
ABSTRACT
Purpose
Insulin lispro is a rapid-acting insulin analogue produced by recombinant DNA technology. As a biosynthetic drug, the protein undergoes strict monitoring aiming for detection and characterization of impurities. The goal of this study was to isolate and identify a derivative of insulin lispro formed during biosynthesis.
Methods
For this purpose, ion exchange chromatography in combination with endoproteinase Glu-C digestion, MALDI-TOF/TOF mass spectrometry and Edman sequencing were employed.
Results
Ion exchange chromatography analysis of related proteins in development batches of recombinant insulin lispro revealed the existence of unknown derivative in excess of the assumed limit. Its molecular mass was 42 Da higher than the theoretical mass of Lys(B31) insulin lispro—one of the expected process-related intermediates. Endoproteinase Glu-C cleavage enabled indication of the modified peptide. Tandem mass spectrometry (MS/MS) allowed to explore the location and type of the modification. The 42 amu shift was present in the mass of y-type ions, while b-type ions were in agreement with theoretical values. It suggested that the modification is present on B31 lysine. Further inquiry revealed the presence of two diagnostic ions for lysine acetylation at m/z 143.1 and 126.1. In addition, the peptide was isolated and sequenced by Edman degradation. Standards of phenylthiohydantoin derivatives of N-ε-acetyl-L-lysine and N-ε-trimethyl-L-lysine, not available commercially, were synthesized in the laboratory. The retention time of the modified residue confirmed its identity as N-ε-acetyl-L-lysine.
Conclusions
The derivative of insulin lispro formed during biosynthesis of the drug was identified to be N-ε-acetyl-L-lysine (B31) insulin lispro.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In 1980s recombinant human insulin has been implemented as a base treatment for patients suffering from diabetes (1). However, when administered, the drug did not mimic physiological, 24-h long profile of endogenous hormone release by beta-cells. Therefore, in recent years, analogues of human insulin have been engineered to improve insulin therapy through modulation of onset and duration of action after injection. Insulin lispro is a rapid-acting insulin analogue designed by inversion of two penultimate amino acids: proline (B28) and lysine (B29) on the C-terminal end of the B-chain of human insulin. The subtle change in the amino acid sequence does not alter receptor binding, whereas it considerably diminishes the insulin’s natural propensity to self-associate to dimers and higher order oligomers (2). Larger amounts of active monomeric insulin available for postprandial injections enable significantly faster glucose level reduction. As a result, the insulin analogue shortens noticeably the time interval between the injection and a meal, offering a greater flexibility with meal times (3).
The analogue is produced by recombinant DNA technology in E. coli as a fusion protein which is further converted to the functional hormone by selected enzymes. However, inherent feature of all recombinant proteins is the presence of post-translational modifications (PTMs) related to their production system and storage (4–6). All PTMs present in a protein drug product may potentially affect its safety and efficacy and should be evaluated in terms of toxicity, immunogenicity and biological activity. In living cells PTMs result in structural and functional heterogeneity and thus can affect numerous biological processes e.g. DNA replication and repair, cellular signalling, cell growth, metabolism, development and apoptosis (7–9). In broader perspective, understanding of biological implications of PTMs on human health is of significant value as it is still unclear if PTMs are the cause or the consequence of certain diseases (10). One such PTM is lysine acetylation which is gaining increasing attention. The recent reports concerning many thousands of acetylated proteins in both eukaryotes and prokaryotes suggest that lysine acetylation is involved in regulation of diverse substantial cellular processes such as chromatin remodeling, protein synthesis, nuclear transport, cell cycle and the coordination of different metabolic pathways (11–14).
