
Abstract
Knowledge concerning transport of maternally administered drugs across the placental barrier is essential for determining potential toxicity of drugs to the fetus and the value of drug therapy during pregnancy. An important determinant for fetal drug exposure is the expression of efflux transporters in the placenta. Among human tissues, the ATP-binding cassette efflux transporter BCRP (gene symbol ABCG2) is most abundantly expressed in the apical membrane of placental syncytiotrophoblasts. Although the precise physiological role of BCRP in the placenta is still unclear, existing data strongly suggest that BCRP plays an important role in protecting the fetus against the potential toxicity of drugs, xenobiotics, and metabolites by expelling them across the placental barrier. In this review, we summarize the current knowledge with respect to the expression, function, and polymorphisms of BCRP, as well as transcriptional and posttranscriptional regulation of the transporter in the placenta. Finally, clinical significance of BCRP in the placenta for drug therapy in pregnant women is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Pregnant women often need to take medication to treat diseases including viral, fungal, or bacteria infections, epilepsy, hypertension, or pregnancy-induced conditions such as depression, nausea and gestational diabetes. In a study in which 578 pregnant women were interviewed, researchers found that 95.8% of the participants took at least one prescription drug, 92.6% of the participants self-medicated with over-the-counter medications, and 45.2% of the participants used herbal medications during pregnancy (1). More recently, a multi-center study also showed that 64% of all pregnant women in the USA used at least one prescription drug other than a vitamin or mineral supplement in the 270 days before delivery (2). Moreover, a large proportion of pregnant women (5–10%) received FDA category D or X drugs which are potentially teratogens, and the general pattern of drug use has been with higher use in early pregnancy compared to later trimesters (2,3). Thus, a major concern arising from the use of medication by pregnant women is the transfer of drugs across the placental barrier, leading to potential toxicity to the developing fetus, particularly at early gestational stages.
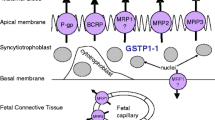
Most maternally administered drugs can cross the placental barrier by passive diffusion to some extent which is primarily determined by the physicochemical properties (e.g., molecular weight, pKa, and lipid solubility) and/or the pharmacokinetic characteristics (e.g., elimination half-life in the mother, protein binding in fetal and maternal compartments, and metabolism in the placenta and fetus) of the drugs (4–6). In recent years, the importance of ATP-binding cassette (ABC) efflux transporters expressed in the apical membrane of placental syncytiotrophoblasts in moderating drug penetration across the placental barrier and thereby limiting fetal drug exposure has been appreciated (7,8). The first ABC transporter that has been recognized to play a significant role in protecting the fetus is P-glycoprotein (P-gp). For example, Lankas et al. (9) showed that the absence of P-gp expression in the placenta of pregnant mice significantly increased the toxicity of the pesticide avermectin to the developing fetus with enhanced placental penetration of the pesticide. The two other major ABC efflux transporters that are expressed in the apical membrane of placental syncytiotrophoblasts and may also be important in protecting the fetus are the multidrug resistance protein 2 (MRP2) and the breast cancer resistance protein (BCRP; Fig. 1). Expression of ABC transporters in the placenta in general has been reviewed recently (8,10,11).

A schematic representation of localization of the major ABC efflux transporters, P-gp, BCRP, and MRP2, in the apical membrane of the placental syncytiotrophoblast. P-gp P-glycoprotein (ABCB1), BCRP breast cancer resistance protein (ABCG2), MRP2 multidrug resistance protein 2 (ABCC2)
This review focuses on BCRP. We will first give a brief overview of BCRP followed by the current knowledge regarding expression and function of the transporter in the placenta. Substantial variation in BCRP expression has been observed in human placenta (12), suggesting that considerable variability could exist in fetal exposure to drugs, xenobiotics, and metabolites. Such variable expression and/or activity may be caused by genetic polymorphisms of BCRP (12). BCRP expression in the placenta is possibly tightly controlled during pregnancy by pregnancy-related steroid hormones, growth factors, and cytokines. We therefore will also review the current understanding concerning single-nucleotide polymorphisms of BCRP in human placenta and the molecular mechanisms by which BCRP expression in the placenta is regulated. Finally, clinical significance of BCRP expression in the placenta will be discussed.
Breast Cancer Resistance Protein (BCRP)
BCRP is an approximately 75 kDa plasma membrane transporter belonging to the subfamily G of the large human ABC transporter superfamily. BCRP is the second member of the subfamily G and hence designated as ABCG2. BCRP was first cloned in 1998 from a breast cancer cell line, MCF-7/AdrVp which is highly resistant to doxorubicin with cross-resistance to daunorubicin and mitoxantrone (13). Shortly after that, an almost identical transporter termed MXR was discovered in a highly mitoxantrone-resistant human colon carcinoma cell line S1-M1–80 (14). Allikmets et al. (15) screened expressed sequence tag database and isolated ABCP cDNA that is essentially identical to the cDNA of BCRP or MXR. ABCP (ABC transporter in placenta) was so named to reflect its high level expression in human placenta. BCRP, MXR and ABCP are essentially the same protein with only a few amino acid differences. It has now been well established that BCRP is an ATP-dependent efflux transporter that is able to expel chemotherapeutic agents out of the cell, and is therefore considered as one of the most important ABC transporters that confer multidrug resistance in cancer cells (16,17).
Functional studies in the last decade indicate that BCRP can transport a broad spectrum of substrates, ranging from chemotherapeutic agents to organic anions. Substrate specificity of BCRP has been extensively reviewed (18,19). Typical chemotherapeutic agents that are transported by BCRP include mitoxantrone, camptothecin derivates (e.g., topotecan and irinotecan), and anthracyclines (e.g., daunorubicin and doxorubicin; 16). Many of these anti-cancer drugs are also P-gp substrates. Nucleoside analogs such as zidovudine (AZT) and lamivudine (3TC) are also BCRP substrates (20,21). BCRP substrates also include, among others, prazosin (22), the tyrosine kinase inhibitors CI1033 and STI571 (23,24), dipyridamole (25), and phytoestrogens (26). Of particular interest is that various drugs commonly administered to pregnant women, including nitrofurantoin (27), cimetidine (28) and glyburide (29), are BCRP substrates. Thus, a large of number of drugs from various therapeutic categories, including anti-cancer drugs, antibiotics, anti-hypertensive, and anti-diabetics, have been identified as BCRP substrates. BCRP can also transport a wide variety of organic anions, conjugated or unconjugated, including estrone-3-sulfate, 17β-estradiol 17-(β-d-glucuronide), dehydroepiandrosterone (30), and methotrexate (31). Collectively, BCRP displays an extremely broad spectrum of substrate specificity that is overlapping, but distinct from that of P-gp or MRP2. In general, P-gp preferentially transports uncharged hydrophobic compounds and some weakly basic substances (32); however, BCRP can transport both hydrophobic and hydrophilic substrates which are conjugated or unconjugated (16,18,32). MRP2 mainly transports organic anions, and glutathione, sulfate, and glucuronate conjugates (33). While BCRP can transport both sulfate and glucuronide conjugates, sulfate conjugates seem to be the preferred substrates (34). However, MRP2 seems to preferentially transport glutathione conjugates (with lower K m values; 33), and BCRP has so far not been shown to transport glutathione conjugates.
