
Abstract
Purpose
In order to clarify the effect of rifampicin on the bioavailability of the P-glycoprotein substrate talinolol, its absorption kinetics was modeled after multiple-dose oral administration of talinolol in healthy subjects.
Methods
A sum of two inverse Gaussian functions was used to calculate the time course of the input rate into the systemic circulation.
Results
The estimated rate of drug entry into the systemic circulation revealed two distinct peaks at 1 and 3.5 h after administration. Rifampicin did not affect bioavailability of talinolol, but did shift the second peak of the input function by 1.3 h to later times. Elimination clearance and one of the intercompartmental distribution clearances increased significantly under rifampicin treatment.
Conclusions
Rifampicin changes the time course of absorption rate but not the fraction absorbed of talinolol. The model suggests the existence of two intestinal absorption windows for talinolol.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although the oral route is the most convenient and most used method of drug delivery, pharmacokinetic modeling is often based on noncompartmental methods (numerical integration) or oversimplified models like the first order absorption model. For the more complex absorption behaviors of extended release formulations or to study food effects, flexible empirical input rate models, such as the inverse Gaussian density function [1,2,3,4,5], or a sum of inverse Gaussian functions [6,7,8,9,10], have been successfully applied. The latter was capable of fitting double-peak data [11] and has some advantages over models based on transit compartments (gamma density) or a Weibull function [8]. All of the aforementioned studies involve applications of absorption models to single dose data.
In this work, we describe the use of the sum of two inverse Gaussian functions (2IG) as oral input rate function in a multiple dosing situation, namely after repeated oral administration of talinolol. Talinolol is a substrate of the efflux transporter P-glycoprotein (P-gp) that has been extensively used to investigate P-gp-mediated drug transport [12, 13] and is recommended by FDA as a probe substrate. The pharmacokinetics of talinolol was evaluated before and after rifampicin administration in order to analyze the effect of P-gp induction on the disposition and absorption of talinolol. In contrast to the previous analysis that was based on noncompartmental methods [14], the approach we present provides an estimate of the rate of talinolol absorption.
Thus, the first purpose was to estimate the time course of rate of talinolol absorption, to determine the effects of rifampicin which remained undetected when using noncompartmental analysis. Our reevaluation of the data by Westphal et al. [14] suggests that rifampicin-mediated P-gp induction affects the intestinal site of talinolol absorption but not bioavailability. Furthermore, reasons for the observed double-peak phenomenon [15] can now be interpreted in terms of the absorption rate profile. Our results shed new light on the role of P-gp–mediated intestinal transport of talinolol and the role of the P-gp-inducer rifampicin [16,17,18]. A second purpose of this study was demonstrate how multiple dose data can be analyzed with the ADAPT 5 software [19] using a complex absorption model.
Methods
Clinical Study Data
We reevaluated the pharmacokinetic data of a previously performed repeated dose drug-drug interaction study with talinolol and rifampicin [14]. Healthy human subjects were treated with talinolol for 14 days (100 mg/day, 07:00 h a.m.). Beginning with the 9th treatment day, rifampicin (600 mg/day, 06:00 h p.m.) was co-administered for 9 days. Concentrations-time curves of talinolol during repeated-dosing were measured at the 7th and 14th treatment day. 8 days before and 3 days after the last oral treatment with talinolol, the serum concentration–time curves of the drug were measured after short-time intravenous infusion (30 mg within 30 min). The data were stored in our databank and used for re-evaluation by pharmacokinetic modeling in fully anonymized manner in agreement with the written informed consent as given by the healthy subjects included in the study (8 males, age 22—26 years; body weight 67–84 kg). The study had been approved by the Independent Ethics Committee of the University Medicine of Greifswald.
Pharmacokinetic Modeling
The time course of the absorption rate (rate of drug input into the central compartment), I(t) was described as a sum of two IGs [7, 10, 11]
where D is dose, F is bioavailability, fi(t) denotes the IG function below and p is a nonnegative quantity that defines the relative contribution of each IG to the input function I(t).
where MTi and \(R{D}_{i}^{2}\) are the scale and shape parameters, respectively, of the ith IG function. The mean input time (MIT) is then given by
This two IG input model above can be extended to represent a mixture of multiple IG. For multiple doses, the input function can be written as:
where \({D}_{j}\) and \(d{t}_{j}\) are the amount and time of the jth dose, and ND is the number of doses.
