
Abstract
Natural products chemistry has witnessed many new developments in the last 5 years like extractions with subcritical water and ionic liquids, LC/HRMS and LC/SPE/cryo-NMR, UHPLC, TLC/MS, MS-based preparative HPLC, comprehensive chromatography (GC × GC, LC × LC), high-throughput screening, introduction of monolithic columns, miniaturisation, and automated structure identification. Nevertheless identifying bioactive constituents in complex plant extracts remains a tedious process. The classical approach of bioassay guided fractionation is time-consuming while off-line screening of extracts does not provide information on individual compounds and sometimes suffers from false positives or negatives. One way out of this is by coupling chromatography with chemical or biochemical assays, so called high resolution screening. An example is the development of HPLC on-line assays for antioxidants. By the post-column addition of a relatively stable coloured radical like DPPH• or ABTS•+, radical scavengers are detected as negative peaks because in a reaction coil they reduce the model radical to its reduced, non-coloured form. When combined with LC/DAD/MS and LC/SPE/NMR, reliable identification of active constituents becomes possible without the necessity of ever isolating them in a classical sense. Also for finding leads for new drugs, combining HPLC with biochemical assays is interesting but technically more difficult. Most enzymes do not work at the organic modifier concentrations commonly encountered in RP-HPLC and the reaction time is often longer requiring dilution and lengthy coils respectively. Therefore, new techniques have to be implemented to gain the required sensitivity for on-line enzyme assays. For stable analytes, high temperature LC offers a solution to the organic modifier problem. When enzymes are highly expensive, like those used in the screening for Cytochrome P450 inhibitors, miniaturisation to chip format may offer a way out. Microreactors (chips) are not only useful for miniaturising larger assays but also offer completely new prospects in phytochemical analysis. One such application is in the sample clean-up of acids and bases like alkaloids. In a lay-out of three parallel channels of 100 μm width with the middle one containing organic phase and the two outer ones water of high pH (feed phase) and low pH (trapping phase) such a chip replaces two classical LLE steps but is much faster and requires less solvents and less manpower input.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The search for bioactive constituents from plants, microbial sources and marine sources has been a challenging but at the same time rewarding task which has led to many new drugs and other potent natural products. In its most classical form this is performed by bioassay guided fractionation. Usually in this line of research there is a cooperation between biologists or medical doctors who are responsible for the biochemical or pharmacological testing, and phytochemists or natural product chemists who take care of the extraction, purification and structure elucidation. The isolation of pure plant constituents started about 200 years ago and 150 years ago the term “phytochemistry” was coined (Rochleder 1854). Since then we have come a long way and many new techniques have been introduced, most noticeably HPLC and various powerful spectroscopic techniques making the identification of new compounds ever faster. However not all of these developments are always obvious in recent papers. Many extractions, purifications, separations and bioassays are still slow and labour-intensive. After giving an overview of the current exciting developments in this field, the paper zooms in on three of them. It is hoped that the paper will stimulate modern phytochemists or natural product chemists to implement some of the new techniques in their own labs.
In Table 1 a number of recent developments in natural products analysis have been summarised. In the field of extraction the replacement of organic solvents by water is environmentally benign and thus interesting. At high temperature the dielectric constant of water drops considerably, making water less polar. Of course this methodology is only suitable and useful for thermostable phytochemicals which are not soluble in water at 100°C or lower. The technique requires high pressure vessels (Ibañez et al. 2003). The higher temperatures can also lead to improved extraction and thus higher yields (Lang and Wai 2003). Ionic liquids are special organic non-volatile solvents (liquid salts). They are expensive but on the other hand can be recycled and have special dissolving properties. An example of their application in phytochemistry is the extraction of the antimalarial drug artemisinin (Lapkin et al. 2006). Another powerful development is the large scale introduction of high-resolution MS (HRMS). Fourier Transform MS with 1 ppm or better mass accuracy provides directly the elemental composition of analytes and especially for unknown compounds this can save a lot of time (Angelova et al. 2008; Wang et al. 2009). Developments in NMR continue to take place. Hyphenation with HPLC via an SPE trap and cooling of the measuring coils to ultra low temperatures (so called “cryo probes”) leads to cleaner samples and an increase of the concentration sensitivity with a factor 10 to 100 depending on the HPLC and LC/NMR probe parameters (Exarchou et al. 2003). Additionally the SPE step allows the use of normal non-deuterated HPLC eluents and the recording of NMR spectra in 100% deuterated solvents. Combining LC/SPE with off-line capillary NMR is not trivial but ideally leads to a much improved mass sensitivity allowing the recording of the 2D spectra of as little as 25 μg and 1D spectra of sub μg quantities (Lambert et al. 2007; Glauser et al. 2008). Interestingly Lin et al. (2008) do not use SPE in their LC/UV/MS/NMR system but collect the fractions in 96 well plates. After evaporation the samples are redissolved in 2–4 μL of NMR solvent which is then transferred to the microcoil NMR probe by an immiscible fluorocarbon-segmented flow with little dispersion. In this way they were able to record 1D spectra of 300 ng of paclitaxel and identify several indole alkaloids after injecting 30 μg of a bioactive cyanobacterial extract (Lin et al. 2008).