A variety of BCRP inhibitors have already been identified, including, among others, GF120918 (a second-generation P-gp inhibitor which belongs to the acridone carboxamide derivatives; 35), fumitremorgin C (FTC; a natural product secreted from the fungi Aspergillus fumigatus) and its analogs (36), tyrosine kinase inhibitors (e.g., CI1033 and STI571; 37,38), anti-HIV protease inhibitors (e.g., nelfinavir and ritonavir; 39), calcium channel blockers (25), immunosuppressants (40), and a variety of food dietary flavonoids (41). More detailed information on BCRP inhibitors can be found in recent reviews (18,42). Identification of BCRP substrates and inhibitors provides the opportunity to pharmacologically modulate BCRP function as a tool of drug therapy (e.g., circumvention of drug resistance in cancers) and allows understanding the molecular mechanisms underlying potential BCRP-mediated drug–drug interactions.
Tissues Expression and Membrane Localization of BCRP
BCRP expression in human tissues has been extensively investigated. Doyle et al. (13) assessed BCRP mRNA expression using commercially prepared human multi-tissue Northern blots. The highest level of BCRP mRNA was seen in placenta, which was approximately 100 times greater than that in liver, small intestine, colon, brain, liver, ovary, testis, or prostate. BCRP mRNA levels were also high in liver and small intestine. There was little or no BCRP mRNA expression in heart, lung, skeletal muscle, kidney, pancreas, spleen, thymus, or peripheral-blood leukocytes. BCRP was named ABCP because of its extremely high level expression in the placenta compared with other tissues (15). Subsequently, the immunohistochemical study by Maliepaard et al. with two different BCRP-specific monoclonal antibodies revealed that BCRP protein was most strongly expressed in the colon epithelium, the placental syncytiotrophoblasts, the small intestinal epithelium, liver canaliculi, and blood vessel capillaries in the brain (43). BCRP protein levels in other tissues were low. In general, the pattern of BCRP protein expression in human tissues determined by immunohistochemistry matches well with that of BCRP mRNA expression obtained in Northern blotting studies. BCRP expression in normal tissues may play a protective role for these tissues. The tissue distribution pattern of Bcrp1, the murine homolog of human BCRP, highly resembles that of human protein with substantial Bcrp1 mRNA and protein expression in the placenta, small intestine, and liver, except that there is little expression of BCRP in human kidney whereas Bcrp1 expression in mouse kidney is substantial (44–46). BCRP gene has also been cloned in rat (46,47), rhesus macaque (48), and porcine (49). The tissue distribution pattern of BCRP in rhesus macaque and porcine has yet to be determined.
With respect to membrane localization of BCRP in human tissues, Maliepaard et al. (43) showed that BCRP is primarily expressed in the apical membrane of the placental syncytiotrophoblasts, in the apical membrane of the epithelium in small intestine and colon, and in the liver canalicular membrane. BCRP is also highly expressed at the luminal surface of the microvessel endothelium of human brain (50). The substantial expression of BCRP in organs important for drug disposition (e.g., small intestine, liver, blood-brain barrier, and placenta) implies that BCRP could play a significant role in the absorption, distribution, and elimination of drugs that are BCRP substrates. Indeed, the importance of BCRP in drug disposition has now been demonstrated in numerous studies (51–54). The pharmacological significance of BCRP in drug transport has been extensively reviewed elsewhere (18,19,27,35,55–57). The apical side localization of human BCRP in placental syncytiotrophoblasts has been confirmed in various studies (58–61). We have shown that mouse Bcrp1 is also localized in the apical membrane of placental syncytiotrophoblasts of the pregnant mouse (45). Similarly, rat Bcrp has been shown to be strongly expressed in the rat placental labyrinth zone in which maternal blood is separated from fetal blood vessels by trophoblasts (62). A schematic representation of BCRP localization in the placenta is shown in Fig. 1.
Gestational Change of BCRP Expression in the Placenta
Recent studies indicate that BCRP expression in the placenta changes with gestational age. Meyer zu Schwabedissen et al. demonstrated that the BCRP mRNA levels in human placenta at preterm (28 ± 1 weeks, 15 placenta) were approximately two times greater (p < 0.05) than that at term (39 ± 2 weeks, 29 placenta), and BCRP protein expression showed the same pattern as that of BCRP mRNA (59). These authors did not measure BCRP expression in human placenta earlier than 28 weeks. However, another study with smaller sample size (six to eight placenta at each gestational age) did not show significant change in BCRP mRNA levels in human placenta with advancing gestation, but BCRP protein levels slightly increased towards the end of gestation (61). Mathias et al. also reported that the BCRP protein and mRNA levels in human placenta did not significantly change with gestational ages, but again these data were obtained with a limited sample size (four to six placenta at each gestational age) with substantial variations (63). The reason for this apparent discrepancy remains to be investigated. The data from rodent studies seem to be consistent with those reported by Meyer zu Schwabedissen et al. (59). In rat, Yasuda et al. found that rat Bcrp protein levels in the placenta at gestation day 14 were significantly higher than those at gestation day 20 (term in rat is approximately 21 days; 64). Likewise, our laboratory also demonstrated that Bcrp1 expression (protein and mRNA) in the placenta of pregnant mice peaked at gestation day 15 (term in mice is approximately 20–21 days; 45). Kalabis et al. reported that Bcrp1 mRNA levels in the placenta of pregnant mice decreased progressively from gestation day 9.5 toward term; however, Bcrp1 protein expression did not change significantly during gestation (65). Overall, BCRP/Bcrp1 expression in the placenta may be under tight control during pregnancy. Taken together, the substantial expression in the apical membrane of placental syncytiotrophoblasts indicate that BCRP/Bcrp1 may play an important role in protecting the fetus by expelling drugs, xenobiotics, and metabolites across the placental barrier, particularly at mid-gestational ages. We note that, while BCRP expression in the placenta peaks at mid-gestation as discussed above, P-gp (63,66) and MRP2 (67) expression in the placenta progressively decreases and increases, respectively, with gestational age towards term. It is possible that the differential placental expression of BCRP, P-gp, and MRP2 over the course of pregnancy provides a compensatory mechanism for protection of the fetus at different gestational stages.
Transport Function of BCRP in the Placenta
The activity of BCRP/Bcrp1 in placental transport has been analyzed in studies using in vitro cell culture, ex vivo perfused placenta, and in vivo animal models. The data published thus far are summarized in Table I. Kolwankar et al. (68) reported BCRP-mediated mitoxantrone transport in microvillus membrane vesicles isolated from human term placenta. Most recently, Gedeon et al. (69) showed that uptake of glyburide into the inside-out membrane vesicles isolated from human term placenta was significantly increased by the BCRP inhibitor novobiocin, but not by the P-gp inhibitor verapamil and the MRP inhibitor indomethacin, suggesting that glyburide was transported by BCRP. Transport activity of BCRP has also been illustrated in vitro with the “model” human placental BeWo cell line or primary trophoblasts in various studies using mitoxantrone or Hoechst 33342 as a BCRP substrate (70–72). Staud et al. (62) examined the transport activity of Bcrp1 in perfused rat placenta using cimetidine as a Bcrp1 substrate. Rat Bcrp1 was shown to significantly limit the maternal-to-fetal transport of cimetidine. When fetal perfusate was recirculated, rat Bcrp1 could actively transport cimetidine from the fetal to the maternal compartment against a concentration gradient. The first study demonstrating in vivo transport activity of Bcrp1 in the placenta was performed in P-gp-knockout pregnant mice (51). Using P-gp-deficient pregnant mice, Jonker et al. (51) demonstrated that co-administration of the Bcrp1 inhibitor GF120918 increased the fetal concentration of topotecan (a BCRP/Bcrp1 substrate) twofold at 30 min after drug administration in the pregnant mice, compared with that in the pregnant vehicle-treated control animals. In vivo studies demonstrating transport activity of placental Bcrp1 using Bcrp1-knockout mice have been reported for topotecan and phytoestrogens (26,44). These studies demonstrated increased fetal/maternal plasma concentration ratios in Bcrp1-knockout pregnant mice compared with those in wild-type pregnant mice, at only one time point after drug administration as a measure of fetal drug exposure. Most recently, our laboratory, for the first time, reported detailed transplacental pharmacokinetics of a model BCRP/Bcrp1 substrate, nitrofurantoin, in the pregnant mouse, and examined the role of Bcrp1 in determining fetal exposure of nitrofurantoin (73). We measured the maternal plasma and fetal AUCs of nitrofurantoin in pregnant mice after drug administration by retro-orbital injection. After 60 min of drug administration, nitrofurantoin in the systemic circulation was nearly completely eliminated in both the wild-type and Bcrp1-knockout pregnant mice. While the maternal plasma AUC of nitrofurantoin was only slightly increased in Bcrp1-knockout pregnant mice compared with that in wild-type pregnant mice, the fetal AUC in Bcrp1-knockout pregnant mice was approximately 5 times greater than that in wild-type pregnant mice. These results clearly suggest that Bcrp1 significantly limits fetal distribution of nitrofurantoin in the pregnant mouse. Such in vivo transplacental pharmacokinetic studies in animal models are particularly valuable, as similar studies of direct placental drug transport in humans are not feasible due to ethical reasons.
