combination of transcriptomics and metabolomics is an emerging strategy to explain molecular mechanisms involved in the antibacterial activity of plant secondary metabolites. Such a systems biology-oriented approach was successfully applied to investigate the mode of action of persimmon tannin against methicillin-resistant Staphylococcus aureus (Liu et al. 2020). Liu et al. (2020) detected 370 genes and 19 metabolites that were differentially expressed under the persimmon tannin treatment and related to osmotic regulation, intracellular pH regulation, amino acid synthesis and metabolism, glycolysis, TCA cycle and iron metabolism. Based on the gene-metabolite network, authors suggested that antibacterial action of persimmon tannin includes cell membrane damage, amino acids limitation, energy metabolism disorder and iron deprivation. The transcriptome and metabolome changes were in agreement and proved that although transcriptional analyses offer an incomplete view of the metabolic changes due to posttranscriptional regulation, metabolomics is supplementary and detects fluctuations which occurs in last step (Liu et al. 2020).
The other interesting approach aiming to investigate the modes of action is the combination of metabolomics and chemogenomics. Such strategy was developed by Ziamperi et al. (2018) to monitor the metabolic response of Mycobacterium smegmatis to new antibacterial compounds. The metabolome profiling strategy was combined with calculations on the genome-scale metabolic model of M. smegmatis. The authors calculated the distance between each enzyme-metabolite pair as the minimum number of reactions connecting these two elements and created the net of similarity and proximity of metabolic changes to drug-target. The enzymes with the highest probability to exhibit local metabolic changes were further correlated with genes, which interestingly showed strong functional dependencies for the drug mode of action in most antibiotic classes. The results of this metabolomics/genomics approach showed that only 8% of the new compounds targeted unconventional cellular processes. More than 70% of studied compounds induced metabolic responses in M. smegmatis characteristic for known modes of action with strong tendency to elicit similar metabolic responses within a drug-group (Ziamperi et al. 2018). The same innovative strategy was adopted to investigate the influence of pharmacologically diverse drugs on metabolic response of Escherichia coli. The mapping of enzymes to drug-induced metabolic changes using a genome-scale metabolic network revealed that carbon metabolism and signalling were the most impacted suggesting new targets of antibacterial compounds (Campos and Zampieri 2019). Although metabolomics/chemogenomics approach focused on synthetic compounds this strategy can be easily applied to determination of the modes of action of food-based secondary metabolites, which presents a vast structural variety and can hit different targets in bacteria cells. With the current availability of genome-scale metabolic models of different bacteria and good accuracy metabolite annotations in untargeted metabolomics this combinational approach can be very effective.
Revealing the mechanisms of resistance
The main tool used to reveal the mechanisms of bacterial resistance was functional metagenomics, which enabled generation of mutants and characterization of resistance genes. However, widely available genomic sequencing made sequence-based approach reliable and accurate strategy to identify antimicrobial resistance. It provided much greater precision in comparison with phenotypic tests, revealed co-carriage of specific genes responsible for multidrug resistance patterns and gave the insight into possibility of genes horizontal transfer, and their distribution by source. It also opened the way to detect the presence of co-resistances not assayed on standard drug panels (Hendriksen et al. 2019). These numerous advantages of genomics in understanding of the mechanisms of bacterial resistance are, however, limited. Genomics does not show the final metabolic state of the cell, which is influenced by posttranscriptional modifications or bacterial signalling cascades, being also important response to external stimuli (Lin et al. 2018). The missing knowledge about bacteria interactions with environment can be successfully described by metabolomics. Metabolomics can enrich the understanding of the mechanisms of bacteria resistance and may contribute to the development of more effective future treatments (Eswara Rao and Kumavath 2017).