The disposition of talinolol, both with and without rifampicin, was described using linear compartment models.
Parameter Estimation
A stepwise estimation process was followed. First, the intravenous (iv) data obtained from the experiments with and without rifampicin were each analyzed using two and three compartment models. From these results, the disposition parameters of the resulting three compartment model were fixed, and the parameters of the absorption model (input to the central compartment) were then estimated from the respective concentration time data of talinolol after oral administration. Note that six parameters, namely F, MT1,\(R{D}_{1}^{2}\), MT2,\(R{D}_{2}^{2}\) and p were estimated from the oral data. Both estimation steps were performed by population analysis (nonlinear mixed-effects modeling) using the maximum likelihood expectation maximization (MLEM) application in the ADAPT (Version 5) software [19]. See Supplemental Material for the code of the ADAPT file implementing the model. All model parameters (compartment disposition model and inverse Gaussian mixture model) were assumed to follow a multivariate log-normal distribution. The residual errors for both the iv and oral modeling analyses were assumed to be normally distributed with proportional and additive variance terms. Model selection (number of compartments in the disposition model) was based on the likelihood-derived Akaike information criterion (AIC). Plots of conditional standardized residuals and the relative standard errors of estimated parameters were also examined.
For the visual predictive check, plasma concentration versus time profiles were simulated for 1000 virtual subjects, using the estimated parameter mean values and variances (Monte Carlo simulation, population simulation without process noise). Lognormal parameter distributions were assumed. The 5th percentile, 50th and 95th percentile of model-predicted concentrations at each time point were extracted from the simulated data.
From the estimated parameters describing the time course of talinolol absorption before and during coadministration of rifampicin, the time points of the first and second peak and the maximum input rates were calculated using the statistical program package Maple (https://de.maplesoft.com). This software was also used to simulate the time course of the amount absorbed with the cumulative IG [20]. The differences between pre- and post-rifampicin treatment were analyzed by the paired t-test. We evaluated correlations between Imax,1 and tmax,1 and between tmax,2 and tmax,1 using linear regression analysis.
Results
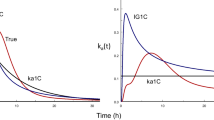
Talinolol iv data were best described (AIC criterion) by a 3-compartment model (Table 1). Rifampicin caused a significant increase in both elimination clearance and intercompartmental clearance 1 (p < 0.01). Renal clearance remained unchanged. Inspection of the oral data indicated a mixture of two IG functions would be appropriate for each subject. Our multiple dose approach with a 2IG input model allowed an excellent fit to all talinolol serum concentration data, obtained before and after coadministration of rifampicin. In one subject, the model could not be fitted to the data because of an unexplained discrepancy between the trough and peak concentration. This subject was excluded during the analysis. The quality of fit is illustrated for three subjects with prototypical time courses of input rate in Fig. 1, by the goodness of fit plots (Fig. 2) and a visual predictive check (Fig. 3). The input function (Fig. 1) reflects the characteristic double peak phenomena observed in the serum concentration–time curves. No significant changes in the parameters of the input function were detected in the presence of rifampicin, except for an increase in the time at which the second peak appears, namely from 3.47 to 4.74 h (p < 0.05). A shift to the right of the second peak was observed in all subjects, albeit with varying degree (Fig. 1). The mean bioavailability of talinolol was not significantly changed by rifampicin, despite a considerable reduction in two subjects (e.g. subject 8 in Fig. 1). The difference in the time courses of the cumulative amount absorbed are clearly visible in Fig. 4. Interestingly, the maximum of the first peak, Imax,1, decreased with increasing tmax,1 (p < 0.05) and there was a significant correlation between tmax,2 and tmax,1 (p < 0.05) (Fig. 5). All these correlations disappeared under rifampicin.

Fits of talinolol data after 100 mg/24 h talinolol in 3 subjects before (left) and after coadministration of rifampicin (middle). The resulting time courses of input rate are shown on the right hand side. The first dose was given at Time 0.

Goodness-of-fit plots showing the observed concentration data of talinolol after multiple oral dosing versus the individual model-predicted values for all 7 subjects without (left) and with rifampicin (right). The regression lines are nearly coincident with the lines of identity.

Visual predictive checks showing the 90% prediction interval (5th to 95th percentiles, dashed lines) and the median on the simulated data (solid line) together with the observed data (open dots).

