
Abstract
Chemical-looping combustion (CLC) of biomass has the potential to facilitate negative CO2 emission in heat and power production when combined with a carbon capture technique. However, typical biomass contains alkali metals and chlorine compounds, such as potassium chloride, which can lead to corrosion of heat-transfer surfaces in the reactors. The combined influence of potassium chloride, hydrochloric acid, and oxygen on the corrosion of five typical heat-transfer materials, which are potential candidates for use in the fuel reactor in a CLC process, was studied using one-week laboratory-scale experiments. The results suggested that potassium chloride, especially in the presence of HCl and O2, greatly affects the corrosion of lower-alloyed heat-transfer materials. The outcome of this study can provide valuable information for selecting suitable heat-transfer materials for CLC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Combusting biomass instead of fossil fuels is a viable option to reduce CO2 emissions into the atmosphere. However, the need and requirements to limit atmospheric temperature increase below 1.5 °C compared to the preindustrial times call for changes in sustainable combustion technologies [1]. One potential option could be to capture and store or utilize CO2 emitted from biomass combustion (bioenergy with carbon capture and storage, BECCS) [2]. In traditional combustion technologies, CO2 emissions are in flue gases mainly containing nitrogen from the combustion air making it challenging to be later separated from one another. In chemical-looping combustion (CLC), this problem is avoided as the oxygen needed for the fuel conversion is brought to the reactor as metal oxides. Hence, the non-condensing part of the flue gas will mainly contain CO2 [3].
A CLC setup consists of two looping reactors, i.e., an air reactor and a fuel reactor. The bed material typically consists of redox-active metal oxides, which allow the shift between different oxidation states and enable oxygen transport between the two reactors. The bed material is first oxidized in the air reactor. Then, the bed material is circulated into the fuel reactor where it is reduced as the fuel is oxidized. It then recirculates back to the air reactor for re-oxidation. If the oxidation is complete, the flue gas will principally consist of pure CO2 and H2O. The principle of CLC is presented in Fig. 1.

The main principle of chemical-looping combustion with two intertwined reactors, where the oxygen carrier is, respectively, oxidized and reduced. This study focuses on the fuel reactor only (red square) (Color figure online)
Lewis and Gilland initially proposed the idea of chemical looping for producing pure CO2 in 1954 [4]. The method has been improved over the years [5,6,7]. Today, chemical looping focuses on combustion processes as a step to allow for simple CO2 capture (CCS) from flue gases [7,8,9,10,11,12]. When using renewable fuels such as biomass, carbon capture, sequestration potential may result in negative CO2 emissions with CLC [13].
In the first-generation CLC designs, traditional heat-exchanger solutions could be used in the air reactor, and only minor corrosion was expected since the flue gases from the fuel reactor and air stream remained separated. Therefore, no corrosion-inducing elements, such as chlorine from the fuel, could enter the air reactor.
Recently, a second-generation CLC setup was proposed by Alstom [14]. This new type of setup allows the use of two sets of heat-exchangers, one in each reactor, raising the boiler efficiency considerably. On the other hand, this increases the risk of severe corrosion of heat-transfer materials, especially when using biomass as fuel. The challenges with biomass in CLC originate from the chemical heterogeneity and large quality variations of the feedstocks [15]. In addition, the ash-forming matter of biomass typically contains considerable amounts of K and Cl, which are particularly reactive and, when forming deposits on the heat-transfer surfaces, may lead to high-temperature corrosion at surface temperatures of 400–600 °C [16,17,18,19,20]. Furthermore, some air leakages may be expected in the fuel reactor because the reactor is operated under negative pressure conditions. In addition to the aggressive K and Cl species, small amounts of oxygen could further accelerate the corrosion of heat-transfer materials.
As reported in earlier studies, different mechanisms for chlorine-induced corrosion have been proposed for biomass combustion:
-
1.