From comparative metabolomics to integration of genomics and metabolomics
The simplest approach leading to understanding of the bacterial resistance is comparative metabolomics aiming to describe the changes between metabolites produced by different single- or multi-drug resistant bacteria in relation to sensitive strains. Such strategy for prediction of multi-drug resistant bacteria characteristics and mechanisms of resistance at the metabolite level was applied by Lin et al. (2019), who clearly discriminated drug sensitive and multi-drug resistant strains and indicated some functional metabolites. The authors confirmed that the profile of antibiotic resistance affects the metabolite set produced by bacteria and that biosynthesis of amino acids, biosynthesis of phenylpropanoids and purine metabolism may be related to multi-drug resistance of E. coli to antibiotics (Lin et al. 2019). Besides the changes is small metabolites set, the changes in proteome and phosphoproteome in E. coli were also detected. The later reflected the correlations between signalling and antibiotic susceptibility in bacteria, indicating that changes in signalling precede resistance development (Lin et al. 2018). This proteomics aspect is however out of the scope of this review. Nevertheless, investigation of metabolome level changes resulting from adaptations occurring in antibiotic resistant mutants can reveal production of specific secondary metabolites being the outcome of activation of poorly expressed or silent gene clusters. Likewise, the de-repression of biosynthetic processes in antibiotics resistant bacteria may be a survival strategy as was shown by Derewacz et al. (2013), who observed the upregulation of number of features shared throughout the diverse mutants in antibiotics resistant Nocardiopsis. The majority of these upregulated metabolites were not found in the progenitor strain confirming their role in new cell state. Interestingly, the metabolic changes appearing in resistant bacteria depict not only mechanisms of resistance but also the impact of environmental conditions on the development of resistance. This aspect was examined by Zampieri et al. (2017b) who introduced novel approach correlating changes of metabolite abundances with potential functional flux rearrangements during evolution of antibiotic resistance in E. coli under two different nutritional conditions. The subsequent genome sequence analysis of evolved populations supported the hypothesis that environmental nutrient composition can directly affect the selection of resistance and compensatory mechanisms such as the shift from respiratory to fermentative metabolism of glucose upon overexpression of efflux pumps (Zampieri et al. 2017b).
The complementary approach combining gene transcription and translation quantification data with identification of accumulated metabolites can provide fully comprehensive view on the molecular changes in bacteria resistance mechanisms. Metabolomics can be useful in evaluation of the response of antibiotic resistant bacteria to the plant-based natural products and the prediction of the possibility to restore the bacteria sensitivity to antibiotics. The combinational therapy including antibiotics and natural products may reveal some functional biomarkers indicating new targets in bacteria cells (Mohana et al. 2018).
Biotransformation reactions
The other important aspect related to bacteria response to antimicrobials of different origin and contributing to resistance mechanisms is a biotransformation. Bacteria possess the ability to metabolize xenobiotics through number of metabolic reactions. They are the only living organisms, which can use potentially harmful compounds as their nutrients or to convert them to nontoxic or less toxic products (Arora 2019). The enzymatic modifications of antimicrobials may result in deactivation, or on the contrary, in activation of the drug, like it works in case of pro-drugs (Chakraborty and Rhee 2015). Biotransformation products can be monitored by metabolomics. The anterior approach was based on determination of products of extracellular enzymatic reactions, whereas new one involves elucidation of intrabacterial fate of antibacterials (Awasthi and Freundlich 2017). Awasthi and Freundlich (2017) presented the use of metabolomics alone or in combination with genomics and biochemical assays, as a powerful platform to characterize chemical transformations performed on small molecules by Mycobacterium tuberculosis. The described transformations were categorized as hydrolysis, N–alkylation, amidation, and nitro group reduction, which result in activation or deactivation of the xenobiotic. The important conclusions were drawn, that metabolites of antimicrobials used can also contribute to bacteria growth inhibition and that both, parent and derived molecule may be the subject of mechanism of action (Awasthi and Freundlich 2017). The similar biotransformation reactions can be expected in bacteria under the influence of plant-based bioactive secondary metabolites, therefore the determination of mechanism of action should not be limited to parent compound. Although the identification of products of metabolic transformations of complex plant extracts may be challenging, it is without doubt the important step in the workflow of antimicrobials discovery.
Limitations of metabolomics
Although we described above numerous applications of metabolomics in antimicrobial drug leads finding, this methodology has also its limitations. The majority of research studying response of bacteria to plant secondary metabolites describes in vitro testing, however investigation of the in vivo mechanism of action is crucial for the development of new drugs (Domínguez et al. 2017). There are several reasons, explaining this situation. The first one is the higher uniformity of samples obtained in vitro comparing to in vivo. Both kinds of sampled should capture the true state of the metabolome, but any variation in the procedure may influence the obtained results, and in vitro conditions are much more predictable, and exclude variations between animals (Halouska et al. 2013). The other aspect is the discrimination between bacterial and host metabolites. If the determination of mode of action requires analysis of metabolites produced by both, bacteria and host, it may be challenging to correctly assign the observed changes. Therefore, to overcome this limitation metabolomics has to be supported by other complementary “OMICS” to study in vivo activity of antimicrobials.