Mean cumulative amount of talinolol absorbed as fraction of dose. The curves were simulated using the mean parameter estimates of the input function.

The first peak of the input rate decrease linearly (p < 0.05) with the time at which peak concentrations are reached (left) and the time point of the second peak increases with that of the first peak (p < 0.05) (right).
Discussion
The modeling approach used in this work provides an estimate of the time course of talinolol’s rate of absorption under multiple dosing, an advantage compared to previously used noncompartmental analysis, which allowed us to quantify the effects of rifampicin on talinolol’s absorption. From our modeling analysis, the double peak in plasma concentrations is obviously due to a bimodal pattern of input rate. Note that we did not observe a decrease in bioavailability in contrast to the initial noncompartmental evaluation by Westphal et al. 2000 [14], who erroneously used iv control instead iv treatment data as previously noted by Chiou et al. 2003 [21]. This correction, however, has been overlooked in later publications citing the original article [16, 22]. Thus, coadministration of rifampicin neither influenced the amount of talinolol absorbed significantly, nor the mean absorption time; solely the second absorption peak was shifted to later times, ie, from 3.47 to 4.74 h (Figs. 1 and 4). The heights of both peaks did not change under rifampicin. The dip of the curve under rifampicin nearly coincidences with the second peak under control conditions. In other words, just at the time point when the input rate is maximal under control conditions, it is minimal after rifampicin.
What remains to be explained, however, are the processes underlying the first peak (after about one hour), which is not affected by P-gp induction, and the second higher peak (between 2 and 4 h after administration), which is shifted to later times after P-gp induction. Here we offer an alternative hypothesis to the “intestinal storage model” originally suggested by Weitschies et al. 2005 [15], which is based on more recent results on the abundance of multidrug transporters along the small and large intestine [23,24,25]. Intestinal P-gp is a high-affinity efflux carrier which counteracts the uptake. Therefore, the highest input-rates are expected in regions with lowest P-gp abundance and carriers saturated with the substrate talinolol. This occurs in the duodenum/jejunum and cecum/ascending colon where the P-gp abundance is lower compared to more distal regions [24] and indicates the existence of two absorption windows for talinolol. The first peak of the input rate occurred in about one hour after oral dosing, i.e., immediately after gastric emptying of water swallowed for drug administration in fasting healthy subjects [26]. Thus, tmax,1 is not much different from the meta-mean of the gastric transit time in the fasted state of 1.37 h [27]. The large intersubject variability is due to one subject with a high tmax,1 value of 2.84 h (and a low Imax,1 of 4.20 mg/h, cf. Figure 5). The high variability of gastric emptying has been discussed elsewhere [28].The emptied water undergoes rapid absorption which generates high drug concentrations in the proximal small intestine [28, 29]. Therefore, the first peak is most likely caused by uptake in the duodenum/proximal jejunum and is limited by the relative short residence time in this region. Thus, the reduction of the first peak with increasing values of tmax,1 (Fig. 5) may reflect the decrease in drug uptake after passing through the absorption window. Although only ~ 10% of the dose is absorbed from the proximal absorption window (Fig. 4), there is a significant correlation between F and Imax,1 (r = 0.68, p < 0.01). Another ~ 40% is subsequently absorbed from a second absorption window with a maximum input rate after more than 3 h (Fig. 1). The correlation between F and Imax,2 was less expressed (r = 0.60) but also significant (P < 0.05). This second window can likely be located to the cecum/ascending colon for the following reasons: Firstly, the P-gp abundance in the cecum/ascending colon is many times lower than in the ileum. Secondly, the time of maximum input rate was similar to the oro-cecal transit time as assessed with the sulfasalazine/sulfapyridine method (3 – 6 h) under identical protocol conditions [30, 31]. Interestingly, our estimate of 3.47 h is nearly identical with the meta-mean small intestinal transit time of 3.49 h [27]. Variability in oro-cecal transit time may be mainly due to the variability in gastric transit time, since the time point of the second peak increases linearly with that of the first peak (Fig. 5).