Gaseous chlorine-induced corrosion, also called active oxidation, occurs when either gaseous HCl or Cl2 interacts with the heat-transfer surface by diffusing into the material and reacting with the alloy to form iron chlorides FeClx, e.g., FeCl2. These chlorides may vaporize and diffuse to the surface toward a higher oxygen partial pressure, where the chlorides oxidize, forming an iron-rich oxide layer at the alloy–gas interface. During the oxide formation, chlorine is released and may react again with the alloy, thus sustaining the corrosion process [21, 22].
-
2.
Alkali chloride-induced corrosion: potassium chloride (KCl) may react directly with iron in low-alloyed steels, forming iron chlorides. Similarly, to the first suggested mechanism, formed iron chlorides diffuse to the surface toward a higher oxygen partial pressure and ultimately oxidize, releasing chlorine, which may continue the corrosion reaction [23, 24].
With higher-alloyed steels, the protective Cr2O3 layer on the alloy surface may react with KCl, resulting in potassium chromate (K2CrO4) formation and loss of the protective properties of the oxide layer [25,26,27]. Simultaneously with K2CrO4 formation, the chlorine is released and may thus sustain the corrosion process described above.
-
3.
Molten salt corrosion: this mechanism involves the direct interaction between the metal oxide scale and the molten salt at the oxide/melt interface, leading to rapid corrosion [28].
The form of chlorine diffusing into the alloys and details of the chlorine-related high-temperature corrosion are not fully understood. In addition to the abovementioned mechanisms, another proposed alternative for chlorine transportation is an electrochemical mechanism, where chloride ions diffuse to the oxide/alloy interface instead of the transport of gaseous chlorine atoms/molecules through the oxide scale [29, 30].
The presence of chlorine can accelerate corrosion and result in oxides that are often very porous by nature and give no protection against continuous attack and re-chlorination of the metal. These oxides are typically poorly attached to metal surface, leading to easy spallation [21, 31].
It has been reported by Eriksson et al. [32] that corrosion is not a noteworthy concern in the CLC of biomass. However, the effect of more reactive atmospheres in fuel reactors has not been studied yet. The question was approached with 20 new laboratory-scale experiments imitating different harsh gas-phase conditions in a fuel reactor. These results were compared to results from an earlier study with different atmospheres [32]. In the past, similar laboratory-scale corrosion experiments carried out in controlled atmospheres have produced valuable results that have been translated to large-scale processes [17,18,19, 27, 32, 33]. The present study aims to investigate whether corrosion can be expected to be a significant problem in the second-generation CLC fuel reactors when firing biomass and thus exposed to potentially more corrosive atmospheres.
Materials and Methods
Materials
Analytical grade potassium chloride (KCl) from Sigma Aldrich was used as a synthetic deposit on heat-transfer samples to represent the most critical ash component in high-temperature corrosion in a biomass-fired CLC boiler.
The five heat-transfer materials chosen to simulate the fuel reactor heat-exchanger surfaces were low-Cr ferritic steels 10CrMo9-10, X10, and high-Cr austenitic stainless steels TP347, TP310, and Sanicro28. In this paper, the steels are referred to as low or high Cr. All tested materials were acquired from boiler manufacturers. The high-Cr samples were cold-finished descaled, while the low-Cr sample qualities were normalized and tempered. The compositions of the alloys were measured with SEM-EDXA and are presented in Table 1.
After cutting (20 × 20 mm), the sample pieces were first pre-treated by polishing in ethanol with P1000, cleaned, and pre-oxidized at 200 °C for 24 h as described by Skrifvars et al. [17]. The deposits were weighed on top of the samples before they were transferred to furnace in a special sample holder holding five samples. The sample holder was equipped with thermocouples which ensured accurate control of temperature as described in more detail by Eriksson et al. [32].
Corrosion Experiments
The corrosion experiments were carried out with 40 samples in total. The five steel types (Table 1) were exposed to two temperatures in four different atmospheres: 50/50 vol% CO2/H2O representing normal conditions in the fuel reactor, CO2/H2O with 500 ppm HCl resembling harsh conditions, CO2/H2O + 1% O2 simulating a slip of oxygen in the reactor, and CO2/H2O + 500 ppm HCl + 1% O2 mimicking the most severe conditions in the reactor. The total exposure time was 168 h at either 450 °C or 550 °C. These temperatures are typical for heat-exchanger surfaces in biomass-fired boilers. A summary of the test conditions is shown in Table 2. The results of the first two conditions have been previously published by Eriksson et al. [32] and will be used as references.