Ultra high pressure LC (UHPLC) enables the use of sub 2 μm particles and thus very high chromatographic efficiencies (small H value). This leads to two interesting possibilities: either very fast separations (1 min) by using short columns at high flow rates or ultra high resolutions (high plate numbers) by combining two 15 cm columns at a standard flow rate with a flat gradient. An example of the latter can be seen in Fig. 1 which shows a 5 h separation of an extract of wounded Arabidopsis thaliana leaves (Grata et al. 2008). An example of the former is depicted in Fig. 2 which shows a RP-HPLC separation of several corn oil triacylglycerols within 1 min on a 3 cm 1.8 μm C18 column (van der Klift et al. 2008). Another way to obtain very high resolution is 2-dimensional HPLC, also called comprehensive LC or LC × LC. After a slow high-resolution separation in the first dimension, 1 min portions of analytes are rapidly separated in real-time in the second dimension on a short column of different selectivity. While this happens, a new 1 min portion is collected in a loop waiting to be analysed. After proper chemometric treatment more peaks can be observed than with either of the two columns when used alone (Dugo et al. 2004; van der Klift et al. 2008). Multiple detectors can be used including MS. Two-dimensional GC (GC × GC) is even more powerful than LC × LC and is theoretically capable of separating > 10,000 volatile constituents. This technique is well adapted to essential oil analysis where overlap between peaks is frequent (Shellie and Marriott 2003; van Mühlen et al. 2008). Data processing remains a bottleneck. In preparative HPLC, MS-based instead of UV-based fraction collection is being introduced and makes it possible to selectively collect sub mg quantities of the desired metabolite even if larger amounts of undesired compounds coelute (Glauser et al. 2008; Thiocone et al. 2008).

UHPLC profile of a wounded pooled extract of Arabidopsis thaliana on a 1.7 μm C18 column of 300 mm × 2.1 mm. Eluent: gradient from water-acetonitrile (95:5) containing 0.1% formic acid to water-acetonitrile (2:98) containing 0.1% formic acid at 0.2 mL/min. Detection: TOF-MS in negative ionisation mode (Reprinted from J Chromatogr B, vol. 871, Grata E, Boccarda J, Guillarme D, Glauser G, Carrupt P-A, Farmerd EE, Wolfender J-L, Rudaz S, UPLC–TOF-MS for plant metabolomics: a sequential approach for wound marker analysis in Arabidopsis thaliana, 261–270. Copyright (2008), with permission from Elsevier.)

UHPLC profile of corn oil triacylglycerols on a 1.8 μm C18 column of 30 mm × 4.6 mm. Eluent: methanol-methyl tert-butyl ether (70:30) at 3.0 mL/min. Detection: UV 210 nm (Reprinted from J Chromatogr A, vol. 1112, van der Klift EJC, Vivó-Truyols G, Claassen FW, van Holthoon FL, van Beek TA, Comprehensive two-dimensional liquid chromatography with ultraviolet, evaporative light scattering and mass spectrometric detection of triacylglycerols in corn oil, 43–55. Copyright (2008), with permission from Elsevier.)
Thin layer chromatography has been an all-time favourite of phytochemists and is still much used. Direct interfacing of HPTLC with MS has always been difficult but nowadays this becomes possible. A natural products example is the analysis of an HPTLC separation of glycosphingolipids by IR-MALDI-MS (Distler et al. 2008). DESI-MS is a relatively new technique that can also be combined with HPTLC (Takáts et al. 2004; Van Berkel et al. 2005). Additionally DESI-MS allows for the first time, the direct probing of analytes in intact living plants by MS under ambient conditions without any sample preparation. Examples include alkaloids in Datura stramonium roots, Atropa belladonna seeds, and various parts of Conium maculatum and lycopenes in tomato (Takáts et al. 2004; Talaty et al. 2005). Ultra-thin layer chromatography (UTLC) can also be combined with MS. UTLC is performed on 10 μm non-particulate planar stationary phases. Claimed benefits encompass faster development (1–6 min) and less solvent consumption (1–4 mL) at a similar or slightly lower resolution than with HPTLC (Salo 2007; Salo et al. 2007). Although––as far as we know––this technique has not yet been applied to natural products, it seems worthwhile to explore its usefulness in phytochemical analysis.
Monolithic materials as a stationary phase are being introduced as HPLC columns or as a flexible matrix to attach specific groups for affinity chromatography. A claimed advantage of monolithic materials is their lower back pressure at higher flow rates resulting in significant time gain. Maruška and Kornyšova (2006) reviewed their application in phytochemical analysis. Similar to UHPLC phases, either multiple columns can be linked to obtain very high plate numbers for fingerprints of complex mixtures (metabolomics) or very short columns can generate 1 min separations. Because they can be made in situ from many different components and can be easily modified afterwards, especially polymeric monoliths in capillaries are useful for affinity chromatography. An example is the attachment of different sugars to acrylate-based monoliths to selectively capture lectins (Tetala et al. 2007).