We also performed similar in vivo transplacental pharmacokinetic studies for glyburide, an antidiabetic drug commonly used to treat gestational diabetes. We found that while the maternal plasma AUCs of glyburide in the wild-type and Bcrp1-knockout pregnant mice were comparable, the fetal AUC of glyburide in the Bcrp1-knockout pregnant mice was two times greater than that in the wild-type pregnant mice (74). Recently, Kraemer et al. (75) performed ex vivo perfusion studies of human placenta to quantify placental transfer of glyburide. These authors have excluded albumin from the perfusion buffer so that net transfer of glyburide could be measured without the effect of high protein binding. Substantial fetal-to-maternal transfer of glyburide against a concentration gradient was observed. Furthermore, the transfer of glyburide across the placental barrier could not be inhibited the addition of verapamil, a P-gp inhibitor. These data suggest that glyburide was possibly effluxed by a transporter other than P-gp or P-gp is not a major player in the glyburide transport by human term placenta. We have previously shown that verapamil is not an effective inhibitor for BCRP (25). Thus, BCRP is likely a major player in the efflux of glyburide by human term placenta. The results of our animal studies provided further evidence to support this conclusion. However, since glyburide is also a P-gp substrate (76) and verapamil used in the study of Kraemer et al. is a relatively weak P-gp inhibitor, the role of P-gp in placental transport of glyburide cannot be excluded. Nevertheless, since it has been shown that the BCRP mRNA level was approximately ten times greater than that of P-gp in human term placenta (70), it is reasonable to assume that BCRP likely plays a much greater role than P-gp in transport of glyburide by human term placenta. High plasma protein binding may also contribute to the minimal fetal distribution of glyburide in vivo (77). Taken together, the in vitro transport, ex vivo perfusion, and in vivo transplacental pharmacokinetic studies all point to the unique role of BCRP/Bcrp1 in limiting fetal exposure of glyburide.
Physiological Role of BCRP in the Placenta
Although it is now quite clear that BCRP plays an important role in protecting the fetus against potential toxicity of drugs and xenobiotics, few studies have been done to demonstrate the physiological role of BCRP in the placenta. Placenta is the organ involved in the synthesis of steroid hormones such as progesterone and estrogens during pregnancy. Dehydroepiandrosterone (DHEA) and its sulfate conjugate (DHEAS) are important precursor molecules for placental estrogen synthesis. Estrone-3-sulfate (E3S) is a metabolite of estrogens but can function as a pool of inactive estrogens. Both DHEAS and E3S are effective substrates of BCRP (30,60); therefore, BCRP in human placenta is likely involved in the elimination of these endogenous sulfate conjugates from the fetal compartment into the maternal circulation. Thus, it has been suggested that BCRP may regulate placental estrogen synthesis through modulating intracellular DHEAS and E3S concentrations (60).
Recently, Evseenko et al. (58,78) showed that inhibition of BCRP activity by a BCRP-specific inhibitor Ko143 increased cytokine-induced apoptosis in primary trophoblasts and BeWo cells. Decreasing BCRP expression in BeWo cells by transfection of BCRP siRNA significantly increased the cell sensitivity to cytokine- and exogenous C6 and C8 ceramide-induced apoptosis. Silencing of BCRP expression also increased intracellular ceramide levels after cytokine exposure, but did not affect cellular protoporphyrin IX concentrations or sensitivity to activators of intrinsic apoptosis pathway. These authors further demonstrated that BCRP expression in placenta from pregnancies with idiopathic fetal growth restriction (IUGR) was significantly lower than that from normal pregnancies. Based on these data, the authors proposed that BCRP may play a hitherto unrecognized role in the placenta, protecting trophoblasts against apoptosis induced by cytokines or other activators via modulation of ceramide signaling, and BCRP is likely a survival factor in differentiation of placental trophoblasts. These data support the hypothesis that decreased BCRP expression in the placenta from IUGR pregnancies may result in placental function deficit, thus contributing to fetal growth restriction. Further studies are needed to confirm this hypothesis.
Genetic Polymorphisms of BCRP in the Placenta
Several laboratories have reported naturally occurring variants of BCRP. Notably, the single nucleotide polymorphisms (SNPs) G34A and C421A, resulting in alterations of BCRP protein at position 12 (V12M) and 141 (Q141K), respectively, occur at a relatively high frequency in most ethnic populations. For example, in the Japanese population, 39–50% are hetetozygous and 7% are homozygous for C421A (79, 80). In the Chinese population, 60% are hetetozygous for C421A (80). Other BCRP SNPs including A616C and A1768T are much less frequent with allele frequencies less than 1% (79–82). In addition, a polymorphism that results in a substitution of stop codon for Gln at position 126 has been identified (79). BCRP SNPs may result in variable expression of the transporter in tissues. Recently, Kobayashi et al. (12) examined the relationship between BCRP gene polymorphisms and protein expression in human placenta of the Japanese population. They showed that G34A and C421A occur at allele frequencies of 18% and 35%, respectively. Also, C376T which results in a stop codon at position 126 occurs at <1% frequency. This is consistent with the data reported by others (79,80). Kobayashi et al. also identified various SNPs with low allele frequencies (<1%), such as G1322A and T1465C, in placenta of the Japanese population, which were not reported by others. The study by Imai et al. (79) showed that the C421A polymorphism was associated with significantly lower expression and activity of the Q141K variant compared with the wild-type protein. Similarly, in placenta of the Japanese population, Kobayashi et al. illustrated that the mean BCRP protein level of the A421 homozygotes was approximately 50% of that of the C421 allele, and heterozygotes displayed an intermediate level. They further showed that this difference is likely caused by posttranscriptional regulation rather than changes in mRNA expression because polymorphism-dependent changes in BCRP mRNA expression were not observed (12). These data suggest that the C421A polymorphism could potentially decrease BCRP expression and activity in human placenta, leading to increased fetal drug exposure.
