
Abstract
Purpose
PAXLOVID™ is nirmatrelvir tablets co-packaged with ritonavir tablets. Ritonavir is used as a pharmacokinetics (PK) enhancer to reduce metabolism and increase exposure of nirmatrelvir. This is the first disclosure of Paxlovid physiologically-based pharmacokinetic (PBPK) model.
Methods
Nirmatrelvir PBPK model with first-order absorption kinetics was developed using in vitro, preclinical, and clinical data of nirmatrelvir in the presence and absence of ritonavir. Clearance and volume of distribution were derived from nirmatrelvir PK obtained using a spray-dried dispersion (SDD) formulation where it is considered to be dosed as an oral solution, and absorption is near complete. The fraction of nirmatrelvir metabolized by CYP3A was estimated based on in vitro and clinical ritonavir drug-drug interaction (DDI) data. First-order absorption parameters were established for both SDD and tablet formulation using clinical data. Nirmatrelvir PBPK model was verified with both single and multiple dose human PK data, as well as DDI studies. Simcyp® first-order ritonavir compound file was also verified with additional clinical data.
Results
The nirmatrelvir PBPK model described the observed PK profiles of nirmatrelvir well with predicted AUC and Cmax values within ± 20% of the observed. The ritonavir model performed well resulting in predicted values within twofold of observed.
Conclusions
Paxlovid PBPK model developed in this study can be applied to predict PK changes in special populations, as well as model the effect of victim and perpetrator DDI. PBPK modeling continues to play a critical role in accelerating drug discovery and development of potential treatments for devastating diseases such as COVID-19. NCT05263895, NCT05129475, NCT05032950 and NCT05064800.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The COVID-19 pandemic was the result of a global outbreak of coronavirus, an infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus. COVID-19 has resulted in more than 6.6 million death and 638 million confirmed cases globally (Coronavirus Death Toll and Trends—Worldometer (worldometers.info)). Alongside vaccines, antiviral treatment is another effective approach to combat COVID-19. PAXLOVID™ is the first oral Mpro (main protease, i.e., SARSCoV-2 3-chymotrypsin–like cysteine protease enzyme) inhibitor that was granted emergency use authorization (EUA) in the United States, and it is currently approved or authorized for conditional or emergency use in more than 65 countries across the globe to treat high-risk COVID-19 patients. Paxlovid is nirmatrelvir tablets co-packaged with ritonavir tablets (Fig. 1). Nirmatrelvir is predominantly metabolized by CYP3A4 [1] and coadministrated ritonavir inhibits CYP3A, which reduces the clearance and prolongs half-life of nirmatrelvir. This strategy has been successfully applied in various HIV treatments [2,3,4].

Structure of PAXLOVID™ – nirmatrelvir tablets co-packaged with ritonavir tablets.
PBPK modeling and simulation has been widely used to accelerate drug discovery and development [5,6,7]. Analysis of historical regulatory submissions showed growing applications of PBPK and 45% of the new drug approvals containing PBPK modeling and simulation in 2019 [5, 8]. Some major applications of PBPK modeling in regulatory submission include predictions of drug-drug interactions (DDI), assessment of PK changes in special populations such as pediatrics and hepatic and renal impairment, prediction of absorption with formulation changes, food effect and others [8, 9]. This study reports a PBPK model of Paxlovid for the first time using first-order absorption kinetics that was developed and verified with in vitro absorption, distribution, metabolism and excretion (ADME), and clinical data, some of which have been previously published [1, 10,11,12,13]. An Advanced Dissolution, Absorption and Metabolism (ADAM) model for Paxlovid focusing on a more mechanistic description of nirmatrelvir dissolution and absorption will be discussed in a subsequent publication due to its complexity. The Paxlovid PBPK models developed are intended to be used to ensure product quality and desired exposure in in special populations (e.g., pediatrics and pregnancy) to inform dose adjustment recommendations if necessary, as well as to predict DDI potential with comedications.
Materials and Methods
Clinical Studies
Conduct of the studies were in accordance with ethical principles derived from the Declaration of Helsinki and in compliance with International Conference on Harmonization Guidelines for Good Clinical Practice. All regulatory requirements were followed, including those affording greater protection to the safety of trial participants. The study protocols and any amendments, as well as informed consent documents were approved by the institutional review board/ethics committee. Written informed consent was obtained from all participants before any study activity. The details of the study design have been reported previously [11]. The study protocols were published in ClinicalTrials.gov with the following identifiers: NCT05263895 [relative bioavailability (Dataset #1 SDD without ritonavir, Dataset #2 SDD with ritonavir, Dataset #3 tablet with ritonavir)], NCT05129475 (food effect, Dataset #4 tablet with ritonavir), NCT05032950 (midazolam DDI, Dataset #5, tablet with ritonavir) and NCT05064800 (dabigatran etexilate DDI, Dataset #6, tablet with ritonavir). The demographic and dosing information are summarized in Table II.