Automated identification of natural products has been a long-standing wish and work in this field has been on-going since the 1970s. Problems include variable HPLC retention times, lack of reference substances, poor ionization in MS, variable field strength of NMR spectrometers, impurities in spectra, and lack of proper software to combine all the data sets. Still some of these problems are being solved and building large NMR data sets for certain classes of compounds seems the way to go (Steinbeck 2004; Moco et al. 2006). When metabolites are only present in the low nanogram range, NMR is impossible but even then structure elucidation by GC/MS after derivatisation is possible (van Beek et al. 2005). High throughput screening allows the automated screening of an ever greater number of plant extracts in a short amount of time and is especially important for the lead finding in the pharmaceutical industry (Ausseil et al. 2007). Another powerful new tool for speeding up lead-finding is high resolution screening. This technique together with high temperature HPLC and chip-based sample preparation of crude alkaloid extracts is discussed in greater detail below.
High-resolution screening
In this chapter we will mainly focus on on-line activity screening or as it is also called “high resolution screening”. It involves the coupling of a chromatographic separation with a chemical or biochemical assay. As each separated compound is measured individually, in many cases it allows the rapid pinpointing of active constituents in complex mixtures. This in turn saves a lot of time as not every compound needs to be purified to homogeneity. One can focus exclusively on the active constituents. A well-known example of a biochemical assay coupled to GC is GC/EAD (electroantennographic detection). In this case the antenna of an insect is used as detector. It can detect for instance sex pheromones very sensitively (Roelofs 1984; Drijfhout et al. 2002). Coupling to HPLC is however more common and of wider applicability.
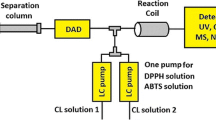
A typical example developed in our group is on-line radical scavenging detection (Dapkevicius et al. 1999; Koleva et al. 2000; Koleva et al. 2001; Dapkevicius et al. 2001; Niederländer et al. 2008). Phenolic antioxidants can terminate radical chain processes in lipid rich foods by reacting with reactive oxygen species (ROS) forming more stable resonance-stabilised radicals or quinone-like structures (Fig. 3). Although they can be used, ROS themselves are not very practical and stable model radicals are easier to work with and lead to more robust on-line systems. The most used model radicals are DPPH• and ABTS•+. In Fig. 4 a scheme of an HPLC on-line radical scavenging assay is presented. In such an assay a relatively stable coloured model radical replaces for instance a peroxyl radical. When an antioxidant is present, mostly a phenolic, the model radical is scavenged (reduced) under the formation of an oxidised antioxidant (e.g. a quinone-like structure as in Fig. 3). As the reduced model radical is not coloured, this leads to disappearance of colour in an off-line assay, on a TLC plate with phenolics when sprayed with model radical or in an on-line HPLC assay as in Fig. 4. Thus with one injection two HPLC profiles are obtained: the “normal” one with positive peaks (e.g. by UV or MS detection) and a “negative” one where negative peaks indicate disappearance of colour and thus the presence of radical scavengers. In this way it is easy to rapidly pinpoint antioxidants in a complex mixture. This saves not only expensive chemicals and time but also avoids possible decomposition of relatively instable antioxidants. An application is the identification of rosmarinic acid and a new caffeic acid trimer in a methanolic extract of thyme leaves (Dapkevicius et al. 2002).

Consecutive radical scavenging of two peroxyl radicals by a diphenol leading first to an intermediate stabilized radical and then to a non-radical ortho-quinone molecule

Lay-out of high-resolution screening assay for radical scavengers in complex mixtures. After the DAD, a pump adds a coloured radical (here: purple DPPH•) which is only reduced to non-coloured DPPHH when radical scavengers elute from the column. Thus a negative peak indicates the presence of a radical scavenger (Reprinted in slightly modified form from J Chromatogr A, vol. 912, Dapkevicius A, van Beek TA, Niederländer HAG, Evaluation and comparison of two improved techniques for the on-line detection of antioxidants in liquid chromatography eluates, 73–82. Copyright (2001), with permission from Elsevier.)
Although high resolution screening speeds up the identification of active constituents as no time is wasted on the purification of inactive constituents, still much time is lost if active constituents need to be isolated for the determination of their structure. Hyphenation with MS and/or NMR is also possible and allows on-line structure elucidation. A nice example is the investigation of a Lonicera extract (Exarchou et al. 2006). For this investigation a two-way valve in between the diode-array detector and the DPPH syringe pump was mounted. In run 1 the valve caused the flow to go to the reaction coil, i.e. exactly as in Fig. 4. This run yielded information on which peaks contain the active radical scavengers (Fig. 5). In run 2 the flow was switched with the flow going to an SPE/NMR unit. Only peak L1 and L2 were measured and for instance no time was spent on studying the main UV-active peak in between L1 and L2 (Fig. 5). After recording of the NMR spectra, the content of the LC/NMR probe was collected and infusion ESI-MS and MS/MS was performed yielding molecular weights and fragmentation patterns. With this information the two peaks could be identified as the known chlorogenic acid and 3′-caffeoyl-chlorogenic acid respectively (Exarchou et al. 2006). In the whole process no preparative separation step occurred. The same technique was used to detect the radical scavengers present in Rhaponticum carthamoides (Miliauskas et al. 2005). A new flavonoid was identified.