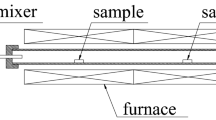
The samples were placed on a ceramic sample holder inside a custom-made gastight quartz reactor and heated in a horizontal alumina tube furnace (Carbolite) (Fig. 2). The setup with mass flow controllers allowed the accurate control of the atmosphere under the test condition. A total gas flow rate of 2000 cm3/min at NTP was used in all experiments. Furthermore, the sample holder was equipped with thermocouples for accurate temperature monitoring and control.

A schematic of the quartz reactor used in the experiments
The samples were inserted into the furnace at room temperature on a ceramic sample holder fitting five samples. Then, the samples were heated to the target temperature with a heating rate of 10 °C min−1. For the first 200 °C, an inert gas flow of 1000 cm3/min of CO2 mixed with 1000 cm3/min of N2 was used. Then, N2 was replaced by either water vapor or a mixture of water vapor with HCl and/or 1% O2, depending on the test, after which the samples were heated to the target temperature, 450 °C or 550 °C.
Analyses
After the 168-h exposure, the samples were cooled down to room temperature, and a small amount of epoxy resin was put over the samples to fix the deposit and left to harden. After 24 h, the samples were cast in epoxy resin and left to harden overnight; the epoxy-embedded pieces were cut in half with a precision saw (Struers Accutom 5) to expose the cross-sectional surface. The cross-section was then polished with gradually finer SiC papers (Struers Tegramin 30) using water-free mineral oil to preserve the salt deposits and eventually carbon-coated for imaging and elemental analysis. The elemental composition analysis and the imaging were done with a scanning electron microscope equipped with an energy-dispersive X-ray (SEM-EDXA, Leo 1530 Gemini, Thermo Scientific Ultra Dry SDD X-ray detector).
The thickness of a corrosion scale was determined from colored panoramic backscattering SEM images using image analysis. The SEM images were colored manually to distinguish the epoxy resin and surface deposit, the unoxidized metal, and the oxide scale. The mean and maximum oxide scale thickness and thickness distribution were calculated from the colored image with analyzing software. A 4-mm-wide colored panoramic SEM image was analyzed using 10,000 thickness measurement points by custom-made software.
The elemental mapping was carried out with the EDXA, and the data were studied using two software packages. The elemental maps were first analyzed with Thermo Scientific Pathfinder software. Then, the data were treated with novel in-house software to compare the amount and/or share of selected elements. The X-Ray Map Analyzer software was created at Åbo Akademi University as a WPF application (windows presentation foundation) in the programming language C# with Visual Studio 2022 using the framework.NET 6.0 (LTS).
The software uses spectral imaging (X-ray maps) element maps exported as CSV files from the Pathfinder analysis software and allows the element contents to be presented as maps in either weight or atomic percentages. This makes it possible to create images where only certain elemental percentages or ratios are shown. The software creates an image by mapping the percentages or ratios into a color gradient.
Results and Discussion
The Effect of the Atmosphere on Scale Thickness
Figure 3 compares the corrosion scale mean thicknesses for the samples exposed to KCl deposit in the four different atmospheres at 450 and 550 °C. The local maximum scale thickness for a particular low-Cr sample was typically 1.5–2x thicker than the mean value, while for the high-Cr samples with a thin scale, the maximum thickness was 3x thicker but still low. The thickness distribution correlated with the most occurring value, which was for all samples of the same order as the mean value. The dotted line in the figure gives the oxide scale thickness of 20 µm after the one-week test. This corrosion layer thickness can be considered significant. It should be observed that this value is arbitrary and does not necessarily directly correlate with the overall corrosion rate in boiler conditions. Scale is a tool for assessing a material's suitability to particular conditions.