Regarding the effect of rifampicin on the absorption process, it appears that the first peak remained unaffected due to the relatively high, P-gp saturable talinolol concentration. P-gp inducing concentrations are also expected in the cecum/ascending colon by entero-hepatic recirculation of rifampicin after glucuronide cleavage [32]. However, talinolol concentrations are much lower than in the proximal small intestine. Thus, P-gp induction may avoid uptake in cecum/ascending colon leading to a shift of the second absorption window to a more distal site, with a delay of the second peak time by 1.27 h. This is based on the assumption that in deeper parts of the colon P-gp cannot be induced because the systemic concentrations after 600 mg rifampicin daily are too low to induce P-gp function [14, 33].
It remains unclear why the expected reduction in bioavailability is seen just in subjects 5 (F = 0.30) and 8 (F = 0.34, Fig. 1), i.e., in two out of seven subjects. These low values are the reason for the observed tendency to a bioavailabiity reduction (Table 1, Fig. 4).
The increase in talinolol clearance after rifampicin can be attributed to an increase of Pgp mediated intestinal secretion [14, 17], but we have no explanation for the significant increase in distribution clearance (Table 1). While a rifampicin mediated inhibition of P-gp in capillary endothelium of certain organs (e.g. brain, testes) [34] could increase tissue distribution, the intercompartmental clearance 1 exceeds blood flow to relevant organs. Thus we leave it as an open question for future research.
It should be added that physiologically-based pharmacokinetic modeling was used to predict rifampicin-mediated drug interaction [35] including talinolol [36, 37]. These modeling results suggested that this interaction can be explained by an increase of intestinal P-gp activity, which is in accordance with our hypothesis.
Conclusions
These results show that more detailed information about the absorption process cannot be obtained from global parameters like bioavailability and mean absorption time. If they remain unchanged under coadministration of a P-gp inductor, this does not necessarily mean that drug absorption is not affected. By evaluating the time course of the input rate, we can get more insight into the region-specific absorption of talinolol. The results clearly demonstrate the advantages of our approach compared to the noncompartmental analysis.
Change history
06 October 2022
The original article has been updated with the correct supplementary information.
Abbreviations
- \(R{D}_{i}^{2}\) :
-
Shape parameter of the ith inverse Gaussian function
- D :
-
Dose
- F :
-
Bioavailability
- f i(t):
-
iTh inverse Gaussian function
- I(t):
-
Input (absorption) rate
- IG:
-
Inverse Gaussian function
- I max ,1 :
-
Maximum of the first peak of the input rate
- I max ,2 :
-
Maximum of the second peak of the input rate
- MIT :
-
Mean input time
- MT i :
-
Scale parameter of the ith inverse Gaussian function
- ND :
-
Number of doses
- P-gp:
-
P-glycoprotein
- t max, 1 :
-
Time of the first peak
- t max, 2 :
-
Time of the second peak
References
Brvar N, Mateovic-Rojnik T, Grabnar I. Population pharmacokinetic modelling of tramadol using inverse Gaussian function for the assessment of drug absorption from prolonged and immediate release formulations. Int J Pharm. 2014;473(1–2):170–8. https://doi.org/10.1016/j.ijpharm.2014.07.013.
Delavenne X, Ollier E, Basset T, Bertoletti L, Accassat S, Garcin A, et al. A semi-mechanistic absorption model to evaluate drug-drug interaction with dabigatran: application with clarithromycin. Br J Clin Pharmacol. 2013;76(1):107–13. https://doi.org/10.1111/bcp.12055.
Wang J, Weiss M, D’Argenio DZ. A note on population analysis of dissolution-absorption models using the inverse Gaussian function. J Clin Pharmacol. 2008;48(6):719–25. https://doi.org/10.1177/0091270008315956.
Weiss M. A novel extravascular input function for the assessment of drug absorption in bioavailability studies. Pharm Res. 1996;13(10):1547–53.
Weiss M, Sermsappasuk P, Siegmund W. Modeling the kinetics of digoxin absorption: enhancement by P-glycoprotein inhibition. J Clin Pharmacol. 2012;52(3):381–7. https://doi.org/10.1177/0091270010396711.
Abebe BT, Weiss M, Modess C, Tadken T, Wegner D, Meyer MJ, et al. Pharmacokinetic drug-drug interactions between trospium chloride and ranitidine substrates of organic cation transporters in healthy human subjects. J Clin Pharmacol. 2020;60(3):312–23. https://doi.org/10.1002/jcph.1523.
Tadken T, Weiss M, Modess C, Wegner D, Roustom T, Neumeister C, et al. Trospium chloride is absorbed from two intestinal “absorption windows” with different permeability in healthy subjects. Int J Pharm. 2016;515(1–2):367–73. https://doi.org/10.1016/j.ijpharm.2016.10.030.