Apart from the problem of in vitro/in vivo testing, metabolomics in some cases might be not sufficient to give the satisfactory answer about the action of antimicrobials. For example, basic chemometrics and molecular networking helpful in making correlations between chemical composition of the crude plant mixtures and its antimicrobial activity do not explain their mechanism of action. These methodologies tentatively identify the active compounds, however for the understanding of their influence on bacteria we still need targeted isolation and undoubtful identification, followed by monitoring of bacteria metabolome under the influence of active compounds (Allard et al. 2017). What is more, the metabolomic classification strategy does not allow to describe unknown mechanisms of action (Dos Santos et al. 2016). Similarly, pathway analysis applied alone does not explore the regulation of bacteria response to stress factors, what can be obtained in more comprehensive gene-metabolite approach (Domínguez et al. 2017).
The other issue which should be challenged in the future is the limited number of bacterial model organisms widely available for metabolic changes evaluation. The most studied models include: Escherichia coli, Bacillus subtilis, Pseudomonas putida, Staphylococcus aureus N315 (MRSA/VSSA), Thermotoga maritima, Synechococcus elongatus PCC7942, Mesorhizobium japonicum MAFF 303,099, Klebsiella pneumoniae MGH 78,578 (serotype K52), Klebsiella variicola At-22, and Streptococcus pyogenes M1 476 (serotype M1)(Pang et al. 2020), however in some cases it is not enough, and data have to be analyzed on the phylogenetically closest organisms. Of course it is not possible to identify all metabolites in all sample types, what is considered as weaknesses of metabolomics science, however mapping metabolites onto metabolic pathways for many model organisms is necessary (Edison et al. 2016).
Conclusions and perspectives
Metabolomics finds a number of new applications in searching new antibacterial leads. One of them is the utilization of metabolomics to select antimicrobial plant secondary metabolites and to understand their antimicrobial action (Fig. 3). The literature data, summarized above, confirm that recent metabolomic studies add new knowledge to the modes of action of plant-based secondary metabolites against bacteria. They reveal that also other than known antibacterial-drugs targets may be involved when bacteria are treated with natural products. Metabolomics can enrich the understanding of the mechanisms of bacteria resistance and may contribute to the development of more effective future treatments, including the prediction of the possibility to restore the bacterial sensitivity to antibiotics. The combinational therapy, including antibiotics and natural products, may reveal some functional biomarkers indicating new targets in bacteria cells. However, metabolomics does not explore the regulation of bacteria response to stress factors. This can be obtained by more comprehensive gene-metabolite approach, combining gene transcription and translation quantification data with identification of accumulated metabolites. Future research on this field should explore possibilities of the integration of transcriptomics, genomics and metabolomics. The constantly developing bioinformatics tools, supported by growing gene and metabolite databases, provide a powerful tools to monitor bacterial metabolism on different levels. Correlation of transcriptomics and metabolomics through the pathway analysis gives further possibilities to indicate new enzymatic targets in bacterial cells, which should be experimentally validated in the next step. Metabolomics along with other “omics” approaches shed a light on the bacterial response to plant secondary metabolites treatment, which influence not only pathogenic bacteria but also human microbiota. The impact of the plant-based food on intestinal bacteria is positively correlated with some health benefits and with the application of “omics”, especially metabolomics, it can be further successfully explored. Therefore, the likely future metabolomics development should include the integrative, comprehensive approach to study food and plant-based secondary metabolites and their impact on human health.