HPLC-UV (upper) and on-line DPPH• and ABTS•+ radical scavenging profiles of a crude methanolic extract of Lonicera periclymenum. L1 and L2 were identified on-line as chlorogenic acid and 3′-caffeoyl-chlorogenic acid respectively (Reprinted from J Chromatogr A, vol. 1112, Exarchou V, Fiamegos YC, van Beek TA, Nanos C, Vervoort J. (2006) Chromatographic hyphenated techniques for the rapid screening and identification of antioxidants in traditional pharmaceutical plant extracts, 293–302. Copyright (2006), with permission from Elsevier.)
Thus one can conclude that the on-line detection of radical scavenging antioxidants is a simple and robust tool rapidly providing information about the activity of individual compounds. However proper evaluation of promising extracts and compounds remains necessary (efficacy in real-life food systems, taste, odour, thermal stability, toxicity). The on-line detection of radical scavenging activity should only be considered a screening tool. And finally on-line identification techniques like LC/DAD/MS and LC/UV/SPE/NMR (Exarchou et al. 2005) especially in combination with high-resolution screening have the future. They allow rapid dereplication of known active compounds which is of vital importance for the pharmaceutical industry.
Also more complicated high-resolution assays making use of enzymes exist. Such biochemical assays are of more interest to the pharmaceutical industry as many drugs target enzymes as part of their mode of action (van Elswijk and Irth 2003; Ingkaninan et al. 2000; Schenk et al. 2003; Schobel et al. 2001). Within our group in cooperation with the Department of Toxicology of Wageningen University, we have developed an on-line HPLC system for detection of Cytochrome P450 (CYP450) 1A2 inhibitors. CYP450 1A2 is involved in the bioactivation of various human procarcinogens such as the herbal constituents methyleugenol and estragole. These compounds occur in low concentrations in certain plants like basil and tarragon and thus enter the human food chain via nowadays popular products like pesto. Via a CYP450 mediated pathway these compounds are metabolically activated and can eventually bind covalently to DNA which in turn could lead to cancer (Fig. 6). Therefore, we were interested whether these herbal products also contain other constituents which, through inhibition of CYP450 1A2, could reduce the metabolic activation and thus toxicity (Fig. 6). The classic approach would be to perform bioassay guided fractionation. A potentially faster method is high resolution screening with the set-up as depicted in Fig. 7.

Bioactivation pathway of methyleugenol. ST = sulfotransferases

Scheme of high resolution screening assay for CYP450 1A2 inhibitors (Jeurissen et al. 2007)
A constraint when using enzymes is the fact that most enzymes are not well compatible with organic solvents. As the use of organic solvents in both normal- and reversed phase chromatography is inevitable for most analytes, a solution to reduce the percentage organic solvent in the post-column reaction coil must be found. At concentrations >15%, CYP1A2 activity dropped due to enzyme inhibition and/or denaturation, whereas with concentrations of 5–15% methanol, a substrate dependent decrease in sensitivity of the assay was observed (Jeurissen et al. 2007). Acetonitrile was less well tolerated. As the analytes in plant extracts are unknown, a maximum methanol percentage should be set, e.g. 5%. The simplest way to realise this, is a 15-fold dilution and the use of a C8 instead of C18 column as less methanol is required to elute analytes from a C8 column. The exact configuration is shown in Fig. 7. After a split and 12-fold dilution, the column effluent is mixed with CYP450 1A2, NADPH (co-factor) and a model substrate for CYP450 1A2 (methoxyresorufin). When no inhibitors are present the enzyme converts the model substrate to the fluorescent resorufin which can be detected by its fluorescence. When an inhibitor elutes this will cause a decrease in the formation of resorufin and thus a decrease in fluorescence (negative peak in HPLC profile) similar to the decrease in absorbance with the radical scavenging assay (vide supra). Proof-of-principle was obtained with the set-up shown in Fig. 7 and CYP450 inhibitors were shown to be present in a kava kava and a basil extract. However, further optimization is required before the high resolution screening of herbal extracts for CYP450 inhibitors can be routinely used. The main problems included peak broadening in the reaction coil due to the necessary long residence time of 4 min and poor sensitivity due to the 15-fold dilution step. Downscaling to a 0.15 mm i.d. LC column with a flow of 1 μL/min may solve some of these issues. The mixing and post-column reaction coil can be constructed on a chip (de Boer et al. 2005a). In addition, the much lower total flow will allow higher enzyme concentrations to be used at reduced costs. To further increase the sensitivity capillary laser-induced fluorescence detection can be considered.