Weiss M. Empirical models for fitting of oral concentration time curves with and without an intravenous reference. J Pharmacokinet Pharmacodyn. 2017;44(3):193–201. https://doi.org/10.1007/s10928-017-9507-3.
Weiss M, Siegmund W. Pharmacokinetic modeling of ketamine enantiomers and their metabolites after administration of prolonged-release ketamine with emphasis on 2,6-Hydroxynorketamines. Clin Pharmacol Drug Dev. 2021. https://doi.org/10.1002/cpdd.993.
Wendling T, Ogungbenro K, Pigeolet E, Dumitras S, Woessner R, Aarons L. Model-based evaluation of the impact of formulation and food intake on the complex oral absorption of mavoglurant in healthy subjects. Pharm Res. 2015;32(5):1764–78. https://doi.org/10.1007/s11095-014-1574-1.
Csajka C, Drover D, Verotta D. The use of a sum of inverse Gaussian functions to describe the absorption profile of drugs exhibiting complex absorption. Pharm Res. 2005;22(8):1227–35. https://doi.org/10.1007/s11095-005-5266-8.
Oswald S, Terhaag B, Siegmund W. In vivo probes of drug transport: commonly used probe drugs to assess function of intestinal P-glycoprotein (ABCB1) in humans. Handb Exp Pharmacol. 2011;201:403–47. https://doi.org/10.1007/978-3-642-14541-4_11.
Shi JG, Zhang Y, Yeleswaram S. The relevance of assessment of intestinal P-gp inhibition using digoxin as an in vivo probe substrate. Nat Rev Drug Discov. 2011;10(1):75. https://doi.org/10.1038/nrd3028-c1 (author reply).
Westphal K, Weinbrenner A, Zschiesche M, Franke G, Knoke M, Oertel R, et al. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin Pharmacol Ther. 2000;68(4):345–55. https://doi.org/10.1067/mcp.2000.109797.
Weitschies W, Bernsdorf A, Giessmann T, Zschiesche M, Modess C, Hartmann V, et al. The talinolol double-peak phenomenon is likely caused by presystemic processing after uptake from gut lumen. Pharm Res. 2005;22(5):728–35. https://doi.org/10.1007/s11095-005-2588-5.
Elmeliegy M, Vourvahis M, Guo C, Wang DD. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug-drug interaction studies. Clin Pharmacokinet. 2020;59(6):699–714. https://doi.org/10.1007/s40262-020-00867-1.
Gramatte T, Oertel R, Terhaag B, Kirch W. Direct demonstration of small intestinal secretion and site-dependent absorption of the beta-blocker talinolol in humans. Clin Pharmacol Ther. 1996;59(5):541–9. https://doi.org/10.1016/S0009-9236(96)90182-4.
Kagan L, Dreifinger T, Mager DE, Hoffman A. Role of p-glycoprotein in region-specific gastrointestinal absorption of talinolol in rats. Drug Metab Dispos. 2010;38(9):1560–6. https://doi.org/10.1124/dmd.110.033019.
D’Argenio D, Schumitzky A, Wang X. User´s guide: Pharmacokinetic/pharmacodynamic systems analysis software. Los Angeles: Biomedical Simulations Resource; 2009.
Weiss M, Kriangkrai W, Sungthongjeen S. An empirical model for dissolution profile and its application to floating dosage forms. Eur J Pharm Sci. 2014;56:87–91. https://doi.org/10.1016/j.ejps.2014.02.013.
Chiou WL, Ma C, Wu TC, Jeong HY. Unexpected lack of effect of the rifampin-induced P-glycoprotein on the oral bioavailability of its substrate, talinolol, in humans: implication in phenotyping. J Pharm Sci. 2003;92(1):4–7. https://doi.org/10.1002/jps.10261 (discussion 8-9).
Zamek-Gliszczynski MJ, Patel M, Yang X, Lutz JD, Chu X, Brouwer KLR, et al. Intestinal P-gp and putative hepatic OATP1B induction: international transporter consortium perspective on drug development implications. Clin Pharmacol Ther. 2021;109(1):55–64. https://doi.org/10.1002/cpt.1916.
Drozdzik M, Busch D, Lapczuk J, Muller J, Ostrowski M, Kurzawski M, et al. Protein abundance of clinically relevant drug transporters in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther. 2019;105(5):1204–12. https://doi.org/10.1002/cpt.1301.