In vitro Regulation of BCRP Expression in Placental Cells
Pregnancy is one of the major physiologically stressful events, during which hormone concentrations are drastically changed. Therefore, it is not surprising that the expression of drug transporters and metabolic enzymes in organs important for drug disposition may be altered during pregnancy. As mentioned above, several laboratories have demonstrated that the expression of human BCRP as well as mouse and rat Bcrp1 in the placenta is gestational age-dependent (45,59,64). Likewise, Bcrp1 protein levels in the liver and kidney of pregnant mice reached a maximum at gestation day 15, but there was no significant change in Bcrp1 protein levels in small intestine over gestational age (45). At present, no data are available regarding the effect of pregnancy on BCRP/Bcrp1 expression in the liver, kidney, and small intestine of species other than mice.
The molecular mechanisms by which pregnancy affects BCRP expression in the placenta and other organs is yet unknown. The promoter of human BCRP gene has been cloned (83), permitting studies on regulation of BCRP gene expression at the transcriptional level. The BCRP promoter lacks a TATA-box and contains several putative Sp sites which are downstream from a putative CpG island. Whether Sp nuclear transcriptional factors are involved in the control of constitutive expression of the BCRP gene is not known. Recently, other nuclear receptors such as peroxisome proliferators-activated receptor gamma (PPARγ; 84), hypoxia-inducible factor 1 (HIF-1; 85), and aryl hydrocarbon receptor (AhR; 86,87) have been shown to be involved in the induction of BCRP gene transcription. Factors other than nuclear receptors have also been reported to affect BCRP expression. For example, To et al. (88) illustrated that aberrant methylation in the predicted CpG island in the BCRP promoter region suppresses transcription of the BCRP gene. Alternative promoter usage has also been demonstrated to cause differential expression of BCRP mRNA in drug-resistant cells and normal tissues (89,90). Whether the above mentioned regulation pathways possibly play a role in the control of BCRP expression in the placenta is yet to be determined.
The concentrations of pregnancy-related hormones such as progesterone and 17β-estradiol rise significantly throughout pregnancy (91). Therefore, it is reasonable to hypothesize that BCRP expression in the placenta may be increased in association with increased concentrations of these hormones. We have recently shown that progesterone receptor isoforms, PRA and PRB, differentially regulate expression of BCRP in the model human placental cell line, BeWo (92). We found that progesterone up-regulated BCRP expression in BeWo cells through PRB, but not PRA. In addition, PRA repressed the PRB activity. We also identified a progesterone response element (PRE) in the BCRP promoter region (92). Evseenko et al. reported that progesterone treatment did not affect BCRP expression in primary trophoblasts (93). It is possible that the trophoblasts secret a high level of endogenous progesterone by which the progesterone receptors are saturated; therefore, further addition of exogenous progesterone could not have an effect. We observed this phenomenon in BeWo cells (92). Progesterone is highly produced by the placenta after weeks 10 in pregnancy (94). Also, progesterone receptor expression has been demonstrated in human placenta (95). Thus, placenta is very likely a target tissue for the action of progesterone. It is therefore reasonable to hypothesize that progesterone may induce BCRP expression in the placenta through PRB and augment the protective role of the transporter during pregnancy. We will test this hypothesis in future study. Kalabis et al. examined the effect of progesterone on Bcrp1 expression in the placenta of pregnant mice, and found that the daily progesterone treatment starting at gestation day 14.5 until gestation day 18.5 significantly increased maternal progesterone concentrations, but did not affect Bcrp1 mRNA and protein expression in the placenta at gestation day 18.5 as compared with vehicle-treatment controls (65). It has been shown that the maternal plasma concentrations of endogenous progesterone at gestation day 14.5 were approximately 60 ng/ml (~190 nM; 96) and the progesterone concentrations in placenta tissues could be even greater. The progesterone receptors have possibly already been saturated at such high progesterone concentrations. Moreover, we have shown that the progesterone receptor expression in the placenta of pregnant mice is significantly decreased from gestation day 10 towards term (term in mice is approximately 20–21 days; 45). Therefore, an induction of placental Bcrp1 expression by additional exogenous progesterone from gestation day 14.5 to gestation day 18.5 may not occur. Collectively, the possibility that progesterone regulates BCRP/Bcrp1 expression in the placenta in vivo over the course of pregnancy warrants further investigation.
The effect of 17β-estradiol on BCRP expression is conflicting. It has been shown that 17β-estradiol induces BCRP expression in various cancer cell lines including the BeWo cells via estrogen receptor α (ERα), and an estrogen response element (ERE) in the BCRP promoter region has also been identified (97,98). In contrast, Imai et al. reported that 17β-estradiol decreased BCRP expression in ERα-positive cancer cell lines through posttranscriptional regulation (e.g., decreased protein biosynthesis and maturation; 99). As to why the posttranscriptional down-regulation of BCRP by 17β-estradiol requires the presence of a nuclear receptor ERα is not known. We also found that 17β-estradiol down-regulated BCRP expression in BeWo cells (72). The reason for this apparent discrepancy remains to be determined. We note that the PRE in the BCRP promoter region is exactly the same as the ERE published by Ee et al. (97). That progesterone and estrogen receptors share the same or similar response elements is possible, as earlier studies suggest that the regulatory elements for different steroids, including progesterone, 17β-estradiol, and glucocorticoids, are either similar or at least share structural features (100). Thus, progesterone and 17β-estradiol could make an impact on each other in regulation of BCRP, when the two hormones are combined. The real situation could be very complex, as 17β-estradiol by itself can induce PRB expression (72,101) and down-regulate BCRP expression (99), and on the other hand, PRA can repress the estrogen receptor activity (102). In BeWo cells, we showed that the 17β-estradiol treatment alone down-regulated BCRP expression (72), presumably due to posttranscriptional regulation as demonstrated by Imai et al. (99); however, the combined treatment of BeWo cells with 17β-estradiol and progesterone significantly increased BCRP expression compared with progesterone treatment alone (72). We have hypothesized that this combined effect is likely due to induction by 17β-estradiol of PRB in BeWo cells which then induces BCRP expression through progesterone (72). The results that progesterone can up-regulate BCRP expression support this hypothesis. We noted that the combined effect of 17β-estradiol and progesterone on BCRP expression in BeWo cells was dependent on concentrations of the two hormones used. For example, the inductive effect of progesterone on BCRP expression was inhibited when progesterone was combined with a high concentration of 17β-estradiol (72). This is likely due to the effect of down-regulation on BCRP expression by 17β-estradiol at a high concentration which overrides the inductive effect of progesterone.
We also observed that estriol, human placental lactogen, and human prolactin could also induce BCRP expression in BeWo cells at physiological concentrations (103). Further studies are needed to elucidate the molecular mechanisms by which estriol, placental lactogen, and prolactin up-regulate BCRP expression. Testosterone by itself did not affect BCRP expression at physiological concentrations. However, testosterone together with 17β-estradiol increased BCRP expression, and this induction was abolished by ERα antagonist ICI-182,780 or the testosterone receptor antagonist flutamide or knock-down of ERα (103). Further analysis revealed that 17β-estradiol increased testosterone receptor mRNA approximately 5.9-fold, suggesting that testosterone in combination with 17β-estradiol increases BCRP expression, possibly through 17β-estradiol-mediated up-regulation of the testosterone receptor.