Mean thickness [µm] of corrosion oxide scales on steels exposed to a KCl deposit in all studied atmospheres at a 450 °C and b 550 °C for 168 h. The dotted line indicates the 20 µm line above which corrosion is considered significant. Data for the CO2/H2O and CO2/H2O + 500 ppm HCl atmospheres are from Eriksson et al. [32]
In the CO2/H2O atmosphere at 450 °C, minor corrosion was observed in alloys 10CrMo9-10 and X10 (6 and 8 µm, respectively), while no corrosion was detected in the high-Cr stainless steel samples. In the presence of water vapor and HCl, the corrosion oxide scale was thicker, reaching up to 13 µm for 10CrMo9-10 and X10. As a comparison, Eriksson et al. [32] reported 4–7 µm oxide scale thicknesses for 10CrMo9-10 and X10 without the KCl deposit in both atmospheres. This is in line with previous studies [e.g., 34], showing that the presence of a KCl deposit increases the corrosion compared to the steel without deposits.
An O2 addition of 1 vol% to the CO2/H2O atmosphere nearly doubled the corrosion to 23 µm while 1% O2 combined with HCl resulted in severe corrosion with an oxide thickness of 45 µm. The presence of a KCl deposit with HCl in the flue gas has been reported to induce more chlorination and more corrosion [34]. According to Zahs et al. [20], relatively high oxygen partial pressures are needed to oxidize FeClx. Thus, adding oxygen to the CLC system will likely keep the corrosion process on-going by allowing more chlorinated metal to be oxidized at higher O2 partial pressures.
At a higher exposure temperature of 550 °C, significant corrosion was observed for 10CrMo9-10 and X10 in all atmospheres, indicating that these alloys cannot withstand corrosion at this temperature, even in the mildest, CO2/H2O conditions. The oxide scale thicknesses in the water vapor atmosphere were 26 and 20 µm, respectively. Again, no corrosion was detected for the high-Cr stainless steels in oxygen-free atmospheres. In the presence of water vapor and HCl, the corrosion scales were significantly thicker, up to 40 µm for 10CrMo9-10 and X10. When the same steels were exposed without the KCl deposit, scale thicknesses of ~ 18 µm in water vapor and 16–23 µm in water vapor + HCl atmospheres were measured [32]. The difference in oxide thicknesses with and without the KCl deposit indicates that the combined effect of KCl and HCl in these harsh conditions leads to the observed extensive corrosion. This originates most likely from a higher concentration of reactive Cl facilitating more chlorination of the elements, in this case, iron.
Adding 1% of O2 to the CO2/H2O atmosphere induced corrosion scales similar to those after the HCl addition. The corrosion scale thicknesses were 43 µm for 10CrMo9-10 and 50 µm for X10 for the atmosphere with 1% O2. Interestingly, the same O2 concentration in the HCl-containing atmosphere led to markedly thicker corrosion scales, i.e., 88 µm for 10CrMo9-10 and 97 µm for X10.
The corrosion was assumed to be the net result of more available Cl originating from both the KCl deposit and HCl in the flue gas and an increase in the partial pressure of oxygen, allowing more chlorinated metal to be oxidized and thus releasing Cl back to the chlorination loop. However, the exact corrosion mechanisms and chlorine transport processes cannot be verified from the data available from this work.
In general, a very minor attack was observed for the high-Cr steels. A thin and homogeneous chromium oxide-rich layer (3–5 µm) had formed on samples exposed to O2 and HCl at 550 °C. The results in the present work suggest that oxygen in the gas atmosphere in the fuel reactor at studied temperatures increases the corrosion in the combustion of biomasses with chloride-containing feedstocks.
Corrosion Oxide Scale Compositions and Morphologies
Since the measured thicknesses, chemical composition, and morphologies of the corrosion oxide scales were almost identical with 10CrMo9-10 and X10, only 10CrMo9-10 will be addressed from this point onward to avoid repetition. When considering these low-Cr steels, the thickness and morphology of the corrosion oxide scale are highly dependent both on the temperature and atmosphere as seen in Fig. 4 below. Surprisingly, the samples exposed to 1% O2 at 450 °C showed signs of sintered KCl particles close to alloy surfaces. Instead of sharp edges, typical to KCl crystals, the particles were rounded and partly bridged to one another (Fig. 5a). This was unexpected because pure KCl has a melting point of 770 °C.