High-temperature HPLC
An elegant option to reduce the percentage organic modifier and thus the dilution step is high temperature HPLC. As mentioned before, at higher temperatures water becomes appreciably more non-polar by itself so at temperatures of 150–200°C much less organic modifier is necessary and correspondingly less dilution is required (de Boer et al. 2005b). Of course this demands some adaptations of the technical part and the method only works for natural products which are stable in water at the chosen temperature. The technical changes consist of a larger column oven, a preheater for the mobile phase before the column and a cooler for the mobile phase after the column (Fig. 8). Standard C18 columns cannot be used at such elevated temperatures and they have to be replaced by more rugged but less efficient columns. De Boer et al. showed nevertheless that the technique works and retention times with the eluent methanol–water (10:90) decreased from 24 to 2 min when the temperature was increased from 90 to 205°C (Fig. 9). The high temperature LC did not affect the biochemical high-resolution assay for cathepsin B inhibitors (de Boer et al. 2005b). An overview of the pros and cons of HT-HPLC was presented by Heinisch and Rocca (2009).

Scheme of high resolution screening assay for enzyme inhibitors with high-temperature HPLC. Key: 1, sample introduction; 2, HTLC oven containing the heating coil and column; 3, cooler; 4, superloop with enzyme solution; 5, bioreactor A; 6, superloop with substrate solution; 7, bioreactor B; 8, ESI-MS. The enzyme continuously converts the substrate into products (eq 2) if no bioactive compounds are eluting from the column. Bioactive compounds present in the effluent bind to the enzyme (eq 1) resulting in a decrease of product turnover (eq 3). The reaction products are continuously detected by ESI-MS (Reprinted from Anal Chem, vol. 77, de Boer AR, Alcaide-Hidalgo JM, Krabbe JG, Kolkman J, van Emde Boas CN, Niessen WMA, Lingeman H, Irth H, High-temperature liquid chromatography coupled on-line to a continuous-flow biochemical screening assay with electrospray ionization mass spectrometric detection, 7894–7900. Copyright (2005), with permission from the American Chemical Society.)

Screening of 3 inhibitors (55–65 μmol/L) of cathepsin B at various column temperatures by means of the system depicted in Fig. 8. a–c mass chromatograms of three products d control mass chromatograms; e–g three analytes. Injection volume, 0.5 μL; column, PRP-1; flow rate LC, 300 μL/min water-methanol (90–10) (Reprinted from Anal Chem, vol. 77, de Boer AR, Alcaide-Hidalgo JM, Krabbe JG, Kolkman J, van Emde Boas CN, Niessen WMA, Lingeman H, Irth H, High-temperature liquid chromatography coupled on-line to a continuous-flow biochemical screening assay with electrospray ionization mass spectrometric detection, 7894–7900. Copyright (2005), with permission from the American Chemical Society.)
Miniaturisation of liquid–liquid extraction (LLE)
LLE is one of the oldest and most used sample preparation techniques. It is especially valuable for the isolation of organic acids and bases. Natural bases like alkaloids can be purified to a great extent by two separate LLE steps. When the pH is alkaline the alkaloids are non-polar and dissolve readily in organic solvents leaving behind polar impurities like sugars, salts and amino acids. When the organic layer is subsequently extracted with acidified water the alkaloids are protonated, become water-soluble and partition into the aqueous layer leaving behind non-polar neutral impurities like sterols, waxes and other fats (Fig. 10). Unfortunately this method is labour-intensive, solvent intensive, slow, and if emulsions are formed it becomes even more tedious. Therefore we made an attempt to miniaturise and combine these two separate LLE steps by carrying them out on a microfluidic glass chip. The general three-phase microfluidic chip design is shown in Fig. 11. The three solvents (basic aqueous phase with crude alkaloids, organic phase in the middle, and acidic aqueous phase, all enter the chip on one side) form parallel streams in the core part of the chip. Due to a selective non-polar coating of the middle channel, some small pillars and the curvature of the channels, the three phases remain each in their own channel, i.e. the flows remain laminar. In Fig. 12 a photo of the three flows can be seen. For greater clarity the dye Sudan red has been added to the chloroform phase in the middle channel. After some optimisation of various parameters with strychnine as model compound, an extract of Strychnos seeds was infused into the chip. All three phases were collected and investigated by off-line HPLC. At flow rates of 0.25 μL/min for both basic and acidic aqueous phases and 0.5 μL/min for the organic phase more than 90% of the present strychnine and brucine was extracted in 25 sec into the acidic aqueous phase showing rapid transport in the 3.5 cm chip. The transport is so fast because diffusion is fast at these small distances. According to HPLC, polar impurities remained in the basic aqueous phase (Tetala et al. 2009). Thus proof of principle was obtained that this lab-on-a-chip device can combine two separate LLE steps. In future the wider applicability will be investigated. Most interesting would be hyphenation with an extraction cell, a separation step (nanoLC or CE) or spectroscopic devices (capillary DAD or nanoMS).