Drozdzik M, Groer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, et al. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm. 2014;11(10):3547–55. https://doi.org/10.1021/mp500330y.
Takayama K, Ito K, Matsui A, Yamashita T, Kawakami K, Hirayama D, et al. In vivo gene expression profile of human intestinal epithelial cells: from the viewpoint of drug metabolism and pharmacokinetics. Drug Metab Dispos. 2021;49(3):221–32. https://doi.org/10.1124/dmd.120.000283.
Koziolek M, Grimm M, Schneider F, Jedamzik P, Sager M, Kuhn JP, et al. Navigating the human gastrointestinal tract for oral drug delivery: Uncharted waters and new frontiers. Adv Drug Deliv Rev. 2016;101:75–88. https://doi.org/10.1016/j.addr.2016.03.009.
Abuhelwa AY, Foster DJR, Upton RN. A quantitative review and meta-models of the variability and factors affecting oral drug Absorption-Part II: gastrointestinal transit time. AAPS J. 2016;18(5):1324–33. https://doi.org/10.1208/s12248-016-9953-7.
Grimm M, Koziolek M, Kühn JP, Weitschies W. Interindividual and intraindividual variability of fasted state gastric fluid volume and gastric emptying of water. Eur J Pharm Biopharm. 2018;127:309–17. https://doi.org/10.1016/j.ejpb.2018.03.002.
Lavin JH, French SJ, Read NW. Comparison of oral and gastric administration of sucrose and maltose on gastric emptying rate and appetite. Int J Obes Relat Metab Disord. 2002;26(1):80–6. https://doi.org/10.1038/sj.ijo.0801828.
Mudie DM, Murray K, Hoad CL, Pritchard SE, Garnett MC, Amidon GL, et al. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol Pharm. 2014;11(9):3039–47. https://doi.org/10.1021/mp500210c.
Gramatte T, Terhaag B. The variability of orocecal transit time evaluated by the salicylazosulphapyridine/sulphapyridine method. Int J Clin Pharmacol Ther Toxicol. 1991;29(4):147–50.
Kolbow J, Modess C, Wegner D, Oswald S, Maritz MA, Rey H, et al. Extended-release but not immediate-release and subcutaneous methylnaltrexone antagonizes the loperamide-induced delay of whole-gut transit time in healthy subjects. J Clin Pharmacol. 2016;56(2):239–45. https://doi.org/10.1002/jcph.624.
Acocella G. Clinical pharmacokinetics of rifampicin. Clin Pharmacokinet. 1978;3(2):108–27. https://doi.org/10.2165/00003088-197803020-00002.
Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104(2):147–53. https://doi.org/10.1172/JCI6663.
Asaumi R, Toshimoto K, Tobe Y, Hashizume K, Nunoya KI, Imawaka H, et al. Comprehensive PBPK model of rifampicin for quantitative prediction of complex drug-drug interactions: CYP3A/2C9 induction and OATP inhibition effects. CPT Pharmacometrics Syst Pharmacol. 2018;7(3):186–96. https://doi.org/10.1002/psp4.12275.
Qian CQ, Zhao KJ, Chen Y, Liu L, Liu XD. Simultaneously predict pharmacokinetic interaction of rifampicin with oral versus intravenous substrates of cytochrome P450 3A/Pglycoprotein to healthy human using a semi-physiologically based pharmacokinetic model involving both enzyme and transporter turnover. Eur J Pharm Sci. 2019;134:194–204. https://doi.org/10.1016/j.ejps.2019.04.026.
Yamazaki S, Costales C, Lazzaro S, Eatemadpour S, Kimoto E, Varma MV. Physiologically-based pharmacokinetic modeling approach to predict rifampin-mediated intestinal P-Glycoprotein induction. CPT Pharmacometrics Syst Pharmacol. 2019;8(9):634–42. https://doi.org/10.1002/psp4.12458.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest Statement
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1
(TXT 9.43 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weiss, M., D’Argenio, D.Z. & Siegmund, W. Analysis of Complex Absorption After Multiple Dosing: Application to the Interaction Between the P-glycoprotein Substrate Talinolol and Rifampicin. Pharm Res 39, 3293–3300 (2022). https://doi.org/10.1007/s11095-022-03397-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-022-03397-6