It was previously acknowledged that lysine acetylation was regulated by enzymes such as lysine acetyltransferases (KATs) and deacetylases (KDACs) (15). However, B. N. Violand et al. suggested that acetylation of lysines may occur by chemical mechanism with acetyl-CoA or another metabolic intermediate providing the source of the acetyl group (16). Indeed, recent studies have shown that lysine acetylation can also proceed non-enzymatically using acetyl-CoA (17) and acetyl phosphate (AcP) as the acetyl donor (18). Interestingly, an analogous AcP-dependent non-enzymatic reaction was observed earlier for phosphorylation (19,20). AcP is a small-molecule metabolite in the phosphotransacetylase (Pta)—acetate kinase (AckA) pathway, one of the basic metabolic networks in E. coli (19,21). In light of the recent explosion of basic research on lysine acetylation (22–24), it seems important to recall previous studies on acetylation of recombinant therapeutic proteins produced in E. coli: neurotropin-3 (25), somatotropin (16), interleukin-10 (26), interleukin-2 (27), chemokine RANTES (regulated on activation, normal T cell expressed and secreted) (17), interferon alfa (28) and gamma (29), stathmin-like subdomains (30), human basic fibroblast growth factor mutein (31) and prochymosin (32). These results support current attempts to understand the mechanism of acetylation and its implications on cell life. Moreover, new discoveries should be expected in this filed since E. coli is still one of the most widely used expression hosts in the biopharmaceutical industry. Nearly 30% of currently approved recombinant therapeutic proteins are produced in this system (33). In concern for patients, the drug approval by regulatory authorities is dependent on still growing demands related to its efficacy, quality and safety (34). Impurities and derivatives of therapeutic proteins should be removed in a purification step or, if present in acceptable quantities, characterized to the greatest possible extent.
The purpose of this work was to isolate and identify an acetylated derivative formed during biosynthesis of insulin lispro. The main difficulty in characterization of the derivative was its limited availability. Moreover, a mass coincidence between acetylated and tri-methylated protein derivatives (see Fig. 1) was a challenge for mass spectrometry analysis.

Structures of acetylated and trimethylated lysine residues.
We applied ion exchange chromatography to isolate the derivative from insulin lispro substance. The derivative was further characterized by combination of enzymatic digestion, mass spectrometry and Edman sequencing. The results confirmed interchangeably that the protein isolated from a production batch is an acetylated derivative of recombinant lispro insulin.
MATERIALS AND METHODS
Source of Insulin Lispro
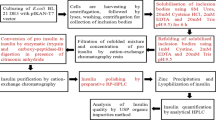
Insulin lispro was produced by recombinant DNA technology essentially as described previously (35). Shortly, insulin lispro precursor was expressed in E. coli in the form of insoluble inclusion bodies. After separation from cell debris, the precursor was transformed into insulin lispro by enzymatic digestion with trypsin and carboxypeptidase B. Finally, insulin was purified by chromatography, dialysis and crystallization.
Isolation of the Insulin Lispro Acetylated Derivative
The derivative was isolated from a development batch of insulin lispro substance by repetitive ion exchange chromatography (IEC) using ICS-5000 system with a PDA detector (Dionex, Sunnyvale, USA). 0.1 mg of the insulin lispro containing 0.8% of the derivative was injected on a DNA Pac PA-100, 250 × 4.0 column (Dionex, Sunnyvale, USA). The separation was carried out at 35°C with a linear gradient elution from 0% to 25% eluent B in 30 min at the flow rate 0.5 ml/min. Eluent A was 8 mM phosphate buffer, 67% ethanol pH 7.6 and eluent B was 0.3 M NaCl, 8 mM phosphate buffer, 67% ethanol pH 7.6. The peak of the derivative (relative retention time 0.8; see Fig. 3) was collected. This procedure was repeated several times to obtain sufficient amount of the derivative. All fractions of the derivative were pooled and evaporated to dryness using Concentrator Plus vacuum centrifuge (Eppendorf, Hamburg, Germany) and kept at −20°C until use.
Peptide Mapping and Isolation of Peptide B22-B31
The derivative was dissolved in 0.5 ml of 0.1 M HEPES buffer pH 7.5 and mixed with 10 μl of 10 μg/ml endoproteinase Glu-C (Protease S. aureus V8; Thermo Scientific, Rockford, USA). The mixture was incubated for 4 h at 37°C. Then the HPLC system (Alliance 2695, Waters, Milford, USA) with UV detector (2489, Waters, Milford, USA), equipped with a Zorbax SB-C18 1.8, 4.6 × 50 mm column (Agilent, Palo Alto, USA), was employed to separate and isolate the peptides. The separation was carried out at 40°C with a linear gradient elution from 10% to 50% eluent B in 20 min at a flow rate 1 ml/min. Eluent A was 0.1% trifluoroacetic acid (TFA) and eluent B was 0.1% TFA with 90% acetonitrile (ACN) (both vol./vol.). The peak of the peptide B21-B31 was collected with Fraction Collector III (Waters, Milford, USA) and eluent was evaporated as mentioned above.