PBPK Model Development and Verification
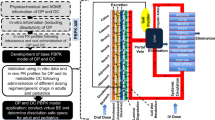
A nirmatrelvir PBPK model was developed and verified with Simcyp® version 21 release 1 (Certara, Sheffield, UK). The model was developed with in vitro [1], preclinical [1, 10], and human clinical data (ClinicalTrials.Org identifiers NCT05263895, NCT05129475, NCT05032950, NCT05064800) [11, 12], where nirmatrelvir was administered with and without ritonavir. The present study focuses on model development and verification of nirmatrelvir and Paxlovid (nirmatrelvir co-dosed with ritonavir). Ritonavir compound file was developed and verified by Simcyp® using ritonavir data across multiple doses and studies [14,15,16,17,18,19,20,21,22,23]. The ritonavir first-order compound file (SV-ritonavir_FO) from the Simcyp® compound library was used for simulations without any modifications. Additional verifications of ritonavir PBPK file were performed with Paxlovid clinical data. Simcyp® midazolam (Sim-Midazolam), dabigatran etexilate (SV-Dabigatran etexilate) and dabigatran (SV-dabigatran) substrate files were used for DDI simulations without any modifications. Dabigatran etexilate substrate file was verified by Simcyp® using thirteen clinical studies. Simulations were performed with 10 trials under the fasted condition using a virtual population library of healthy volunteers in Simcyp® (Sim-Healthy Volunteers) keeping the number of subjects, age range and gender ratio consistent with the clinical trials. The scheme of nirmatrelvir model development and verification is shown in Fig. 2. A stepwise approach was taken to build and establish the first-order absorption nirmatrelvir PBPK model. Firstly, nirmatrelvir systemic parameters (clearance and volume of distribution), as well as fraction absorbed (Fa) and first-order absorption rate constant (ka) were estimated using PK data obtained by dosing a spray dried dispersion (SDD) formulation without ritonavir. Secondly, nirmatrelvir CYP3A fm was estimated using SDD formulation co-dosed with ritonavir. Lastly, Fa and Ka of nirmatrelvir commercial tablet was obtained using PK data of Paxlovid. The first-order absorption Paxlovid PBPK model developed was then verified using single and multiple dosing data of nirmatrelvir tablets co-administered with ritonavir, as well as both the control arms and the interactions in the DDI studies with midazolam and dabigatran etexilate.

Nirmatrelvir PBPK model development and verification scheme.
The details of nirmatrelvir PBPK model development is discussed here. An amorphous solid dispersion formulation of nirmatrelvir, using spray drying process, was developed to enable oral solution dosing. Due to the lack of intravenous (IV) PK data, oral solution data (Dataset #1 and #2, data in Fig. 3) where healthy volunteers were dosed with nirmatrelvir SDD formulation with and without co-administration of ritonavir was used for the PBPK model building to define nirmatrelvir absorption and systemic PK parameters. Clinical data with SDD formulation was treated as oral solution data. The oral clearance of nirmatrelvir (CLPO = 28.36 L/h) and oral steady state volume of distribution (V/F = 1 L/kg) of nirmatrelvir were estimated from SDD solution formulation (NCT05263895, relative bioavailability, Dataset # 1, data in Fig. 3A) by fitting the clinical data. The steady state volume of distribution Vss of nirmatrelvir was described using the full PBPK model in Simcyp® with prediction “Method 2”. “Method 2” is based upon the mechanistic approaches developed by Rodgers and Rowland [24,25,26,27], which considers partitioning of compounds into neutral lipids and phospholipids, electrostatic interactions of strong bases with acidic phospholipids, and binding to extracellular proteins of acids and bases and to lipoproteins of neutral compounds. The method calculates Vss from the partition coefficient (Kp) into different tissues based on physicochemical properties (pKa, LogD, plasma protein binding and blood-to-plasma ratio). The Vss value was adjusted to fit the observed volume by using a global ‘Kp scalar’ of the different tissues. Nirmatrelvir is mainly metabolized by CYP3A4 based on in vitro reaction phenotyping data [1]. The fm,CYP3A4 value was optimized by using ritonavir DDI data with nirmatrelvir SDD formulation (Dataset #2, data in Fig. 3B). The intrinsic clearance by CYP3A4 (CLint,CYP3A4) was derived using the optimized fm,CYP3A4 value, observed oral clearance (CLPO) and renal clearance with the retrograde function in Simcyp®. The remaining of the clearance was assigned to other metabolic clearance. Renal clearance was estimated from multiple ascending dose (MAD) study where participants received BID dosing of 75 mg, 250 mg and 750 mg of nirmatrelvir suspension with 100 mg ritonavir for 10 days [11]. Renal clearance was calculated from urinary excretion data on Day 10, and exhibited no significant dose dependence. Therefore, the observed renal clearance from the aforementioned three doses of nirmatrelvir was averaged and the mean value of 3.4 L/h was used as an input for renal clearance of nirmatrelvir in the model. Biliary clearance of nirmatrelvir was assumed to be minimal based on studies in preclinical species [1]. Nirmatrelvir absorption was modeled using first-order absorption kinetics. The ka for nirmatrelvir SDD tablet and commercial nirmatrelvir tablet co-packaged with ritonavir was estimated with the parameter estimation tool within Simcyp® using the observed mean plasma concentration versus time profiles from Dataset #1 (Data in Fig. 3A) and Dataset #3 (Data in Fig. 4), respectively. Fa of nirmatrelvir in SDD formulation was assumed to be 1 based on its solubility and absorption observed in preclinical species [1]. The fa of nirmatrelvir from the tablet dosage form when co-administered with ritonavir was estimated using the ratio between the observed AUCinf after a single dose of nirmatrelvir tablet and AUCinf after a single dose of nirmatrelvir SDD formulation in presence of ritonavir [Dataset #1 (Data in Fig. 3B) and Dataset #3 (Data in Fig. 4)]. Fu,gut was assumed to be 1 and Qgut was set to 10 L/h. In vitro enzyme and transport inhibition/induction data were also incorporated into the model directly in order to capture any autoinhibition/autoinduction and to enable perpetrator DDI prediction. Methods of these in vitro DDI studies and ADME properties have been published previously [1, 28,29,30,31,32,33]. Verification of CYP3A4 autoinhibition/ autoinduction of nirmatrelvir will be discussed in the ADAM model in a subsequent paper, as single and multiple ascending dose (SAD and MAD) clinical studies used suspension formulation rather than tablet and ADAM model is needed to accurately describe the disposition of nirmatrelvir in suspension dosage form. Fu,inc values for the in vitro DDI parameters were estimated to be about 1 based on fu,mic of 0.824 at 1 mg/mL human liver microsomal protein concentration and the incubation conditions (HLM concentration of 0.01 – 0.3 mg/mL) for the in vitro DDI studies. Induction parameters (Indmax and IndC50) were calibrated against rifampicin.