BCRP expression in the placenta can also be influenced by cytokines and growth factors. Evseenko et al. showed that treatment of primary term trophoblasts with tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) significantly decreased BCRP protein and mRNA expression, but IL-6 had no significant effect (93). On the other hand, epidermal growth factor (EGF) and insulin-like growth factor II significantly increased BCRP protein and mRNA expression (59,93). There is evidence that the concentrations of cytokines such as TNF-α and IL-1β are elevated in some of obstetric disorders such as preeclampsia and gestational diabetes. Thus, the fetal protective function of BCRP against xenobiotics may be decreased in such obstetric disorders. BCRP expression in primary trophoblasts was shown to be increased with trophoblast differentiation (71), which is consistent with the role of EGF that promotes trophoblast differentiation (104). Maternal inflammation could also alter BCRP expression in the placenta. For example, down-regulation of Bcrp1 expression in the placenta of endotoxin-treated near term pregnant rats has recently been indicated (11). The data regarding regulation of BCRP expression as discussed above are summarized in Table II.
Clinical Significance of BCRP Expression in the Placenta and Future Direction
The findings that BCRP plays a significant role in limiting fetal distribution of various drugs (Table I) have important clinical implications. To limit fetal drug exposure, drugs that are actively transported by BCRP may be preferred. On the other hand, if the fetus is the target of drug therapy, drugs that bypass BCRP in human placenta may be optimal. Glyburide is an excellent example of drugs that bypass BCRP-mediated transport by human placenta. Glyburide has been shown to be safe in pregnancy due to minimal fetal exposure (105). We and others have demonstrated that BCRP-mediated placental transport of glyburide is at least one of the mechanisms by which fetal exposure of the drug is limited (69,74). Many drugs are routinely used by pregnant women either intentionally or by misuse; however, for a majority of these drugs, it is still not known if they are substrates of BCRP and other efflux transporters. Much more work is therefore needed in the future to identify substrate drugs of BCRP and other efflux transporters in the placenta, and to determine how high their affinity is to these transporters. Placental drug transport has so far been primarily demonstrated in in vitro transport (placental cell lines or placenta membrane vesicles) or ex vivo placenta perfusion studies. The data obtained from such in vitro and ex vivo studies may not be used to accurately predict what happens in vivo. Thus, in addition to studies using improved in vitro models (e.g., MDCK cells stably expressing BCRP for Transwell transport studies to identify BCRP substrate drugs), more in vivo transplacental pharmacokinetic studies of drugs in animal models are needed in future work to obtain detailed information of fetal drug distribution kinetics during pregnancy. Such information is extremely valuable for drug therapy in pregnancy. Drug-drug interactions at the placental barrier through inhibition of BCRP can occur if a BCRP substrate drug happens to be co-administered with a BCRP inhibitor. Such interactions could lead to increased fetal drug exposure through inhibition of BCRP in the placenta. Thus, caution should be taken when a substrate and an inhibitor of BCRP are concomitantly administered to pregnant women.
Another important area related to BCRP-mediated placental drug transport is the regulation of BCRP during pregnancy. Recent studies indicate that BCRP expression in placental cell lines or primary trophoblasts is affected by pregnancy-related steroid hormones, cytokines, and growth factors. Such findings have significant clinical relevance. For example, progesterone may induce BCRP expression in the placenta and augment the protective role of the transporter during pregnancy. On the other hand, patients with obstetric disorders associated with elevated cytokine levels may be at increased risk to fetal exposure of BCRP substrate drugs due to down-regulation of BCRP expression in the placenta by cytokines. Much more work is needed to elucidate the molecular mechanisms by which BCRP expression is regulated by pregnancy-related steroids, cytokines, and growth factors, and to determine if such regulation pathways contribute to gestational age-dependent BCRP/Bcrp1 expression in the placenta in vivo. In addition, the physiological role of BCRP in the placenta and the impact of BCRP SNPs on fetal drug exposure warrant further investigation.
In summary, significant progress has been made in the past 5 years with respect to the role of BCRP in placental drug transport. Understanding the molecular mechanisms of drug transport across the placental barrier as well as regulation of transporter expression in the placenta can help develop strategies to control fetal drug exposure and optimize drug therapy during pregnancy.
References
D. D. Glover, M. Amonkar, B. F. Rybeck, and T. S. Tracy. Prescription, over-the-counter, and herbal medicine use in a rural, obstetric population. Am. J. Obstet. Gynecol. 188:1039–1045 (2003).
S. E. Andrade, J. H. Gurwitz, R. L. Davis, K. A. Chan, J. A. Finkelstein, K. Fortman, H. McPhillips, M. A. Raebel, D. Roblin, D. H. Smith, M. U. Yood, A. N. Morse, and R. Platt. Prescription drug use in pregnancy. Am. J. Obstet. Gynecol. 191:398–407 (2004).
S. E. Andrade, M. A. Raebel, A. N. Morse, R. L. Davis, K. A. Chan, J. A. Finkelstein, K. K. Fortman, H. McPhillips, D. Roblin, D. H. Smith, M. U. Yood, R. Platt, and J. H. Gurwitz. Use of prescription medications with a potential for fetal harm among pregnant women. Pharmacoepidemiol. Drug Saf. 15:546–54 (2006).
M. R. Syme, J. W. Paxton, and J. A. Keelan. Drug transfer and metabolism by the human placenta. Clin. Pharmacokinet. 43:487–514 (2004).
P. Myllynen, M. Pasanen, and K. Vahakangas. The fate and effects of xenobiotics in human placenta. Expert Opin. Drug Metab. Toxicol. 3:331–46 (2007).
L. S. Hodge, and T. S. Tracy. Alterations in drug disposition during pregnancy. Expert Opin. Drug Metab. Toxicol. 3:557–71 (2007).
M. Ceckova-Novotna, P. Pavek, and F. Staud. P-glycoprotein in the placenta: expression, localization, regulation and function. Reprod. Toxicol. 22:400–10 (2006).
D. Evseenko, J. W. Paxton, and J. A. Keelan. Active transport across the human placenta: impact on drug efficacy and toxicity. Expert Opin. Drug Metab. Toxicol. 2:51–69 (2006).
G. R. Lankas, L. D. Wise, M. E. Cartwright, T. Pippert, and D. R. Umbenhauer. Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod. Toxicol. 12:457–63 (1998).
J. D. Unadkat, A. Dahlin, and S. Vijay. Placental drug transporters. Curr. Drug Metab. 5:125–31 (2004).
J. Behravan, and M. Piquette-Miller. Drug transport across the placenta, role of the ABC drug efflux transporters. Expert Opin. Drug Metab. Toxicol. 3:819–830 (2007).
D. Kobayashi, I. Ieiri, T. Hirota, H. Takane, S. Maegawa, J. Kigawa, H. Suzuki, E. Nanba, M. Oshimura, N. Terakawa, K. Otsubo, K. Mine, and Y. Sugiyama. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab. Dispos. 33:94–101 (2005).
L. A. Doyle, W. Yang, L. V. Abruzzo, T. Krogmann, Y. Gao, A. K. Rishi, and D. D. Ross. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 95:15665–15670 (1998).
K. Miyake, L. Mickley, T. Litman, Z. Zhan, R. Robey, B. Cristensen, M. Brangi, L. Greenberger, M. Dean, T. Fojo, and S. E. Bates. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 59:8–13 (1999).
R. Allikmets, L. M. Schriml, A. Hutchinson, V. Romano-Spica, and M. Dean. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 58:5337–5339 (1998).
L. A. Doyle, and D. D. Ross. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22:7340–58 (2003).
R. W. Robey, O. Polgar, J. Deeken, K. W. To, and S. E. Bates. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 26:39–57 (2007).