Synthesis of Phenylthiohydantoin Amino Acid Standards for Protein Sequencing
The water solutions of N-ε-acetyl-L-lysine (acK) and N-ε-N-ε-N-ε-trimethyl-L-lysine (3meK, both from Sigma, St. Louis, USA), containing 50 nmol each, were evaporated to dryness in Eppendorf tubes and converted into phenylthiocarbamyl (PTC) derivatives essentially as described elsewhere (36). Obtained dry deposits were then converted into phenylthiohydantoin (PTH) derivatives by dissolving them in 100 μl portions of 25% (vol./vol.) TFA, 0.001% (wt./vol.) dithiothreitol in water, flushing with argon, sealing, and incubation for 30 min at 64°C. After this step the PTH derivatives were purified by HPLC on a LC-18-DB 4.6 × 250 mm column (Supelco, Bellefonte, USA). Two solvents were used: A – 0.1% TFA (all vol./vol.) and B – 0.07% TFA, 80% ACN (vol./vol.). The linear gradient 0–100% of the solvent B over 15 min, spectrophotometric detection at 269 nm, and 1 ml/min flow rate were applied. Fractions containing PTH-acK and PTH-3meK were collected, evaporated to dryness, and kept at −20°C until use.
N-terminal Sequencing
Determination of N-terminal sequence of polypeptide chains by Edman degradation was performed on a PPSQ30 (Shimadzu, Kyoto, Japan) automatic protein sequencer. Before analyses the instrument was calibrated using a commercial PTH proteinaceous amino acid standard mixture (Wako, Osaka, Japan) and, separately, using the synthesized PTH-acK as well as PTH-3meK. The calibration was performed by 25 pmol amounts of these compounds, dissolved previously in 37% (vol./vol.) ACN in water. The sequencing was done using polypeptide chains absorbed on TFA-treated glass fiber disks (Wako, Osaka, Japan) coated by polybrene (Sigma, St. Louis, USA).
Mass Spectrometry
The insulin lispro derivative and endoproteinase Glu-C derived peptides were analyzed in a reflector mode with the use of 4800 Plus MALDI-TOF/TOF mass spectrometer (AB SCIEX, Framingham, USA). α-cyano-4-hydroxy-cinnamic acid matrix was exploited. External calibration was performed with a 4700 proteomics analyzer calibration mixture provided by AB SCIEX. Data Explorer Software, Version 4.9, and General Protein/Mass Analysis for Windows, version 8.2, were applied for assignment of a, b and y ions to the MS/MS spectrum of acetylated peptide. DeNovo Explorer (GPS Explorer TM Software, Version 3.6) was exploited to match the MS/MS spectrum to the annotated peptide sequence.
RESULTS AND DISCUSSION
Insulin lispro is a short-acting insulin analogue produced by recombinant DNA technology in E. coli as a precursor which is further converted to the native hormone by enzymes (Fig. 2).

Scheme of enzymatic conversion of precursor during the production process of insulin lispro.
During the production process, different impurities called related proteins are formed at both biosynthesis and purification steps. Related proteins constitute various modifications of a desired protein including deamidated, methylated, oxidized, cleaved, aggregated forms and adducts, just to name a few, and they are present in a final bulk of a drug substance. All can be monitored by various chromatographic methods. Figure 3 illustrates the IEC-chromatogram of insulin lispro manufactured at Institute of Biotechnology and Antibiotics in Warsaw.

IEC chromatogram of insulin lispro.
The main peak in this chromatogram is insulin lispro. Small peaks arising from the baseline (seen in enlargement in Fig. 3) are related proteins. The derivative peak 1 was isolated and subjected for further investigation. Taking into account the retention time relative to insulin lispro, derivative 1 was suspected to be one of intermediates possessing additional basic amino acids at chain B: Lys(B31) insulin lispro or Lys(B31)Arg(B32) insulin lispro, which can appear during enzymatic conversion of the precursor (see Fig. 2). Surprisingly, MALDI analysis of the collected fraction of peak 1 showed a +42 mass increment when compared to the theoretical mass of Lys(B31) insulin lispro (5932.7 Da) (Fig. 4). Such a change suggests trimethylation or acetylation of lysine. On the basis of the recent discoveries of serine and threonine acetylation by a bacterial effector from Yersinia (37), these residues were also considered as suspicious during our study. Moreover, two modifications, methylation and formylation, can occur simultaneously within Lys(B31) insulin lispro molecule resulting in +42 mass shift (14 Da + 28 Da).

MALDI TOF spectrum of derivative 1.