Method development results: Predicted versus observed PK profile of nirmatrelvir SDD formulation with and without ritonavir (300 mg nirmatrelvir and three doses of 100 mg ritonavir at -12, 0, 12 h). Clinical data from NCT05263895 (relative bioavailability, Dataset #1 and #2). Human clinical data are presented as discrete points of mean value and standard deviation. Solid line in the middle is simulated mean concentration from the model. The two boundary lines represent 5th and 95th percentile. A Without ritonavir. B With ritonavir.

Method development result: Predicted versus observed nirmatrelvir PK profile of Paxlovid (300 mg nirmatrelvir tablet and three doses of 100 mg ritonavir at -12, 0, 12 h). Clinical data from NCT05263895 (relative bioavailability, Dataset #3). Human clinical data are presented as discrete points of mean value and standard deviation. Solid line in the middle is simulated mean concentration from the model. The two boundary lines represent 5th and 95th percentile.
The developed Paxlovid first-order PBPK model was verified with single and multiple dosing studies of nirmatrelvir tablets in the presence of ritonavir tablet by comparing the AUC and Cmax values between predicted and observed. All simulations were conducted with 10 trials maintaining the age range, proportion of females, and number of subjects consistent with the clinical studies. DDI prediction simulations of Paxlovid or ritonavir alone as inhibitors were performed with midazolam and dabigatran etexilate prodrug (dabigatran active was monitored) as substrates. Both the control arms and the interactions of DDI studies with midazolam and dabigatran etexilate were used for model verification. Model verification with a wide range of nirmatrelvir doses was conducted using the nirmatrelvir ADAM model, because suspension formulation was used for the MAD studies, and it required the ADAM model to describe the complex dissolution process of the nirmatrelvir crystalline material. The systemic parameters are identical for both the ADAM and the first-order models. The only difference between the two models is the absorption parameters. As such, the systemic parameters are considered verified with various doses by the ADAM model. Because of the complexity of the ADAM model, it will be discussed in a separate paper.
Results
The input parameters of nirmatrelvir first-order PBPK model and in vitro DDI values are summarized in Table I. The data sources for the input parameters of the nirmatrelvir model are also included in Table I. Information pertaining to clinical data used in model verification and model development namely, dose and dose regimen, age range of healthy volunteers, proportion of females included in the trials have been summarized in Table II. The actual clinical data are shown in Figs. 3, 4, 5 and 6 for nirmatrelvir and supplemental material Figures S1-S4 for ritonavir.

Method Verification Result: Predicted versus observed nirmatrelvir PK profile of Paxlovid (300 mg nirmatrelvir tablets and three doses of 100 mg ritonavir at -12, 0, 12 h). Clinical data from NCT05129475 (food effect, Dataset #4). Human clinical data are presented as discrete points of mean value and standard deviation. Solid line in the middle is simulated mean concentration from the model. The two boundary lines represent 5th and 95th percentile.

Method Verification Result: Predicted versus observed nirmatrelvir PK profile of Paxlovid: (A) 300 mg nirmatrelvir tablets and 100 mg ritonavir BID for 5 days, 9 doses total. Clinical data from NCT05032950 (midazolam DDI, Dataset #5). (B) 300 mg nirmatrelvir tablets and 100 mg ritonavir BID for 2 days, 3 doses total. Clinical data from NCT05064800 (dabigatran etexilate DDI, Dataset #6). Human clinical data are presented as discrete points of mean value and standard deviation. Solid line in the middle is simulated mean concentration from the model. The two boundary lines represent 5th and 95th percentile.
Model Development
Absorption
Nirmatrelvir SDD formulation of amorphous material was developed to enable solution dosing of nirmatrelvir. In vitro experiments confirmed that nirmatrelvir from the SDD formulation remained in solution without precipitating for a period of 24 h, significantly longer than the absorption phase in vivo (Pfizer data on file). The fa of nirmatrelvir from the tablet dosage form when co-administered with ritonavir was estimated to be 0.73 using the ratio between the observed AUCinf after a single dose of nirmatrelvir tablet and AUCinf after a single dose of nirmatrelvir SDD formulation in the presence of ritonavir. The estimated ka values of nirmatrelvir tablet and SDD formulation were 0.55 h−1 and 2.63 h−1, respectively.