Utilization of metabolomics in antibacterial plant-based leads discovery
References
Adebo OA, Njobeh PB, Adebiyi JA, Gbashi S, Kayitesi E (2017) Food metabolomics: a new frontier in food analysis and its application to understanding fermented foods. In: Chavarri Hueda M (ed) Functional food—improve health through adequate food. IntechOpen Limited, London
Allard PM, Genta-Jouve G, Wolfender J-L (2017) Deep metabolome annotation in natural products research: towards a virtuous cycle in metabolite Identification. Curr Opin Chem Biol 36:40–49
Alonso A, Marsal S, Julià A (2015) Analytical methods in untargeted metabolomics: state of the art in 2015. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2015.00023
Anand U, Jacobo-Herrera N, Altemimi A, Lakhssassi N (2019) A comprehensive review on medicinal plants as antimicrobial therapeutics: potential avenues of biocompatible drug discovery. Metabolites 9(11):258. https://doi.org/10.3390/metabo9110258
Arora PK (2019) Microbial metabolism of xenobiotic compounds. Springer Nature Singapore Pte Ltd, Singapore
Awasthi D, Freundlich JS (2017) Antimycobacterial metabolism: illuminating mycobacterium tuberculosis biology and drug discovery. Trends Microbiol 25:756–767
Baldwin IT (2010) Plant volatiles. Current Biol. https://doi.org/10.1016/j.cub.2010.02.052
Berg G, Köberl M, Rybakova D, Müller H, Grosch R, Smalla K (2017) Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol Ecology. https://doi.org/10.1093/femsec/fix050
Biancolillo A, Marini F (2018) Chemometric methods for spectroscopy-based pharmaceutical analysis. Front Chem. https://doi.org/10.3389/fchem.2018.00576
Bittencourt MLF, Ribeiro PR, Franco RLP, Hilhorst HWM, Castro RD, Fernandez LG (2015) Metabolite profiling, antioxidant and antibacterial activities of brazilian propolis: use of correlation and multivariate analyses to identify potential bioactive compounds. Food Res Int 76:449–457
Božik M, Cejnar P, Šašková M, Nový P, Maršík P, Klouček P (2018) Stress response of Escherichia coli to essential oil components—insights on low-molecular-weight proteins from MALDI-TOF. Sci Rep. https://doi.org/10.1038/s41598-018-31255-2
Campos AI, Zampieri M (2019) Metabolomics-driven exploration of the chemical drug space to predict combination antimicrobial therapies. Mol Cell 74:1291–1303
Chakraborty S, Rhee KY (2015) Tuberculosis drug development: history and evolution of the mechanism–based paradigm. Cold Spring Harbor Perspect Med. https://doi.org/10.1101/cshperspect.a021147
Chauhan R, Ravi J, Datta P, Chen T, Schnappinger D, Bassler KE, Balazsi G, Gennaro ML (2016) Reconstruction and topological characterization of the sigma factor regulatory network of Mycobacterium tuberculosis. Nat Commun 7:11062. https://doi.org/10.1038/ncomms11062
Chen J, Tang C, Zhang R, Ye S, Zhao Z, Huang Y, Xu X, Lan W, Yang D (2020) Metabolomics analysis to evaluate the antibacterial activity of the essential oil from the leaves of Cinnamomum camphora (Linn.) Presl. J Ethnopharmacol. https://doi.org/10.1016/j.jep.2020.112652
Cruickshank-Quinn CI, Jacobson S, Hughes G, Powell RL, Petrache I, Kechris K, Bowler R, Reisdorph N (2018) Metabolomics and transcriptomics pathway approach reveals outcome-specific perturbations in COPD. Sci Rep. https://doi.org/10.1038/s41598-018-35372-w
de Oliveira Dembogurski DS, da Silva TD, Boaretto AG, Rigo GV, da Silva RC, Tasca T, Macedo AJ, Carollo CA, Silva DB (2018) Brown propolis-metabolomic innovative approach to determine compounds capable of killing Staphylococcus aureus biofilm and Trichomonas vaginalis. Food Res Int 111:661–673
Derewacz DK, Goodwin CR, McNees CR, McLean JA, Bachmann BO (2013) Antimicrobial drug resistance affects broad changes in metabolomic phenotype in addition to secondary metabolism. PNAS 110:2336–2341
Domínguez Á, Muñoz E, López MC, Cordero M, Martínez JP, Viñas M (2017) Transcriptomics as a tool to discover new antibacterial targets. Biotechnol Lett 39:819–828