Q. Mao, and J. D. Unadkat. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J 7:E118–33 (2005).
P. Krishnamurthy, and J. D. Schuetz. Role of ABCG2/BCRP in biology and medicine. Annu. Rev. Pharmacol. Toxicol. 46:381–410 (2006).
G. Pan, N. Giri, and W. F. Elmquist. Abcg2/Bcrp1 mediates the polarized transport of antiretroviral nucleosides abacavir and zidovudine. Drug Metab. Dispos. 35:1165–73 (2007).
X. Wang, T. Furukawa, T. Nitanda, M. Okamoto, Y. Sugimoto, S. Akiyama, and M. Baba. Breast cancer resistance protein (BCRP/ABCG2) induces cellular resistance to HIV-1 nucleoside reverse transcriptase inhibitors. Mol. Pharmacol. 63:65–72 (2003).
T. Litman, M. Brangi, E. Hudson, P. Fetsch, A. Abati, D. D. Ross, K. Miyake, J. H. Resau, and S. E. Bates. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2). J. Cell Sci. 113(Pt 11):2011–2021 (2000).
C. Erlichman, S. A. Boerner, C. G. Hallgren, R. Spieker, X. Y. Wang, C. D. James, G. L. Scheffer, M. Maliepaard, D. D. Ross, K. C. Bible, and S. H. Kaufmann. The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res. 61:739–48 (2001).
H. Burger, H. van Tol, A. W. Boersma, M. Brok, E. A. Wiemer, G. Stoter, and K. Nooter. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 104:2940–2 (2004).
Y. Zhang, A. Gupta, H. Wang, L. Zhou, R. R. Vethanayagam, J. D. Unadkat, and Q. Mao. BCRP transports dipyridamole and is inhibited by calcium channel blockers. Pharm Res 22:2023–34 (2005).
J. Enokizono, H. Kusuhara, and Y. Sugiyama. Effect of breast cancer resistance protein (Bcrp/Abcg2) on the disposition of phytoestrogens. Mol. Pharmacol. 72:967–975 (2007).
G. Merino, J. W. Jonker, E. Wagenaar, A. E. van Herwaarden, and A. H. Schinkel. The breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion, and milk secretion of the antibiotic nitrofurantoin. Mol. Pharmacol. 67:1758–1764 (2005).
P. Pavek, G. Merino, E. Wagenaar, E. Bolscher, M. Novotna, J. W. Jonker, and A. H. Schinkel. Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J. Pharmacol. Exp. Ther. 312:144–152 (2005).
C. Gedeon, J. Behravan, G. Koren, and M. Piquette-Miller. Transport of glyburide by placental ABC transporters: implications in fetal drug exposure. Placenta 27:1096–1102 (2006).
Y. Imai, S. Asada, S. Tsukahara, E. Ishikawa, T. Tsuruo, and Y. Sugimoto. Breast cancer resistance protein exports sulfated estrogens but not free estrogens. Mol. Pharmacol. 64:610–618 (2003).
Z. S. Chen, R. W. Robey, M. G. Belinsky, I. Shchaveleva, X. Q. Ren, Y. Sugimoto, D. D. Ross, S. E. Bates, and G. D. Kruh. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-d-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 63:4048–4054 (2003).
B. Sarkadi, L. Homolya, G. Szakacs, and A. Varadi. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol. Rev. 86:1179–1236 (2006).
A. T. Nies, and D. Keppler. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. 453:643–659 (2007).
S. Matsushima, K. Maeda, C. Kondo, M. Hirano, M. Sasaki, H. Suzuki, and Y. Sugiyama. Identification of the hepatic efflux transporters of organic anions using double-transfected Madin–Darby canine kidney II cells expressing human organic anion-transporting polypeptide 1B1 (OATP1B1)/multidrug resistance-associated protein 2, OATP1B1/multidrug resistance 1, and OATP1B1/breast cancer resistance protein. J. Pharmacol. Exp. Ther. 314:1059–1067 (2005).
C. M. Kruijtzer, J. H. Beijnen, H. Rosing, W. W. ten Bokkel Huinink, M. Schot, R. C. Jewell, E. M. Paul, and J. H. Schellens. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J. Clin. Oncol. 20:2943–2950 (2002).
J. D. Allen, A. van Loevezijn, J. M. Lakhai, M. van der Valk, O. van Tellingen, G. Reid, J. H. Schellens, G. J. Koomen, and A. H. Schinkel. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 1:417–425 (2002).
P. J. Houghton, G. S. Germain, F. C. Harwood, J. D. Schuetz, C. F. Stewart, E. Buchdunger, and P. Traxler. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 64:2333–2337 (2004).
Z. Shi, X. X. Peng, I. W. Kim, S. Shukla, Q. S. Si, R. W. Robey, S. E. Bates, T. Shen, C. R. Ashby Jr., L. W. Fu, S. V. Ambudkar, and Z. S. Chen. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 67:11012–11020 (2007).
A. Gupta, Y. Zhang, J. D. Unadkat, and Q. Mao. HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2). J. Pharmacol. Exp. Ther. 310:334–341 (2004).
A. Gupta, Y. Dai, R. R. Vethanayagam, M. F. Hebert, K. E. Thummel, J. D. Unadkat, D. D. Ross, and Q. Mao. Cyclosporin A, tacrolimus and sirolimus are potent inhibitors of the human breast cancer resistance protein (ABCG2) and reverse resistance to mitoxantrone and topotecan. Cancer Chemother. Pharmacol. 58:374–383 (2006).
S. Zhang, X. Yang, and M. E. Morris. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol. Pharmacol. 65:1208–1216 (2004).
A. Ahmed-Belkacem, A. Pozza, S. Macalou, J. M. Perez-Victoria, A. Boumendjel, and A. Di Pietro. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2). Anticancer Drugs 17:239–243 (2006).
M. Maliepaard, G. L. Scheffer, I. F. Faneyte, M. A. van Gastelen, A. C. Pijnenborg, A. H. Schinkel, M. J. van De Vijver, R. J. Scheper, and J. H. Schellens. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 61:3458–3464 (2001).
J. W. Jonker, M. Buitelaar, E. Wagenaar, M. A. Van Der Valk, G. L. Scheffer, R. J. Scheper, T. Plosch, F. Kuipers, R. P. Elferink, H. Rosing, J. H. Beijnen, and A. H. Schinkel. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U.S.A. 99:15649–15654 (2002).
H. Wang, X. Wu, K. Hudkins, A. Mikheev, H. Zhang, A. Gupta, J. D. Unadkat, and Q. Mao. Expression of the breast cancer resistance protein (Bcrp1/Abcg2) in tissues from pregnant mice: effects of pregnancy and correlations with nuclear receptors. Am. J. Physiol. Endocrinol. Metab. 291:E1295–304 (2006).
Y. Tanaka, A. L. Slitt, T. M. Leazer, J. M. Maher, and C. D. Klaassen. Tissue distribution and hormonal regulation of the breast cancer resistance protein (Bcrp/Abcg2) in rats and mice. Biochem. Biophys. Res. Commun. 326:181–187 (2005).
S. Hori, S. Ohtsuki, M. Tachikawa, N. Kimura, T. Kondo, M. Watanabe, E. Nakashima, and T. Terasaki. Functional expression of rat ABCG2 on the luminal side of brain capillaries and its enhancement by astrocyte-derived soluble factor(s). J. Neurochem. 90:526–536 (2004).