Distribution
The volume of distribution of nirmatrelvir was described using full PBPK model with prediction “Method 2” within Simcyp® as described in the method section. The global Kp scalar was determined by employing the parameter estimation tool using the observed mean PK profile from the participants who received nirmatrelvir SDD formulation without ritonavir [NCT05263895 (relative bioavailability, Dataset # 1)]. The estimated Kp scalar was 0.48 that resulted in a predicted Vss of 0.48 L/kg based on “Method 2”.
Metabolism and Renal Clearance
During the model development, initial simulations were conducted using the in vitro measured fm,CYP3A4 value of 0.99. However, this did not capture the observed PK profile of nirmatrelvir when co-administered with ritonavir. Therefore, fm,CYP3A4 was optimized using the observed PK profile from participants who received 300 mg nirmatrelvir SDD formulation with three doses of 100 mg ritonavir at -12 h, 0 h and 12 h [NCT05263895 (relative bioavailability, Dataset #2)]. The sensitivity analysis was performed using an increment of ~ 0.05 with three fm,CYP3A4 values at 0.80, 0.85 and 0.99 (Supplemental Material Table S1). An fm,CYP3A4 of 0.85 best described the PK profile of nirmatrelvir in the presence of ritonavir with the predicted/observed ratios of 0.92 and 0.87 for Cmax and AUCinf, respectively (Supplemental Material Table S1). The current model with an fm,CYP3A4 of 0.85 described the available clinical data well without any biases. More precise estimate of fm can lead to overfitting of the data considering interindividual variability. CLint,CYP3A4 was estimated to be 0.148 μl/min/pmol using the retrograde function in Simcyp® with the optimized fm,CYP3A4 of 0.85, observed geometric mean of CLpo (28.36 L/h), renal clearance of 3.4 L/h (clinical data), and Fa of 1. The remaining fm of 0.15 was assigned to additional HLM CLint (3.23 μl/min/mg). Renal clearance of 3.4 L/h was obtained from clinical data and entered into nirmatrelvir compound file (see Materials and Methods section). Nirmatrelvir renal clearance did not change in the presence and absence of ritonavir.
Model Development Results
The systemic parameters (clearance and volume of distribution) for the nirmatrelvir PBPK model was successfully developed using data from studies in NCT05263895 (relative bioavailability, Dataset #1 and #2) with SDD formulation (modeled as oral solution). These systemic parameters determined with SDD formulation were used to estimate ka for the nirmatrelvir tablet formulation. The model described the observed clinical PK of nirmatrelvir well for both of the formulations with and without ritonavir (Figs. 3 and 4). In addition, the Simcyp® ritonavir compound file with first-order absorption kinetics adequately described the observed ritonavir plasma concentration profiles (Supplemental Material, Table S2 and Figures S1-S2).
Model Verification
The established Paxlovid PBPK model was verified with data from three single and multiple dose clinical studies for food effect and DDI [NCT05129475 (food effect, Dataset #4), NCT05032950 (midazolam DDI, Dataset #5) and NCT05064800 (dabigatran etexilate DDI, Dataset #6)]. The verification results are shown in Table II and Figs. 5 and 6. The model successfully predicted the nirmatrelvir exposures with predicted/observed ratios ranging from 0.90 to 0.98 for AUCinf and Cmax (Table II). This indicates that the estimated model input parameters were appropriate to predict the nirmatrelvir exposures. The fed state arm of the clinical data was not used in the model verification as it requires the use of the ADAM model, which will be discussed in a subsequent publication. DDI clinical studies with midazolam (CYP3A substrate) and dabigatran etexilate (P-gp substrate) were conducted with both Paxlovid and ritonavir alone as perpetrators for comparison purpose (Table III). Given that the Cmax and AUClast ratios as a result of DDI were similar between Paxlovid and ritonavir alone, it was concluded that perpetrator DDIs of Paxlovid for CYP3A and P-gp were mainly driven by ritonavir rather than nirmatrelvir. The verifications of Simcyp® ritonavir compound file using these studies are summarized in the Supplemental Material (Table S2 and Figures S3-S4). Simcyp® ritonavir compound file described the clinical data well for both PK and DDI with predictions falling within twofold of the observed values (Table III and Supplemental Material Table S2).
Discussion
Nirmatrelvir first-order absorption PBPK model was successfully developed by characterizing the ADME properties of nirmatrelvir and its interaction with ritonavir. The model was developed with nirmatrelvir SDD formulation and tablet with and without ritonavir, and verified with all relevant clinical data including single and multiple dose studies. Certain clinical data were not included in the model verification, such as the SAD and MAD studies with nirmatrelvir suspension, due to complex absorption processes that require ADAM model to mechanistically describe the dissolution/absorption properties, which will be discuss in a subsequent publication.
As a peptide mimetic, nirmatrelvir has many unique ADME properties leading to its PK behavior. Nirmatrelvir has low passive permeability in the MDCK-LE [34] (Papp 1.76 × 10–6 cm/s) and Caco-2 (Papp 0.66–1.19 × 10–6 cm/s) in vitro assays [1]. Despite the low passive permeability in vitro, nirmatrelvir is well absorbed in preclinical species (e.g., rat) [1] and humans. The in vitro-in vivo disconnect on nirmatrelvir oral absorption is currently not well-understood, although similar observations have been reported in the literature [35]. Nirmatrelvir is a P-gp substrate, but not a BCRP substrate [1]. P-gp efflux doesn’t appear to have significant impact on oral absorption of nirmatrelvir at the doses studied as SDD formulation showed near complete absorption, and no significant supra-proportional exposure was observed with increased dose in rat [36]. This is consistent with literature reports of minimal impact of P-gp efflux on oral absorption for compounds with relatively high solubility and permeability [37,38,39,40,41,42,43].