Dos Santos BS, Silva LCN, Silva TD, Rodrigues JFS, Grisotto MAG, Correia MTS, Napoleão TH, Silva MV, Paiva PMG (2016) Application of omics technologies for evaluation of antibacterial mechanisms of action of plant-derived products. Front Microbiol. https://doi.org/10.3389/fmicb.2016.01466
dos Santos FA, Sousa IP, Furtado NAJC, Da Costa FB (2018) Combined OPLS-DA and decision tree as a strategy to identify antimicrobial biomarkers of volatile oils analyzed by gas chromatography–mass spectrometry. Rev Bras 28:647–653
Drapal M, Perez-Fons L, Wheeler PR, Fraser PD (2014) The application of metabolite profiling to Mycobacterium spp.: determination of metabolite changes associated with growth. J Microbiol Methods 106:23–32
Ebrahimabadi EH, Ghoreishi SM, Masoum S, Ebrahimabadi AH (2016) Combination of GC/FID/mass spectrometry fingerprints and multivariate calibration techniques for recognition of antimicrobial constituents of Myrtus communis L. essential oil. J Chromatogr B 1008:50–57
Edison AS, Hall RD, Junot C, Karp PD, Kurland IJ, Mistrik R, Reed LK, Saito K, Salek RM, Steinbeck C, Sumner LW, Viant MR (2016) The time is right to focus on model organism metabolomes. Metabolites. https://doi.org/10.3390/metabo6010008
Eswara Rao T, Kumavath R (2017) Bacterial metabolomics reveals the evolution of antibiotic resistance. J Syst Biol Proteome Res 1:1–2
García-Campos MA, Espinal-Enríquez J, Hernández-Lemus E (2015) Pathway analysis: state of the art. Front Physiol. https://doi.org/10.3389/fphys.2015.00383
Gardinassi LG, Xia J, Safo S, Li S (2017) Bioinformatics tools for the Interpretation of metabolomics data. Current Pharmacol Rep 3:374–383
Gorlenko CL, Kiselev HY, Budanova EV, Zamyatnin AA Jr, Ikryannikova LN (2020) Plant secondary metabolites in the battle of drugs and drug-resistant bacteria: new heroes or worse clones of antibiotics? Antibiotics. https://doi.org/10.3390/antibiotics9040170
Grennan KS, Chen C, Gershon ES, Liu C (2014) Molecular network analysis enhances understanding of the biology of mental disorders. BioEssays 36:606–616
Halouska S, Zhang B, Gaupp R, Lei S, Snell E, Fenton RJ, Barletta RG, Somerville GA, Powers R (2013) Revisiting protocols for the NMR analysis of bacterial metabolomes. J Integr OMICS 3:120–137
Han ML, Zhu Y, Creek DJ, Lin YW, Gutu AD, Hertzog P, Purcell T, Shen HH, Moskowitz SM, Velkov T, Li J (2019) Comparative metabolomics and transcriptomics reveal multiple pathways associated with polymyxin killing in Pseudomonas aeruginosa. Msystems. https://doi.org/10.1128/mSystems.00149-18
Hendriksen RS, Bortolaia V, Tate H, Tyson GH, Aarestrup FM, McDermott PF (2019) Using genomics to track global antimicrobial resistance. Front Public Health. https://doi.org/10.3389/fpubh.2019.00242
Hintz T, Matthews KK, Di R (2015) The use of plant antimicrobial compounds for food preservation. Biomed Res Int. https://doi.org/10.1155/2015/246264
Hoerr V, Duggan GE, Zbytnuik L, Poon KKH, Große C, Neugebauer U, Methling K, Löffler B, Vogel HJ (2016) Characterization and prediction of the mechanism of action of antibiotics through NMR metabolomics. BMC Microbiol. https://doi.org/10.1186/s12866-016-0696-5
Hong J, Hu J, Ke F (2016) Experimental induction of bacterial resistance to the antimicrobial peptide tachyplesin I and investigation of the resistance mechanisms. Antimicrob Agents Chemother 60:6067–6075
Hossain SM, Bojko B, Pawliszyn J (2013) Automated SPME–GC–MS monitoring of headspace metabolomics responses of E. coli to biologically active components extracted by the coating. Anal Chim Acta 776:41–49
Khatri P, Sirota M, Butte AJ (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1002375
Koen N, van Bred SV, Loots DT (2018) Elucidating the antimicrobial mechanisms of colistin sulfate on Mycobacterium tuberculosis using metabolomics. Tuberculosis 111:14–19
Kumar N, Bansal A, Sarma GS, Rawal RK (2014) Chemometrics tools used in analytical chemistry: an overview. Talanta 123:186–199