T. Ueda, S. Brenner, H. L. Malech, S. M. Langemeijer, S. Perl, M. Kirby, O. A. Phang, A. E. Krouse, R. E. Donahue, E. M. Kang, and J. F. Tisdale. Cloning and functional analysis of the rhesus macaque ABCG2 gene. Forced expression confers an SP phenotype among hematopoietic stem cell progeny in vitro. J. Biol. Chem. 280:991–998 (2005).
T. Eisenblatter, S. Huwel, and H. J. Galla. Characterisation of the brain multidrug resistance protein (BMDP/ABCG2/BCRP) expressed at the blood-brain barrier. Brain Res. 971:221–231 (2003).
H. C. Cooray, C. G. Blackmore, L. Maskell, and M. A. Barrand. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 13:2059–2063 (2002).
J. W. Jonker, J. W. Smit, R. F. Brinkhuis, M. Maliepaard, J. H. Beijnen, J. H. Schellens, and A. H. Schinkel. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J. Natl. Cancer Inst. 92:1651–1656 (2000).
G. Merino, A. E. van Herwaarden, E. Wagenaar, J. W. Jonker, and A. H. Schinkel. Sex-dependent expression and activity of the ATP-binding cassette transporter breast cancer resistance protein (BCRP/ABCG2) in liver. Mol. Pharmacol. 67:1765–1771 (2005).
G. Merino, A. I. Alvarez, M. M. Pulido, A. J. Molina, A. H. Schinkel, and J. G. Prieto. Breast cancer resistance protein (BCRP/ABCG2) transports fluoroquinolone antibiotics and affects their oral availability, pharmacokinetics, and milk secretion. Drug Metab. Dispos. 34:690–695 (2006).
W. Zhang, B. N. Yu, Y. J. He, L. Fan, Q. Li, Z. Q. Liu, A. Wang, Y. L. Liu, Z. R. Tan, J. Fen, Y. F. Huang, and H. H. Zhou. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin. Chim. Acta 373:99–103 (2006).
Y. Sugimoto, S. Tsukahara, E. Ishikawa, and J. Mitsuhashi. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmacokinetics/pharmacodynamics. Cancer Sci. 96:457–465 (2005).
C. Q. Xia, J. J. Yang, and L. S. Gan. Breast cancer resistance protein in pharmacokinetics and drug-drug interactions. Expert Opin. Drug Metab. Toxicol. 1:595–611 (2005).
A. E. van Herwaarden, and A. H. Schinkel. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol. Sci. 27:10–16 (2006).
D. A. Evseenko, P. Murthi, J. W. Paxton, G. Reid, B. S. Emerald, K. M. Mohankumar, P. E. Lobie, S. P. Brennecke, B. Kalionis, and J. A. Keelan. The ABC transporter BCRP/ABCG2 is a placental survival factor, and its expression is reduced in idiopathic human fetal growth restriction. FASEB J. 21:3592–3605 (2007).
H. E. Meyer zu Schwabedissen, M. Grube, A. Dreisbach, G. Jedlitschky, K. Meissner, K. Linnemann, C. Fusch, C. A. Ritter, U. Volker, and H. K. Kroemer. Epidermal growth factor-mediated activation of the map kinase cascade results in altered expression and function of ABCG2 (BCRP). Drug Metab. Dispos. 34:524–533 (2006).
M. Grube, S. Reuther, H. Meyer Zu Schwabedissen, K. Kock, K. Draber, C. A. Ritter, C. Fusch, G. Jedlitschky, and H. K. Kroemer. Organic anion transporting polypeptide 2B1 and breast cancer resistance protein interact in the transepithelial transport of steroid sulfates in human placenta. Drug Metab. Dispos. 35:30–35 (2007).
D. Yeboah, M. Sun, J. Kingdom, D. Baczyk, S. J. Lye, S. G. Matthews, and W. Gibb. Expression of breast cancer resistance protein (BCRP/ABCG2) in human placenta throughout gestation and at term before and after labor. Can. J. Physiol. Pharmacol. 84:1251–1258 (2006).
F. Staud, Z. Vackova, K. Pospechova, P. Pavek, M. Ceckova, A. Libra, L. Cygalova, P. Nachtigal, and Z. Fendrich. Expression and transport activity of breast cancer resistance protein (Bcrp/Abcg2) in dually perfused rat placenta and HRP-1 cell line. J. Pharmacol. Exp. Ther. 319:53–62 (2006).
A. A. Mathias, J. Hitti, and J. D. Unadkat. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289:R963–969 (2005).
S. Yasuda, S. Itagaki, T. Hirano, and K. Iseki. Expression level of ABCG2 in the placenta decreases from the mid stage to the end of gestation. Biosci. Biotechnol. Biochem. 69:1871–1876 (2005).
G. M. Kalabis, S. Petropoulos, W. Gibb, and S. G. Matthews. Breast cancer resistance protein (Bcrp1/Abcg2) in mouse placenta and yolk sac: ontogeny and its regulation by progesterone. Placenta 28:1073–1081 (2007).
S. Gil, R. Saura, F. Forestier, and R. Farinotti. P-glycoprotein expression of the human placenta during pregnancy. Placenta 26:268–270 (2005).
H. E. Meyer zu Schwabedissen, G. Jedlitschky, M. Gratz, S. Haenisch, K. Linnemann, C. Fusch, I. Cascorbi, and H. K. Kroemer. Variable expression of MRP2 (ABCC2) in human placenta: influence of gestational age and cellular differentiation. Drug Metab. Dispos. 33:896–904 (2005).
D. Kolwankar, D. D. Glover, J. A. Ware, and T. S. Tracy. Expression and function of ABCB1 and ABCG2 in human placental tissue. Drug Metab. Dispos. 33:524–529 (2005).
C. Gedeon, G. Anger, M. Piquette-Miller, and G. Koren. Breast cancer resistance protein: mediating the trans-placental transfer of glyburide across the human placenta. Placenta in press (2008).
M. Ceckova, A. Libra, P. Pavek, P. Nachtigal, M. Brabec, R. Fuchs, and F. Staud. Expression and functional activity of breast cancer resistance protein (BCRP, ABCG2) transporter in the human choriocarcinoma cell line BeWo. Clin. Exp. Pharmacol. Physiol. 33:58–65 (2006).
D. A. Evseenko, J. W. Paxton, and J. A. Keelan. ABC drug transporter expression and functional activity in trophoblast-like cell lines and differentiating primary trophoblast. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290:R1357–1365 (2006).
H. Wang, L. Zhou, A. Gupta, R. R. Vethanayagam, Y. Zhang, J. D. Unadkat, and Q. Mao. Regulation of BCRP/ABCG2 expression by progesterone and 17beta-estradiol in human placental BeWo cells. Am. J. Physiol. Endocrinol. Metab. 290:E798–807 (2006).
Y. Zhang, H. Wang, J. D. Unadkat, and Q. Mao. Breast Cancer Resistance Protein 1 limits fetal distribution of nitrofurantoin in the pregnant mouse. Drug Metab. Dispos. 35:2154–2158 (2007).
L. Zhou, S. B. Naraharisetti, H. Wang, J. D. Unadkat, M. F. Hebert, and Q. Mao. The Breast Cancer Resistance Protein (Bcrp1/Abcg2) Limits Fetal Distribution of Glyburide in the Pregnant Mouse - An OPRU Network and UW SCOR Study. Mol. Pharmacol. in press (2008).
J. Kraemer, J. Klein, A. Lubetsky, and G. Koren. Perfusion studies of glyburide transfer across the human placenta: implications for fetal safety. Am. J. Obstet. Gynecol. 195:270–274 (2006).