In vitro fm,CYP3A4 of nirmatrelvir was estimated to be 0.99 based on formation rate of the major metabolites with selective inhibitors [1]. In vivo fm,CYP3A4 was estimated to be 0.85 based on ritonavir DDI study, indicating some uncertainties of in vitro assays in estimating in vivo fm. Sensitivity analysis of fm can help further understand the victim DDI risk in vivo. With ritonavir coadministration, the major clearance pathway of nirmatrelvir shifted from CYP3A metabolism to renal elimination [12], with an increased half-life from 2 to 7 hour. At 100 mg dose, ritonavir completely inhibits CYP3A [18]. Ritonavir at 100 mg dose is commonly used as a booster for protease inhibitors in HIV therapy (e.g., in combination with lopinavir, saquinavir, or atazanavir) [4]. Compared to ritonavir, nirmatrelvir is a much weaker CYP3A and P-gp inhibitor based on in vitro inhibition potency. As such, Paxlovid perpetrator DDIs for CYP3A and P-gp are mainly driven by ritonavir. The slight over-prediction of midazolam DDI might be due to variability of the clinical data. The mean and 90% confidence interval of AUClastR are 14.5 (12.2, 17.2) for Paxlovid and 16.8 (14.1, 19.9) for ritonavir DDI with midazolam. An AUC∞R of 23.8 was reported for ritonavir (100 mg BID) with midazolam DDI in the literature [17]. The underprediction of dabigatran etexilate DDI is likely because the in vitro Ki for P-gp might be higher than the in vivo Ki. Scaling factors are usually needed to more accurately predict in vivo transporter-mediated DDI. For perpetrator DDI predictions, sensitivity analysis around Ki values is recommended to account for the uncertainties in experimental Ki values for in vivo translation [44,45,46,47,48,49]. When co-administered with ritonavir, CYP3A4 metabolism of nirmatrelvir is inhibited leading to increased fractional renal clearance (fCL,R). Although nirmatrelvir renal clearance (CLR) remains the same in the presence and absence of ritonavir, the percentage of nirmatrelvir that is eliminated renally significantly increases when co-administered with ritonavir. Consequently, a reduced nirmatrelvir dose is recommended for moderate renal impairment patients, and Paxlovid is not recommended in patients with severe renal impairment (Paxlovid HCP FS 09,262,022 (fda.gov)).
The first-order absorption PBPK model of Paxlovid has been applied to predict the DDI risk with weak and moderate CYP3A inducers and suggested no significant risk of DDI (Supplemental Material, Table S3). These PBPK simulation results were accepted by FDA. In addition, the Paxlovid models are being applied to inform dose selection of special populations (e.g., organ impairments, pediatrics, and pregnancy), DDI predictions and bioequivalence simulations.
Conclusions
A Paxlovid PBPK model with first-order absorption kinetics has been successfully developed and verified. The model describes the nirmatrelvir PK well with and without ritonavir from multiple clinical studies using tablet formulation. The PBPK model not only helps to understand Paxlovid disposition mechanically, but also enables forward predictions of DDI with co-meds, exposure changes in special populations to inform dose adjustment recommendation, and support product label. The Paxlovid PBPK model enables informed internal decisions and regulatory submissions to address various queries. PBPK modeling continues to play a pivotal role in accelerating drug discovery and development of potentially life-saving medicines as has been described herein for Paxlovid.