Lee MS, Yang YL, Wu CY, Chen YL, Lee CK, Tzean SS, Lee TH (2019) Efficient identification of fungal antimicrobial principles by tandem MS and NMR database. J Food Drug Anal 27:860–868
Lin MH, Potel CM, Tehrani KHME, Heck AJR, Martin NI, Lemeer S (2018) A new tool to reveal bacterial signaling mechanisms in antibiotic treatment and resistance. Mol Cell Proteomics 17:2496–2507
Lin Y, Li W, Sun L, Lin Z, Jiang Y, Ling Y, Lin X (2019) Comparative metabolomics shows the metabolic profiles fluctuate in multidrug resistant Escherichia coli strains. J Proteom. https://doi.org/10.1016/j.jprot.2019.103468
Liu M, Feng M, Yang K, Cao Y, Zhang J, Xu J, Hernández SH, Wei X, Fan M (2020) Transcriptomic and metabolomic analyses reveal antibacterial mechanism of astringent persimmon tannin against Methicillin-resistant Staphylococcus aureus isolated from pork. Food Chem. https://doi.org/10.1016/j.foodchem.2019.125692
Ma J, Shojaie A, Michailidis G (2016) Network-based pathway enrichment analysis with incomplete network information. Bioinformatics 32:3165–3174
Mack SG, Turner RL, Dwyer DJ (2018) Achieving a predictive understanding of antimicrobial stress physiology through systems biology. Trends Microbiol 26:296–312
Maree J, Kamatou G, Gibbons S, Viljoen A, Van Vuuren S (2014) The application of GC–MS combined with chemometrics for the identification of antimicrobial compounds from selected commercial essential oils. Chemom Intell Lab Syst 130:172–181
Markley JL, Bruschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS (2017) The future of NMR-based metabolomics. Curr Opin Biotechnol 43:34–40
Mirsaeidi M, Banoei MM, Winston BW, Schraufnagel DE (2015) Metabolomics: applications and promise in mycobacterial disease. Ann Am Thorac Soc 12:1278–1287
Mohana NC, Yashavanth HC, Rakshith RD, Mithun PR, Nuthan BR, Satish S (2018) Omics based approach for biodiscovery of microbial natural products in antibiotic resistance era. J Genet Eng Biotechnol 6:1–8
Nagana Gowda GA, Raftery D (2017) Recent advances in NMR-based metabolomics. Anal Chem 3:490–510
Nandakumar M, Nathan C, Rhee KY (2014) Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat Commun. https://doi.org/10.1038/ncomms5306
Pang Z, Chong J, Li S, Xia J (2020) MetaboAnalystR 3.0: toward an optimized workflow for global metabolomics. Metabolites. https://doi.org/10.3390/metabo10050186
Pinu FR, Villas-Boas SG (2017) Extracellular microbial metabolomics: the state of the art. Metabolites. https://doi.org/10.3390/metabo7030043
Quinn RA, Nothias LF, Vining O, Meehan M, Esquenazi E, Dorrestein PC (2017) Molecular networking as a drug discovery, drug metabolism, and precision medicine strategy. Trends Pharmacol Sci 38:143–154
Rácz A, Andrić F, Bajusz D, Héberger K (2018) Binary similarity measures for fingerprint analysis of qualitative metabolomic profiles. Metabolomics. https://doi.org/10.1007/s11306-018-1327-y
Raheem DJ, Tawfike AF, Abdelmohsen UR, Edrada-Ebel RA, Fitzsimmons-Thoss V (2019) Application of metabolomics and molecular networking in investigating the chemical profile and antitrypanosomal activity of British bluebells (Hyacinthoides non-scripta). Sci Rep. https://doi.org/10.1038/s41598-019-38940-w
Rajoka MSR, Shia J, Mehwish HM, Zhu J, Li Q, Shao D, Huang Q, Yang H (2017) Interaction between diet composition and gut microbiota and its impact on gastrointestinal tract health. Food Sci Human Wellness 6:121–130
Sebak M, Saafan AE, AbdelGhani S, Bakeer W, El-Gendy AO, Espriu LC, Duncan K, Edrada-Ebel R (2019) Bioassay- and metabolomics-guided screening of bioactive soil actinomycetes from the ancient city of Ihnasia, Egypt. PLoS One 14(12):e0226959. https://doi.org/10.1371/journal.pone.0226959
Schelli K, Zhong F, Zhu J (2017) Comparative metabolomics revealing Staphylococcus aureus metabolic response to different antibiotics. Microb Biotechnol 10:1764–1774