General purification scheme for alkaloids (base) from neutral polar and non-polar impurities

Top middle: general lay-out of 3-phase microreactor capable of separating a crude alkaloid fraction into three separate fractions; bottom left: lay-out (side view) with dimensions of the three parallel channels, the solvents in the channels make direct contact; bottom right: real image of 5 × 2 cm glass chip, the holes at the side are for the fluid connections. Five Euro cents coin is for size comparison only

Photo of 3-phase chip showing three laminar parallel flows. The middle chloroform containing channel has been made hydrophobic to maintain a stable phase separation. For better visual clarity (for photo only) the dye Sudan red has been added to the chloroform phase
Conclusion
Phytochemical analysis is still a vibrant field with many new and exciting developments. As highlighted above, high resolution screening, especially when combined with DAD, MS and/or NMR can really lead to not only on-line detection of bioactive compounds but also to their identification without prior isolation. This can speed up the finding of new leads from plants, marine organisms and microbes in pharmaceutical industry. A prerequisite is of course that the bioassay is amenable for post-column application. Other developments focus on improved sample clean-up and especially further hyphenation of often still separate extraction––sample prep––separation––detection steps. Potentially this leads to a huge decrease in necessary time, effort and use of chemicals.
Abbreviations
- ABTS•+ :
-
2, 2′-Azinobis-(3-ethylbenzothiazoline-6-sulfonate) radical cation
- CE:
-
Capillary electrophoresis
- CYP450:
-
Cytochrome P450
- DAD:
-
Diode array detection
- DESI:
-
Desorption electrospray ionisation
- DPPH• :
-
Diphenylpicrylhydrazyl radical
- GC:
-
Gas chromatography
- GC × GC:
-
Two-dimensional gas chromatography
- HPLC:
-
High pressure liquid chromatography
- HPTLC:
-
High performance thin layer chromatography
- HRMS:
-
High resolution mass spectrometry
- HT:
-
High temperature
- LC:
-
Liquid chromatography
- LC × LC:
-
Two-dimensional HPLC
- LLE:
-
Liquid–liquid extraction
- MS:
-
Mass spectrometry
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NMR:
-
Nuclear magnetic resonance
- ROS:
-
Reactive oxygen species
- RP:
-
Reversed phase
- SPE:
-
Solid phase extraction
- UHPLC:
-
Ultra high pressure liquid chromatography
- UTLC:
-
Ultra thin layer chromatography
References
Angelova N, Kong H-W, van der Heijden R, Yang S-Y, Choi YH, Kim HK, Wang M, Hankemeier T, van der Greef J, Xu G, Verpoorte R (2008) Recent methodology in the phytochemical analysis of Ginseng. Phytochem Anal 19:2–16. doi:10.1002/pca.1049
Ausseil F, Samson A, Aussagues Y, Vandenberghe I, Creancier L, Pouny I, Kruczynski A, Massiot G, Bailly C (2007) High-throughput bioluminescence screening of ubiquitin-proteasome pathway inhibitors from chemical and natural sources. J Biomol Screen 12:106–116. doi:10.1177/1087057106296494
Dapkevicius A, van Beek TA, Niederländer HAG, de Groot Æ (1999) On-line detection of antioxidative activity in high-performance liquid chromatography eluates by chemiluminescence. Anal Chem 71:736–740. doi:10.1021/ac9805908
Dapkevicius A, van Beek TA, Niederländer HAG (2001) Evaluation and comparison of two improved techniques for the on-line detection of antioxidants in liquid chromatography eluates. J Chromatogr A 912:73–82. doi:10.1016/S0021-9673(01)00548-9
Dapkevicius A, van Beek TA, Lelyveld GP, van Veldhuizen A, de Groot Æ, Linssen JPH, Venskutonis R (2002) Isolation and structure elucidation of radical scavengers from Thymus vulgaris leaves. J Nat Prod 65:892–896
de Boer AR, Bruyneel B, Krabbe JG, Kolkman J, Lingeman H, Niessen WMA, Irth H (2005a) A microfluidic-based enzymatic assay for bioactivity screening combined with capillary liquid chromatography and mass spectrometry. Lab Chip 5:1286–1292. doi:10.1039/b506559c
de Boer AR, Alcaide-Hidalgo JM, Krabbe JG, Kolkman J, Van Emde Boas CN, Niessen WMA, Lingeman H, Irth H (2005b) High-temperature liquid chromatography coupled on-line to a continuous-flow biochemical screening assay with electrospray ionization mass spectrometric detection. Anal Chem 77:7894–7900. doi:10.1021/ac0510282
Distler U, Hülsewig M, Souady J, Dreisewerd K, Haier J, Senninger N, Friedrich AW, Karch H, Hillenkamp F, Berkenkamp S, Peter-Katalinić J, Müthing J (2008) Matching IR-MALDI-o-TOF mass spectrometry with the TLC overlay binding assay and its clinical application for tracing tumor-associated glycosphingolipids in hepatocellular and pancreatic cancer. Anal Chem 80:1835–1846. doi:10.1021/ac702071x