Data Availability
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Abbreviations
- ADAM:
-
Advanced dissolution, absorption, and metabolism
- ADME:
-
Absorption, distribution, metabolism, and excretion
- AUC:
-
Area under the concentration–time curve
- AUCinf :
-
Area under the concentration–time curve from time 0 to infinite
- B/P:
-
Blood-to-plasma ratio
- BCRP:
-
Breast cancer resistance protein
- BID:
-
Twice a day
- Caco-2:
-
Human colorectal adenocarcinoma cells
- CLint :
-
Intrinsic clearance
- CLint ,CYP3A4 :
-
Intrinsic clearance of cytochrome P450 3A4
- CLpo :
-
Oral clearance
- CLR :
-
Renal clearance
- Cmax :
-
Maximum concentration
- CYP3A:
-
Cytochrome P450 3A
- CYP3A4:
-
Cytochrome P450 3A4
- DDI:
-
Drug-drug interaction
- EUA:
-
Emergency use authorization
- fa :
-
Fraction absorbed
- fm :
-
Fraction metabolized
- fm, CYP3A4 :
-
Fraction metabolized by CYP3A4
- fu :
-
Fraction unbound
- fu, gut :
-
Fraction unbound in enterocytes
- fu, mic :
-
Fraction unbound in microsomal incubation
- fup :
-
Fraction unbound in plasma
- γ:
-
Hill coefficient
- GI:
-
Gastrointestinal
- HIV:
-
Human immunodeficiency virus
- IndC50 :
-
Concentration resulting in half maximal induction
- Indmax :
-
Maximal fold induction over vehicle
- ka :
-
First-order absorption rate constant
- Kapp :
-
The concentration of mechanism-based inhibitor associated with half maximal inactivation rate
- Ki :
-
Concentration of inhibitor that supports half maximal inhibition
- kinact :
-
Maximal rate of enzyme inactivation
- Kp :
-
Tissue partition coefficient
- LogP:
-
Log10 of partition coefficient between octanol and water
- MAD:
-
Multiple ascending dose
- MDCK-LE:
-
Madin-Darby canine kidney low efflux cells
- MW:
-
Molecular weight
- N:
-
Number of subjects
- NCA:
-
Noncompartmental analysis
- NMV:
-
Nirmatrelvir
- Obs:
-
Observed
- Papp :
-
Apparent permeability
- PBPK:
-
Physiologically-based pharmacokinetic
- P-gp:
-
P-glycoprotein
- PK:
-
Pharmacokinetics
- Pred:
-
Predicted
- Qgut :
-
A hybrid term including both villous blood flow and permeability through the enterocyte membrane
- RTV:
-
Ritonavir
- SAD:
-
Single ascending dose
- SD:
-
Single dose
- SDD:
-
Spray-dried dispersion
- Vss :
-
Steady state volume of distribution
References
Eng H, Dantonio AL, Kadar EP, Obach RS, Di L, Lin J, Patel NC, Boras B, Walker GS, Novak JJ, Kimoto E, Singh RSP, Kalgutkar AS. Disposition of nirmatrelvir, an orally bioavailable inhibitor of SARS-CoV-2 3C-like protease, across animals and humans. Drug Metab Dispos. 2022;50(5):576–90.
Cooper CL, van Heeswijk RPG, Gallicano K, Cameron DW. A review of low-dose ritonavir in protease inhibitor combination therapy. Clin Infect Dis. 2003;36(12):1585–92.
De Nicolo A, Simiele M, Calcagno A, Abdi AM, Bonora S, Di Perri G, D’Avolio A. Intracellular antiviral activity of low-dose ritonavir in boosted protease inhibitor regimens. Antimicrob Agents Chemother. 2014;58(7):4042–7.
Hull MW, Montaner JSG. Ritonavir-boosted protease inhibitors in HIV therapy. Ann Med. 2011;43(5):375–88.
Chu X, Prasad B, Neuhoff S, Yoshida K, Leeder JS, Mukherjee D, Taskar K, Varma MVS, Zhang X, Yang X, Galetin A. Clinical Implications of Altered Drug Transporter Abundance/Function and PBPK Modeling in Specific Populations: An ITC Perspective. Clin Pharmacol Ther. 2022;112(3):501–26.
Shebley M, Sandhu P, Emami Riedmaier A, Jamei M, Narayanan R, Patel A, Peters SA, Reddy VP, Zheng M, de Zwart L, Beneton M, Bouzom F, Chen J, Chen Y, Cleary Y, Collins C, Dickinson GL, Djebli N, Einolf HJ, Gardner I, Huth F, Kazmi F, Khalil F, Lin J, Odinecs A, Patel C, Rong H, Schuck E, Sharma P, Wu SP, Xu Y, Yamazaki S, Yoshida K, Rowland M. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin Pharmacol Ther. 2018;104(1):88–110.
Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I, Wang Y. Physiologically Based Pharmacokinetic Modeling in Regulatory Science: An Update From the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–5.
Zhang X, Yang Y, Grimstein M, Fan J, Grillo JA, Huang S-M, Zhu H, Wang Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018–2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60(S1):S160–78.
Grimstein M, Yang Y, Zhang X, Grillo J, Huang S-M, Zineh I, Wang Y. Physiologically Based Pharmacokinetic Modeling in Regulatory Science: An Update From the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–5.
Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, Boras B, Cardin RD, Carlo A, Coffman KJ, Dantonio A, Di L, Eng H, Ferre R, Gajiwala KS, Gibson SA, Greasley SE, Hurst BL, Kadar EP, Kalgutkar AS, Lee JC, Lee J, Liu W, Mason SW, Noell S, Novak JJ, Obach RS, Ogilvie K, Patel NC, Pettersson M, Rai DK, Reese MR, Sammons MF, Sathish JG, Singh RSP, Steppan CM, Stewart AE, Tuttle JB, Updyke L, Verhoest PR, Wei L, Yang Q, Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–93.
Singh RSP, Toussi SS, Hackman F, Chan PL, Rao R, Allen R, Van Eyck L, Pawlak S, Kadar EP, Clark F, Shi H, Anderson AS, Binks M, Menon S, Nucci G, Bergman A. Innovative Randomized Phase I Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID-19 Trial of Nirmatrelvir. Clin Pharmacol Ther. 2022;112(1):101–11.
Singh RSP, Walker GS, Kadar EP, Cox LM, Eng H, Sharma R, et al. Metabolism and excretion of nirmatrelvir in humans using quantitative fluorine nuclear magnetic resonance spectroscopy: a novel approach for accelerating drug development. Clin Pharmacol Ther. 2022;112(6):1201–6.
Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, Baniecki M, Hendrick VM, Damle B, Simon-Campos A, Pypstra R, Rusnak JM. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N Engl J Med. 2022;386(15):1397–408.