Enzymatic digestion and peptide mapping were employed to identify the modification site of the derivative. The experiment was performed in parallel with insulin lispro. The complete digestion of insulin lispro with endoproteinase Glu-C results in four fragments: I (A5-A17, B1-B13) II (A18-A21, B14-B21), III (B22-B30), IV (A1-A4). A small amount of intermediate fragment I + IV might be present due to slow hydrolysis of A4-A5 bond. When the peptide maps of insulin lispro and derivative 1 were compared (see Fig. 5) the difference in the retention time of the peak corresponding to fragment III of the derivative and insulin lispro was observed. It was indicative of a modification within fragment III (peptide B22-B31).

Peptide maps (RP-HPLC chromatograms) of insulin lispro and the derivative 1 after enzymatic digestion with endoproteinase Glu-C; fragment IV eluted at dead time.
Expected peptide fragments resulting from digestion of the derivative with endoproteinase Glu-C are listed in Table I. MALDI analysis of the digest gave a distinct signal at m/z 1286.7 (a sum of 1244.7 + 42; see Fig. 6) which was assigned to peptide B22-B31 (fragment III of the derivative). As this peptide does not contain any serine, further investigation was narrowed down to threonine and lysine residues.

MALDI TOF spectrum of derivative 1 after enzymatic digestion with endoproteinase Glu-C.
Tandem mass spectrometry was performed on this peptide to explore the location and type of the modification. All b-type ions seen in MS/MS spectrum (Fig. 7) were in agreement with theoretical values, whereas y-type ions were 42 amu over the predicted masses. The presence of two ions: b9 [1098.6] and y1 [189.1] confirms that B30 Thr remains unmodified, while B31 Lys is altered. The sequence of the peptide B22-B31 was as follows: RGFFYTKPTKmod. Closer inspection of the MS/MS spectrum revealed the presence of two diagnostic ions for acetylated lysine at m/z 143.1 and m/z 126.1 (Fig. 8). Although the signal at m/z 143.1 can be also associated with mono-ethylated arginine, the co-presence of 143.1 with 126.1 undoubtedly indicates acetylation. The immonium ion at m/z 126.1 is specific for lysine acetylation and enables discrimination between acetylation and trimethylation of lysine (38).

MALDI-TOF/TOF spectrum of peptide B22-B31.

Enlarged MALDI-TOF/TOF spectrum of peptide B22-B31 with diagnostic ions 126.1 and 143.1. Insert – structure of diagnostic ions.
In order to confirm the type of modification in derivative 1, Edman degradation was employed as an orthogonal method. During Edman degradation the automatic protein sequencer consecutively detaches amino acid residues from N-termini of the analyzed polypeptides, converts them into PTH derivatives and performs their identification by HPLC chromatography. Since standards of PTH derivatives of N-ε-acetyl-L-lysine and N-ε-trimethyl-L-lysine are not commercially available, they were synthesized in the laboratory. Analysis of the peptide B22-B31 gave the following 9-mer sequence: RGFFYTKPTKac (N-ε-acetyl-L-lysine was ascertained at 10th position). The chromatograms illustrating the mentioned 10th amino acid residue of this peptide superimposed on acK as well as 3meK standards are presented in Fig. 9. Peaks belonging to the standards of N-ε-acetyl-L-lysine and N-ε-trimethyl-L-lysine differ in retention time sufficiently to allow the exclusion of trimethylation at B31 in the derivative.

Identification of the 10th residue of the peptide B22-B31 performed during Edman degradation. The illustration presents the HPLC chromatogram of the PTH amino acid residue detached during 10th cycle of sequencing, superimposed on the chromatograms of the acK as well as 3meK standards. The retention time of the 10th residue is identical as that of N-ε-acetyl-L-lysine.
On the basis of the presented results it is interesting that a derivative with acetylated lysine at position B28 was dot detected so far. It is possible that acetylation does not take place in other locations than B31 or acetylated derivatives are formed at undetectable levels. A new project is open now in our institute to understand mechanism of acetylation in insulin analogues produced in E. coli and to examine all possible acetylation sites. Details of this work will be published in the future.