Drijfhout FP, Groot AT, Posthumus MA, van Beek TA, de Groot Æ (2002) Coupled gas chromatographic-electroantennographic responses of Lygocoris pabulinus (L.) to female and male produced volatiles. Chemoecology 12:113–118. doi:10.1007/s00049-002-8334-0
Dugo P, Favoino O, Luppino R, Dugo G, Mondello L (2004) Comprehensive two-dimensional normal-phase (adsorption)-reversed-phase liquid chromatography. Anal Chem 76:2525–2530. doi:10.1021/ac0352981
Exarchou V, Godejohann M, van Beek TA, Gerothanassis IP, Vervoort JJM (2003) LC-UV-solid-phase extraction-NMR-MS combined with a new cryogenic flow probe and its application to the identification of compounds present in Greek oregano. Anal Chem 75:6288–6294. doi:10.1021/ac0347819
Exarchou V, Krucker M, van Beek TA, Vervoort JJM, Gerothanassis IP, Albert K (2005) LC-NMR coupling technology: recent advancements and applications in natural products analysis. Magn Reson Chem 43:681–687. doi:10.1002/mrc.1632
Exarchou V, Fiamegos YC, van Beek TA, Nanos C, Vervoort J (2006) Chromatographic hyphenated techniques for the rapid screening and identification of antioxidants in traditional pharmaceutical plant extracts. J Chromatogr A 1112:293–302. doi:10.1016/j.chroma.2005.11.077
Glauser G, Guillarme D, Grata E, Boccard J, Thiocone A, Carrupt P-A, Veuthey J-L, Rudaz S, Wolfender J-L (2008) Optimized liquid chromatography–mass spectrometry approach for the isolation of minor stress biomarkers in plant extracts and their identification by capillary nuclear magnetic resonance. J Chromatogr A 1180:90–98. doi:10.1016/j.chroma.2007.12.021
Grata E, Boccarda J, Guillarme D, Glauser G, Carrupt P-A, Farmerd EE, Wolfender J-L, Rudaz S (2008) UPLC–TOF-MS for plant metabolomics: a sequential approach for wound marker analysis in Arabidopsis thaliana. J Chromatogr B 871:261–270. doi:10.1016/j.jchromb.2008.04.021
Heinisch S, Rocca J-L (2009) Sense and nonsense of high-temperature liquid chromatography. J Chromatogr A 1216:642–658. doi:10.1016/j.chroma.2008.11.079
Ibañez E, Kubátová AF, Señoráns J, Cavero S, Reglero G, Hawthorne SB (2003) Subcritical water extraction of antioxidant compounds from rosemary plants. J Agric Food Chem 51:375–382. doi:10.1021/jf025878j
Ingkaninan K, de Best CM, van der Heijden R, Hofte AJP, Karabatak B, Irth H, Tjaden UR, van der Greef J, Verpoorte R (2000) High-performance liquid chromatography with on-line UV, mass spectrometric and biochemical detection for identification of acetylcholinesterase inhibitors from natural products. J Chromatogr A 872:61–73. doi:10.1016/S0021-9673(99)01292-3
Jeurissen SMF, Claassen FW, Havlik J, Bouwmans EE, Cnubben NHP, Sudhölter EJR, Rietjens IMCM, van Beek TA (2007) Development of an on-line high performance liquid chromatography detection system for human cytochrome P450 1A2 inhibitors in extracts of natural products. J Chromatogr A 1141:81–89. doi:10.1016/j.chroma.2006.12.007
Koleva II, Niederländer HAG, van Beek TA (2000) An on-line HPLC method for detection of radical scavenging compounds in complex mixtures. Anal Chem 72:2323–2328. doi:10.1021/ac9912451
Koleva II, Niederländer HAG, van Beek TA (2001) Application of ABTS radical cation for selective on-line detection of radical scavengers in HPLC eluates. Anal Chem 73:3373–3381. doi:10.1021/ac0013610
Lambert M, Wolfender J-L, Stærk D, Christensen SB, Hostettmann K, Jaroszewski JW (2007) Identification of natural products using HPLC-SPE combined with capNMR. Anal Chem 79:727–735. doi:10.1021/ac0616963
Lang Q, Wai CM (2003) Pressurized water extraction (PWE) of terpene trilactones from Ginkgo biloba. Green Chem 5:415–420. doi:10.1039/b300496c
Lapkin AA, Plucinski PK, Cutler M (2006) Comparative assessment of technologies for extraction of artemisinin. J Nat Prod 69:1653–1664. doi:10.1021/np060375j
Lin Y, Schiavo S, Orjala J, Vouros P, Kautz R (2008) Microscale LC-MS-NMR platform applied to the identification of active cyanobacterial metabolites. Anal Chem 80:8045–8054. doi:10.1021/ac801049k
Maruška A, Kornyšova O (2006) Application of monolithic (continuous bed) chromatographic columns in phytochemical analysis. J Chromatogr A 1112:319–330. doi:10.1016/j.chroma.2006.01.099
Miliauskas G, van Beek TA, de Waard P, Venskutonis RP, Sudhölter EJR (2005) Identification of radical scavenging compounds in Rhaponticum carthamoides by means of LC-DAD-SPE-NMR. J Nat Prod 68:168–172. doi:10.1021/np0496901
Moco S, Tseng L-H, Spraul M, Chen Z, Vervoort J (2006) Building-up a comprehensive database of flavonoids based on nuclear magnetic resonance data. Chromatographia 64:503–508. doi:10.1365/s10337-006-0077-6
Niederländer HAG, van Beek TA, Barsatute A, Koleva II (2008) Antioxidant activity assays on-line with liquid chromatography. J Chromatogr A 1210:121–134. doi:10.1016/j.chroma.2008.09.061