Eichbaum C, Cortese M, Blank A, Burhenne J, Mikus G. Concentration effect relationship of CYP3A inhibition by ritonavir in humans. Eur J Clin Pharmacol. 2013;69(10):1795–800.
Greenblatt DJ, Peters DE, Oleson LE, Harmatz JS, MacNab MW, Berkowitz N, Zinny MA, Court MH. Inhibition of oral midazolam clearance by boosting doses of ritonavir, and by 4,4-dimethyl-benziso-(2H)-selenazine (ALT-2074), an experimental catalytic mimic of glutathione oxidase. Br J Clin Pharmacol. 2009;68(6):920–7.
Ieiri I, Tsunemitsu S, Maeda K, Ando Y, Izumi N, Kimura M, Yamane N, Okuzono T, Morishita M, Kotani N, Kanda E, Deguchi M, Matsuguma K, Matsuki S, Hirota T, Irie S, Kusuhara H, Sugiyama Y. Mechanisms of pharmacokinetic enhancement between ritonavir and saquinavir; micro/small dosing tests using midazolam (CYP3A4), fexofenadine (p-glycoprotein), and pravastatin (OATP1B1) as probe drugs. J Clin Pharmacol. 2013;53(6):654–61.
Mathias AA, German P, Murray BP, Wei L, Jain A, West S, Warren D, Hui J, Kearney BP. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin Pharmacol Ther. 2010;87(3):322–9.
Mathias AA, West S, Hui J, Kearney BP. Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin Pharmacol Ther. 2009;85(1):64–70.
Ouellet D, Hsu A, Granneman GR, Carlson G, Cavanaugh J, Guenther H, Leonard JM. Pharmacokinetic interaction between ritonavir and clarithromycin. Clin Pharmacol Ther. 1998;64(4):355–62.
Greenblatt DJ, von Moltke LL, Harmatz JS, Durol AL, Daily JP, Graf JA, Mertzanis P, Hoffman JL, Shader RI. Alprazolam-ritonavir interaction: implications for product labeling. Clin Pharmacol Ther. 2000;67(4):335–41.
Kirby BJ, Collier AC, Kharasch ED, Dixit V, Desai P, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of HIV protease inhibitors 2: in vivo induction and in vitro to in vivo correlation of induction of cytochrome P450 1A2, 2B6, and 2C9 by ritonavir or nelfinavir. Drug Metab Dispos. 2011;39(12):2329–37.
Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of the HIV protease inhibitors 3: effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab Dispos. 2012;40(3):610–6.
Aarnoutse RE, Kleinnijenhuis J, Koopmans PP, Touw DJ, Wieling J, Hekster YA, Burger DM. Effect of low-dose ritonavir (100 mg twice daily) on the activity of cytochrome P450 2D6 in healthy volunteers. Clin Pharmacol Ther. 2005;78(6):664–74.
Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24(5):918–33.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238–57.
Rodgers T, Leahy D, Rowland M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J Pharm Sci. 2005;94(6):1259–76.
Rodgers T, Rowland M, Rodgers T, Rowland M. Physiologically-based pharmacokinetic modeling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238–57.
Lazzaro S, West MA, Eatemadpour S, Feng B, Varma MVS, Rodrigues AD, et al. Translatability of in vitro inhibition potency to in vivo P-glycoprotein mediated drug interaction risk. J Pharm Sci. 2023;112(6):1715–23.
Costales C, Lin J, Kimoto E, Yamazaki S, Gosset JR, Rodrigues AD, Lazzaro S, West MA, West M, Varma MVS. Quantitative prediction of breast cancer resistant protein mediated drug-drug interactions using physiologically-based pharmacokinetic modeling. CPT: Pharmacometrics Syst Pharmacol. 2021;10(9):1018–31.
Kimoto E, Costales C, West MA, Bi YA, Vourvahis M, David Rodrigues A, Varma MVS. Biomarker-Informed Model-Based Risk Assessment of Organic Anion Transporting Polypeptide 1B Mediated Drug-Drug Interactions. Clin Pharmacol Ther. 2022;111(2):404–15.
Mathialagan S, Feng B, Rodrigues AD, Varma MVS. Drug-Drug Interactions Involving Renal OCT2/MATE Transporters: Clinical Risk Assessment May Require Endogenous Biomarker-Informed Approach. Clin Pharmacol Ther. 2021;110(4):855–9.
Feng B, Hurst S, Lu Y, Varma MV, Rotter CJ, El-Kattan A, Lockwood P, Corrigan B. Quantitative prediction of renal transporter-mediated clinical drug-drug interactions. Mol Pharm. 2013;10(11):4207–15.
Ryu S, Woody N, Chang G, Mathialagan S, Varma MVS. Identification of Organic Anion Transporter 2 Inhibitors: Screening, Structure-Based Analysis, and Clinical Drug Interaction Risk Assessment. J Med Chem. 2022;65(21):14578–88.
Di L, Whitney-Pickett C, Umland JP, Zhang H, Zhang X, Gebhard DF, Lai Y, Federico JJ, Davidson RE, Smith R, Reyner EL, Lee C, Feng B, Rotter C, Varma MV, Kempshall S, Fenner K, El-kattan AF, Liston TE, Troutman MD. Development of a new permeability assay using low-efflux MDCKII cells. J Pharm Sci. 2011;100(11):4974–85.