Senizza A, Rocchetti G, Callegari ML, Lucini L, Morelli L (2020) Linoleic acid induces metabolic stress in the intestinal microorganism Bifidobacterium breve DSM 20213. Sci Rep. https://doi.org/10.1038/s41598-020-62897-w
Shimizu K (2013) Metabolic regulation of a bacterial cell system with emphasis on Escherichia coli metabolism. ISRN Biochem. https://doi.org/10.1155/2013/645983
Sieniawska E, Sawicki R, Golus J, Georgiev MI (2020) Untargetted metabolomic exploration of the Mycobacterium tuberculosis stress response to cinnamon essential oil. Biomolecules. https://doi.org/10.3390/biom10030357
Silva MS, Cordeiro C, Roessner U, Figueiredo A (2019) Metabolomics in crop research—Current and emerging methodologies. Front Plant Sci. https://doi.org/10.3389/fpls.2019.01013
Tuyiringire N, Tusubira D, Munyampundu J-P, Tolo CU, Muvunyi CM, Ogwang PE (2018) Application of metabolomics to drug discovery and understanding the mechanisms of action of medicinal plants with anti-tuberculosis activity. Clin Transl Med 7:1–12
Vincent IM, Ehmann DE, Mills SD, Perros M, Barrett MP (2016) Untargeted metabolomics to ascertain antibiotic modes of action. Antimicrob Agents Chemother 60:2281–2291
Wan KS, Liu CK, Lee WK, Ko MC, Huang CS (2016) Cranberries for preventing recurrent urinary tract infections in uncircumcised boys. Altern Ther Health Med 22:20–23
Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N et al (2016) Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat Biotechnol 34:828–837
Wang ZY, Zhu Q, Zhang HY (2013) Metabolite concentration as a criterion for antibacterial discovery. Curr Comput Aided Drug Des 9:412–416
Wanichthanarak K, Fahrmann JF, Grapov D (2015) Genomic, proteomic, and metabolomic data integration strategies. Biomarker Insights 7:1–6
Worley B, Powers R (2016) PCA as a practical indicator of OPLS-DA model reliability. Current Metabol 4:97–103
Xia J, Sinelnikov IV, Han B, Wishart DS (2015) Metabo Analyst 3.0—Making metabolomics more meaningful. Nucl Acids Res. https://doi.org/10.1093/nar/gkv380
Yang Q, Liang Q, Balakrishnan B, Belobrajdic DP, Feng QJ, Zhang W (2020) Role of dietary nutrients in the modulation of gut microbiota: a narrative review. Nutrients. https://doi.org/10.3390/nu12020381
Zampieri M, Zimmermann M, Claassen M, Sauer U (2017a) Nontargeted metabolomics reveals the multilevel response to antibiotic perturbations. Cell Rep 19:1214–1228
Zampieri M, Enke T, Chubukov V, Ricci V, Piddock L, Sauer U (2017) Metabolic constraints on the evolution of antibiotic resistance. Mole Syst Biol 13:917
Zampieri M, Szappanos B, Buchieri MV, Trauner A, Piazza I, Picotti P, Gagneux S, Borrell S, Gicquel G, Lelievre J, Papp B, Sauer U (2018) High-throughput metabolomic analysis predicts mode of action of uncharacterized antimicrobial compounds. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aal3973
Zhao X, Chen L, Wu J, He Y, Yang H (2020) Elucidating antimicrobial mechanism of nisin and grape seed extract against Listeria monocytogenes in broth and on shrimp through NMR-based metabolomics approach. Int J Food Microbiol. https://doi.org/10.1016/j.ijfoodmicro.2019.108494
Zhi BY, Yan Y, Yi ZL (2008) Investigation of antimicrobial model of Hemsleya pengxianensis W.J. Chang and its main active component by metabolomics technique. J Ethnopharmacol 116:89–95
Funding
MIG acknowledges the financial support of the European’s Union Horizon 2020 research and innovation programme, project PlantaSYST (SGA-CSA No. 739582 under FPA No. 664620).
Author information
Authors and Affiliations
Contributions
ES designed the manuscript, analysed data, and contributed to writing, MIG reviewed the work and obtained funding.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sieniawska, E., Georgiev, M.I. Metabolomics: towards acceleration of antibacterial plant-based leads discovery. Phytochem Rev 21, 765–781 (2022). https://doi.org/10.1007/s11101-021-09762-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-021-09762-4