CONCLUSIONS
Ion exchange chromatography analysis of related proteins in development batches of recombinant insulin lispro revealed the existence of unknown derivative in excess of the assumed limit 0.1%. This derivative was monitored in all development batches of insulin lispro and it occurred in a range of <LOD – 0.8% depending on purification methods. The molecular mass of the derivative was 42 Da higher that the theoretical mass of Lys(B31) insulin lispro—one of the expected process-related intermediates. Both, Edman sequencing and MS/MS fragmentation provided clear-cut evidence of lysine acetylation and exclusion of trimethylation contingency. The derivative was identified to be N-ε-acetyl-L-lysine (B31) insulin lispro. We do not know at present if acetylation of this insulin lispro derivative proceeds enzymatically or with the participation of acetyl phosphate (AcP) as the acetyl donor, but certainly our discovery is another indication that lysine acetylation is a frequently occurring posttranslational modification in E. coli. It is highly probable that our finding does not close the above list of acetylated therapeutic proteins produced in E. coli. A worldwide intensification of research on protein acetylation should bring new discoveries in this field.
Abbreviations
- 3meK:
-
N-ε-N-ε-N-ε-trimethyl-L-lysine
- acK:
-
N-ε-acetyl-L-lysine
- AckA:
-
Acetate kinase
- ACN:
-
Acetonitrile
- AcP:
-
Acetyl phosphate
- IEC:
-
Ion exchange chromatography
- KATs:
-
Lysine acetyltransferases
- KDACs:
-
Lysine deacetylases
- Pta:
-
Phosphotransacetylase
- PTC:
-
Phenylthiocarbamyl
- PTH:
-
Phenylthiohydantoin
- PTM:
-
Post-translational modification
- RANTES:
-
Regulated on activation, normal T cell expressed and secreted chemokine
- TFA:
-
Trifluoroacetic acid
REFERENCES
Heller S, Kozlovski P, Kurtzhals P. Insulin’s 85th anniversary-An enduring medical miracle. Diabetes Res Clin Pract. 2007;78(2):149–58.
Ciszak E, Beals JM, Frank BH, Baker JC, Carter ND, Smith GD. Role of C-terminal B-chain residues in insulin assembly: the structure of hexameric LysB28ProB29-human insulin. Structure. 1995;3(6):615–22.
Bakaysa DL, Radziuk J, Havel HA, Brader ML, Li S, Dodd SW, et al. Physicochemical basis for the rapid time-action of LysB28ProB29-insulin: dissociation of a protein-ligand complex. Protein Sci. 1996;5(12):2521–31.
Klein ML, Bartley TD, Davis JM, Whiteley DW, Lu HS. Isolation and structural characterization of three isoforms of recombinant consensus α interferon. Arch Biochem Biophys. 1990;276(2):531–7.
Bierczyńska-Krzysik A, Łopaciuk M, Pawlak-Morka R, Stadnik D. Investigation of asparagine deamidation in a SOD1-based biosynthetic human insulin precursor by MALDI-TOF mass spectrometry. Acta Biochim Pol. 2014;61(2):349–57.
Walsh G. Post-translational modifications of protein biopharmaceuticals. Drug Discov Today. 2010;15(17–18):773–80.
Abdel-Hafiz HA, Horwitz KB. Post-translational modifications of the progesterone receptors. J Steroid Biochem Mol Biol. 2014;140:80–9.
Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17(6):666–72.
Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12(12):773–86.
Karsdal MA, Henriksen K, Leeming DJ, Woodworth T, Vassiliadis E, Bay-Jensen AC. Novel combinations of Post-Translational Modification (PTM) neo-epitopes provide tissue-specific biochemical markers-are they the cause or the consequence of the disease? Clin Biochem. 2010;43(10–11):793–804.
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327(5968).
Soppa J. Protein acetylation in archaea, bacteria, and eukaryotes. Archaea. 2010;1–9.
Lima BP, Antelmann H, Gronau K, Chi BK, Becher D, Brinsmade SR, et al. Involvement of protein acetylation in glucose-induced transcription of a stress-responsible promoter. Mol Microbiol. 2011;81(5):1190–204.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and Co-regulates major cellular functions. Science. 2009;325(5942):834–40.
Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31(4):449–61.
Violand BN, Schlittler MR, Lawson CQ, Kane JF, Siegel NR, Smith CE, et al. Isolation of Escherichia coli synthesized recombinant eukaryotic proteins that contain epsilon-N-acetyllysine. Protein Sci. 1994;3(7):1089–97.
d’ Alayer J, Expert-Bezançon N, Béguin P. Time- and temperature-dependent acetylation of the chemokine RANTES produced in recombinant Escherichia coli. Protein Expr Purif. 2007;55(1):9–16.
Weinert BT, Iesmantavicius V, Wagner SA, Schölz C, Gummesson B, Beli P, et al. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell. 2013;51(2):265–72.