Price E, Kalvass JC, DeGoey D, Hosmane B, Doktor S, Desino K. Global Analysis of Models for Predicting Human Absorption: QSAR, In Vitro, and Preclinical Models. J Med Chem. 2021;64(13):9389–403.
Sathish JG, Bhatt S, DaSilva JK, Flynn D, Jenkinson S, Kalgutkar AS, Liu M, Manickam B, Pinkstaff J, Reagan WJ, Shirai N, Shoieb AM, Sirivelu M, Vispute S, Vitsky A, Walters K, Wisialowski TA, Updyke LW. Comprehensive Nonclinical Safety Assessment of Nirmatrelvir Supporting Timely Development of the SARS-COV-2 Antiviral Therapeutic. Paxlovid Int J Toxicol. 2022;41(4):276–90.
Chiou WL, Chung SM, Wu TC, Ma C. A comprehensive account on the role of efflux transporters in the gastrointestinal absorption of 13 commonly used substrate drugs in humans. Int J Clin Pharmacol Ther. 2001;39(3):93–101.
Lee YJ, Chung SJ, Shim CK. Limited role of P-glycoprotein in the intestinal absorption of cyclosporin A. Biol Pharm Bull. 2005;28(4):760–3.
Cao X, Yu LX, Barbaciru C, Landowski CP, Shin HC, Gibbs S, Miller HA, Amidon GL, Sun D. Permeability dominates in vivo intestinal absorption of P-gp substrate with high solubility and high permeability. Mol Pharm. 2005;2(4):329–40.
Berggren S, Hoogstraate J, Fagerholm U, Lennernäs H. Characterization of jejunal absorption and apical efflux of ropivacaine, lidocaine and bupivacaine in the rat using in situ and in vitro absorption models. Eur J Pharm Sci. 2004;21(4):553–60.
Chiou WL, Chung SM, Wu TC. Apparent lack of effect of P-glycoprotein on the gastrointestinal absorption of a substrate, tacrolimus, in normal mice. Pharm Res. 2000;17(2):205–8.
Lin JH. How significant is the role of P-glycoprotein in drug absorption and brain uptake? Drugs Today (Barcelona, Spain: 1998). 2004;40(1):5–22.
Toyobuku H, Tamai I, Ueno K, Tsuji A. Limited influence of P-glycoprotein on small-intestinal absorption of cilostazol, a high absorptive permeability drug. J Pharm Sci. 2003;92(11):2249–59.
Bentz J, O’Connor MP, Bednarczyk D, Coleman J, Lee C, Palm J, Pak YA, Perloff ES, Reyner E, Balimane P, Brännström M, Chu X, Funk C, Guo A, Hanna I, Herédi-Szabó K, Hillgren K, Li L, Hollnack-Pusch E, Jamei M, Lin X, Mason AK, Neuhoff S, Patel A, Podila L, Plise E, Rajaraman G, Salphati L, Sands E, Taub ME, Taur JS, Weitz D, Wortelboer HM, Xia CQ, Xiao G, Yabut J, Yamagata T, Zhang L, Ellens H. Variability in P-glycoprotein inhibitory potency (IC50) using various in vitro experimental systems: implications for universal digoxin drug-drug interaction risk assessment decision criteria. Drug Metab Dispos. 2013;41(7):1347–66.
Cook JA, Feng B, Fenner KS, Kempshall S, Liu R, Rotter C, Smith DA, Troutman MD, Ullah M, Lee CA. Refining the In Vitro and In Vivo Critical Parameters for P-Glycoprotein, [I]/IC50 and [I2]/IC50, That Allow for the Exclusion of Drug Candidates from Clinical Digoxin Interaction Studies. Mol Pharm. 2010;7(2):398–411.
Izumi S, Nozaki Y, Maeda K, Komori T, Takenaka O, Kusuhara H, Sugiyama Y. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab Dispos. 2015;43(2):235–47.
Yabut J, Houle R, Wang S, Liaw A, Katwaru R, Collier H, Hittle L, Chu X. Selection of an Optimal In Vitro Model to Assess P-gp Inhibition: Comparison of Vesicular and Bidirectional Transcellular Transport Inhibition Assays. Drug Metab Dispos. 2022;50(7):909–22.
Yoshida K, Guo C, Sane R. Quantitative Prediction of OATP-Mediated Drug-Drug Interactions With Model-Based Analysis of Endogenous Biomarker Kinetics. CPT: Pharmacometrics Syst Pharmacol. 2018;7(8):517–24.
Li R, Barton HA, Varma MV. Prediction of Pharmacokinetics and Drug-Drug Interactions When Hepatic Transporters are Involved. Clin Pharmacokinet. 2014;53(8):659–78.
Acknowledgements
Authors greatly appreciate the help of Demetria Baker DeBartolo in data QC and report formatting; thanks to Rajendra S Kadam and Donna S. Cox for managing the clinical studies; thanks to many Pfizer colleagues for their input and discussion.
Funding
This study was sponsored by Pfizer Inc., New York, NY, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors are employees of Pfizer Inc., New York, NY, USA and may hold stock or stock options.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sagawa, K., Lin, J., Jaini, R. et al. Physiologically-Based Pharmacokinetic Modeling of PAXLOVID™ with First-Order Absorption Kinetics. Pharm Res 40, 1927–1938 (2023). https://doi.org/10.1007/s11095-023-03538-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-023-03538-5