P. E. Golstein, A. Boom, J. van Geffel, P. Jacobs, B. Masereel, and R. Beauwens. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch. 437:652–660 (1999).
T. N. Nanovskaya, I. Nekhayeva, G. D. Hankins, and M. S. Ahmed. Effect of human serum albumin on transplacental transfer of glyburide. Biochem. Pharmacol. 72:632–639 (2006).
D. A. Evseenko, J. W. Paxton, and J. A. Keelan. The xenobiotic transporter ABCG2 plays a novel role in differentiation of trophoblast-like BeWo cells. Placenta 28(Suppl A):S116–S120 (2007).
Y. Imai, M. Nakane, K. Kage, S. Tsukahara, E. Ishikawa, T. Tsuruo, Y. Miki, and Y. Sugimoto. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol. Cancer Ther. 1:611–616 (2002).
C. P. Zamber, J. K. Lamba, K. Yasuda, J. Farnum, K. Thummel, J. D. Schuetz, and E. G. Schuetz. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics 13:19–28 (2003).
A. Tamura, K. Wakabayashi, Y. Onishi, M. Takeda, Y. Ikegami, S. Sawada, M. Tsuji, Y. Matsuda, and T. Ishikawa. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 98:231–239 (2007).
Y. Honjo, K. Morisaki, L. M. Huff, R. W. Robey, J. Hung, M. Dean, and S. E. Bates. Single-nucleotide polymorphism (SNP) analysis in the ABC half-transporter ABCG2 (MXR/BCRP/ABCP1). Cancer Biol. Ther. 1:696–702 (2002).
K. J. Bailey-Dell, B. Hassel, L. A. Doyle, and D. D. Ross. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim. Biophys. Acta 1520:234–241 (2001).
I. Szatmari, G. Vamosi, P. Brazda, B. L. Balint, S. Benko, L. Szeles, V. Jeney, C. Ozvegy-Laczka, A. Szanto, E. Barta, J. Balla, B. Sarkadi, and L. Nagy. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J. Biol. Chem. 281:23812–23823 (2006).
P. Krishnamurthy, D. D. Ross, T. Nakanishi, K. Bailey-Dell, S. Zhou, K. E. Mercer, B. Sarkadi, B. P. Sorrentino, and J. D. Schuetz. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 279:24218–24225 (2004).
B. Ebert, A. Seidel, and A. Lampen. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 26:1754–1763 (2005).
B. Ebert, A. Seidel, and A. Lampen. Phytochemicals induce breast cancer resistance protein in Caco-2 cells and enhance the transport of benzo[a]pyrene-3-sulfate. Toxicol. Sci. 96:227–236 (2007).
K. K. To, Z. Zhan, and S. E. Bates. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Mol. Cell Biol. 26:8572–8585 (2006).
T. Nakanishi, K. J. Bailey-Dell, B. A. Hassel, K. Shiozawa, D. M. Sullivan, J. Turner, and D. D. Ross. Novel 5′ untranslated region variants of BCRP mRNA are differentially expressed in drug-selected cancer cells and in normal human tissues: implications for drug resistance, tissue-specific expression, and alternative promoter usage. Cancer Res 66:5007–5011 (2006).
Y. Zong, S. Zhou, S. Fatima, and B. P. Sorrentino. Expression of mouse Abcg2 mRNA during hematopoiesis is regulated by alternative use of multiple leader exons and promoters. J Biol Chem 281:29625–29632 (2006).
M. C. Neville, T. B. McFadden, and I. Forsyth. Hormonal regulation of mammary differentiation and milk secretion. J. Mammary Gland Biol. Neoplasia 7:49–66 (2002).
H. Wang, E. W. Lee, L. Zhou, P. C. Leung, D. Ross, J. D. Unadkat, and Q. Mao. Progesterone receptor isoforms, PRA and PRB, differentially regulate expression of the breast cancer resistance protein (BCRP) in human placental choriocarcinoma BeWo cells. Mol. Pharmacol. in press (2008).
D. A. Evseenko, J. W. Paxton, and J. A. Keelan. Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab. Dispos. 35:595–601 (2007).
F. S. Khan-Dawood, and M. Y. Dawood. Estrogen and progesterone receptor and hormone levels in human myometrium and placenta in term pregnancy. Am. J. Obstet. Gynecol. 150:501–505 (1984).
C. Cudeville, F. Mondon, B. Robert, R. Rebourcet, T. M. Mignot, C. Benassayag, and F. Ferre. Evidence for progesterone receptors in the human fetoplacental vascular tree. Biol. Reprod. 62:759–765 (2000).
G. M. Kalabis, A. Kostaki, M. H. Andrews, S. Petropoulos, W. Gibb, and S. G. Matthews. Multidrug resistance phosphoglycoprotein (ABCB1) in the mouse placenta: fetal protection. Biol. Reprod. 73:591–597 (2005).
P. L. Ee, S. Kamalakaran, D. Tonetti, X. He, D. D. Ross, and W. T. Beck. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 64:1247–51 (2004).
Y. Zhang, G. Zhou, H. Wang, X. Zhang, F. Wei, Y. Cai, and D. Yin. Transcriptional upregulation of breast cancer resistance protein by 17beta-estradiol in ERalpha-positive MCF-7 breast cancer cells. Oncology 71:446–455 (2006).
Y. Imai, E. Ishikawa, S. Asada, and Y. Sugimoto. Estrogen-mediated post transcriptional down-regulation of breast cancer resistance protein/ABCG2. Cancer Res. 65:596–604 (2005).
D. von der Ahe, S. Janich, C. Scheidereit, R. Renkawitz, G. Schutz, and M. Beato. Glucocorticoid and progesterone receptors bind to the same sites in two hormonally regulated promoters. Nature 313:706–709 (1985).
T. Flototto, D. Niederacher, D. Hohmann, T. Heimerzheim, P. Dall, S. Djahansouzi, H. G. Bender, and B. Hanstein. Molecular mechanism of estrogen receptor (ER)alpha-specific, estradiol-dependent expression of the progesterone receptor (PR) B-isoform. J. Steroid Biochem. Mol. Biol. 88:131–142 (2004).
P. H. Giangrande, and D. P. McDonnell. The A and B isoforms of the human progesterone receptor: two functionally different transcription factors encoded by a single gene. Recent Prog. Horm. Res. 54:291–313 (1999).
H. Wang, J. D. Unadkat, and Q. Mao. Hormonal regulation of BCRP expression in human placental BeWo cells. Pharm Res in press (2008).
D. W. Morrish, J. Dakour, and H. Li. Functional regulation of human trophoblast differentiation. J. Reprod. Immunol. 39:179–195 (1998).
O. Langer, D. L. Conway, M. D. Berkus, E. M. Xenakis, and O. Gonzales. A comparison of glyburide and insulin in women with gestational diabetes mellitus. N. Engl. J. Med. 343:1134–1138 (2000).
Acknowledgement
The author gratefully acknowledges financial support from NIH grants HD044404 and HD047892. The author also greatly thanks Drs. Jashvant D. Unadkat (Department of Pharmaceutics, University of Washington) and Mary F. Hebert (Department of Pharmacy, University of Washington) for collaborations on pregnancy-related studies. Mr. Hsiao Peng (Department of Pharmaceutics, University of Washington) is greatly appreciated for preparing the graphic.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s11095-008-9570-y
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mao, Q. BCRP/ABCG2 in the Placenta: Expression, Function and Regulation. Pharm Res 25, 1244–1255 (2008). https://doi.org/10.1007/s11095-008-9537-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9537-z