McCleary WR, Stock JB, Ninfa AJ. Is acetyl phosphate a global signal in Escherichia coli. J Bacteriol. 1993;175(10):2793–8.
Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, et al. Structural, Kinetic and Proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One. 2014;9(4):e94816.
Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69(1):12–50.
Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, et al. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia Coli. Mol Cell Proteomics. 2009;8(2):215–25.
Thao S, Chen CS, Zhu H, Escalante-Semerena JC. N-ε-lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS One. 2010;5(12):e15123.
Ma Q, Wood TK. Protein acetylation in prokaryotes increases stress resistance. Biochem Biophys Res Commun. 2011;410(4):846–51.
Ross FE, Zamborelli T, Herman AC, Yeh CH, Tedeschi NI, Luedke ES. Detection of acetylated lysine residues using sequencing by Edman degradation and mass spectrometry. In: Marshak D, editor. Techniques in protein chemistry, Vol. VII. New York: Academic; 1996. p. 201–7.
Pflumm MN, Gruber SC, Tsarbopoulos A, Wylie D, Pramanik B, Bausch JN, et al. Isolation and characterization of an acetylated impurity in Escherichia coli-derived recombinant human interleukin-10 (IL-10) drug substance. Pharm Res. 1997;14(6):833–6.
Moya G, González LJ, Huerta V, García Y, Morera V, Pérez D, et al. Isolation and characterization of modified species of a mutated (Cys125 -Ala) recombinant human interleukin-2. J Chromatogr A. 2002;971(1–2):129–42.
Takao T, Kobayashi M, Nishimura O, Shimonishi Y. Chemical characterization of recombinant human leukocyte interferon A using fast atom bombardment mass spectrometry. J Biol Chem. 1987;262(8):3541–7.
Honda S, Asano T, Kajio T, Nishimura O. Escherichia coli-derived human interferon-gamma with Cys-Tyr-Cys at the N-terminus is partially N alpha-acylated. Arch Biochem Biophys. 1989;269(2):612–22.
Charbaut E, Redeker V, Rossier J, Sobel A. N-terminal acetylation of ectopic recombinant proteins in Escherichia coli. FEBS Lett. 2002;529(2–3):341–5.
Suenaga M, Ohmae H, Tsuji S, Tanaka Y, Koyama N, Nishimura O. epsilon-N-acetylation in the production of recombinant human basic fibroblast growth factor mutein. Prep Biochem Biotechnol. 1996;26(3–4):259–70.
Wu J, Chang S, Gong X, Liu D, Ma Q. Identification of N-terminal acetylation of recombinant human prothymosin alpha in Escherichia coli. Biochim Biophys Acta. 2006;1760(8):1241–7.
Huang CJ, Lin H, Yang X. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J Ind Microbiol Biotechnol. 2012;39(3):383–99.
Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15(1–2):40–56.
Bocian W, Borowicz P, Mikołajczyk J, Sitkowski J, Tarnowska A, Bednarek E, et al. NMR structure of biosynthetic engineered human insulin monomer B31(Lys)-B32(Arg) in water/acetonitrile solution comparison with the solution structure of native human insulin monomer. Biopolymers. 2008;89(10):820–30.
White JA, Hart RJ, Fry JC. An evaluation of the Waters Pico-Tag system for the amino-acid analysis of food materials. J Autom Chem. 1986;8(4):170–7.
Mukherjee S, Hao YH, Orth K. A newly discovered post-translational modification–the acetylation of serine and threonine residues. Trends Biochem Sci. 2007;32(5):210–6.
Zhang K, Yau PM, Chandrasekhar B, New R, Kondrat R, Imai BS, et al. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: an application for determining lysine 9 acetylation and methylation of histone H3. Proteomics. 2004;4(1):1–10.
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by EU within the European Regional Development Fund—POIG. 01.01.02-00-007/08-06. The research was partially carried out with equipment purchased on grants POIG.02.01.00-12-064/08 and POIG.02.01.00-12-167/08. Faculty of Biochemistry, Biophysics and Biotechnology is a partner of the Leading National Research Center (KNOW) supported by the Ministry of Science and Higher Education.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Szewczak, J., Bierczyńska-Krzysik, A., Piejko, M. et al. Isolation and Characterization of Acetylated Derivative of Recombinant Insulin Lispro Produced in Escherichia coli . Pharm Res 32, 2450–2457 (2015). https://doi.org/10.1007/s11095-015-1637-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1637-y