Rochleder F (1854) Phytochemie. Verlag W. Engelmann, Leipzig
Roelofs WL (1984) Electroantennogram assays: rapid and convenient screening procedures for pheromones. In: Hummel HA, Miller TA (eds) Techniques in pheromone research. Springer, Berlin, pp 131–160
Salo P (2007) Thin-layer chromatography with ultraviolet and mass spectrometric detection: from preparative-layer to miniaturized ultra-thin-layer technique. Dissertation, University of Helsinki
Salo PK, Vilmunen S, Salomies H, Ketola RA, Kostiainen R (2007) Two-dimensional ultra-thin-layer chromatography and atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry in bioanalysis. Anal Chem 79:2101–2118. doi:10.1021/ac0620359
Schenk T, Breel GJ, Koevoets P, van der Berg S, Hogenboom AC, Irth H, Tjaden UR, van der Greef J (2003) Screening of natural products extracts for the presence of phosphodiesterase inhibitors using liquid chromatography coupled online to parallel biochemical detection and chemical characterization. J Biomol Screen 8:421–429. doi:10.1177/1087057103255973
Schobel U, Frenay M, van Elswijk DA, McAndrews JM, Long KR, Olson LM, Bobzin SC, Irth H (2001) High resolution screening of plant natural product extracts for estrogen receptor a and b binding activity using an online HPLC-MS biochemical detection system. J Biomol Screen 6:291–303
Shellie R, Marriott P (2003) Opportunities for ultra-high resolution analysis of essential oils using comprehensive two-dimensional gas chromatography: a review. Flav Fragr J 18:179–191. doi:10.1002/ffj.1225
Steinbeck C (2004) Recent developments in automated structure elucidation of natural products. Nat Prod Rep 21:512–518. doi:10.1039/b400678j
Takáts Z, Wiseman JM, Gologan B, Cooks RG (2004) Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306:471–473. doi:10.1126/science.1104404
Talaty N, Takáts Z, Cooks RG (2005) Rapid in situ detection of alkaloids in plant tissue under ambient conditions using desorption electrospray ionization. Analyst (Lond) 130:1624–1633. doi:10.1039/b511161g
Tetala KKR, Chen B, Visser GM, van Beek TA (2007) Single step synthesis of carbohydrate monolithic capillary columns for affinity chromatography of lectins. J Sep Sci 30:2828–2835. doi:10.1002/jssc.200700356
Tetala KKR, Swarts JW, Chen B, Janssen AEM, van Beek TA (2009) A three-phase microfluidic chip for rapid sample clean-up of alkaloids from plant extracts. Lab Chip (submitted)
Thiocone A, Farmer EE, Wolfender J-L (2008) Screening for wound-induced oxylipins in Arabidopsis thaliana by differential HPLC-APCI/MS profiling of crude leaf extracts and subsequent characterisation by capillary-scale NMR. Phytochem Anal 19:198–205. doi:10.1002/pca.1051
van Beek TA, Silva IMMS, Posthumus MA, Melo R (2005) Partial elucidation of Trichogramma putative sex pheromone at trace levels by solid-phase microextraction and gas chromatography-mass spectrometry studies. J Chromatogr A 1067:311–321. doi:10.1016/j.chroma.2004.09.030
Van Berkel GJ, Ford MJ, Deibel MA (2005) Thin-layer chromatography and mass spectrometry coupled using desorption electrospray ionization. Anal Chem 77:1207–1215. doi:10.1021/ac048217p
van der Klift EJC, Vivó-Truyols G, Claassen FW, van Holthoon FL, van Beek TA (2008) Comprehensive two-dimensional liquid chromatography with ultraviolet, evaporative light scattering and mass spectrometric detection of triacylglycerols in corn oil. J Chromatogr A 1178:43–55. doi:10.1016/j.chroma.2007.11.039
van Elswijk DA, Irth H (2003) Analytical tools for the detection and characterization of biologically active compounds from nature. Phytochem Rev 1:427–439. doi:10.1023/A:1026089809218
von Mühlen C, Alcaraz Zini C, Bastos Caramão E, Marriott PJ (2008) Comparative study of Eucalyptus dunnii volatile oil composition using retention indices and comprehensive two-dimensional gas chromatography coupled to time-of-flight and quadrupole mass spectrometry. J Chromatogr A 1200:34–42. doi:10.1016/j.chroma.2008.05.070
Wang J, van der Heijden R, Spijksma G, Reijmers T, Wang M, Xu G, Hankemeier T, van der Greef J (2009) Alkaloid profiling of the Chinese herbal medicine Fuzi by combination of matrix-assisted laser desorption ionization mass spectrometry with liquid chromatography-mass spectrometry. J Chromatogr A 1216:2169–2178. doi:10.1016/j.chroma.2008.11.077
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Beek, T.A., Tetala, K.K.R., Koleva, I.I. et al. Recent developments in the rapid analysis of plants and tracking their bioactive constituents. Phytochem Rev 8, 387–399 (2009). https://doi.org/10.1007/s11101-009-9125-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-009-9125-9