Back scattering SEM images of 10CrMo9-10 samples exposed to all tested atmospheres at 450 °C and 550 °C for 168 h. The scale bar can be seen in the images (Color figure online)

1.5-mm-wide SEM images of 10CrMo9-10 samples exposed in O2 and O2 + HCl atmospheres at 450 °C and 550 °C for 168 h. Sintered and bridged KCl particles can be seen in image (a). In b–d, a continuous layer, a clear indication of salt melting can be seen. (Marked with a white arrow) (Color figure online)
When the atmosphere also contained HCl at 450 °C, the melting was more pronounced, resulting in a partly continuous salt layer close to the alloy surface (indicated by the white arrow in Fig. 5b).
At 550 °C, also a continuous layer of salt can be seen close to the alloy and oxide surface in Fig. 5c and d. This indicates that at least part of the salt had been molten during the experiment. The melting behavior of the salt was similar with X10 samples but not with high-Cr steel samples. The salt melting suggests that at these relatively low temperatures, such interactions between the salt and the alloy surface occur that the melting point of pure KCl is decreased.
As described above, the chlorine corrosion process may induce the formation of solid FeCl2 at the metal/oxide interface. This solid FeCl2 may volatilize and then diffuse outward against higher oxygen partial pressure. According to Grabke et al. and others [21, 35], Cl2 and/or HCl can react with iron in the alloy, forming ferrous (Fe2+) chloride according to reactions (1) or (2). Continuous evaporation of iron chloride may take place at the metal-scale interface according to the reaction (3). These volatile iron chlorides may penetrate and diffuse to the scale surface and oxidize because of the increased oxygen partial pressure on the surface. Resulting oxides are often very porous by nature and give no protection to continuous attack and re-chlorination of the metal. The oxidation of FeCl2 could be described with reactions (4) and/or (5) [35]. In the presence of humidity, Cl2 forms HCl, thereby decreasing the Cl2 content [25]. Thus, mainly the reactions 2, 3, 4, and 5 were assumed to occur in this work.
The binary mixture of KCl and FeCl2 has a considerably lower melting point than pure KCl which could cause the melting of the salt close to the metal surface. This was also reported by Okoro et al. [34]. Janz et al. [36] reported a KCl–FeCl2 melting/eutectic temperature of 340–393 °C. The melt fractions of KCl–FeCl2 mixtures were modeled with FactSage calculations of respective conditions in Fig. 6. The analyses show that already a small amount of FeClx in the system will induce a melt of several weight% at both studied temperatures.

Melt fraction in KCl–FeCl2 mixtures at 450 °C and 550 °C calculated with FactSage software
It has also been proposed that the formation of low-melting ferric (Fe3+) chloride is possible through an oxychlorination reaction (6) [37]. After oxychlorination, FeCl3 oxidizes to Fe2O3 in a similar way as FeCl2 to reaction (5). It should be noted that compared to the KCl–FeCl2 mixture, the first melting point of the KCl–FeCl3 mixture is even lower, i.e., 202–220 °C [36]
The continuous partly molten salt layer can be seen in all the low-alloyed steel samples exposed to an O2-containing atmosphere. This melt formation could further be verified with SEM elemental maps showing that the partly molten layer was indeed K–Fe–Cl. The EDX analyses of the dense K–Fe–Cl-containing layer above the FeO next to the original sample surface consisted of 31 mol% K, 8.8 mol% Fe, and 35.6 mol% Cl in the O2-containing atmosphere at 550 °C. Assuming K present as KCl and Fe partly as FeCl2 suggests that the share of FeCl2 in the dense layer contained 10.6 wt% FeCl2. Comparing this with the phase diagram (Fig. 6) suggests 25 wt% melt in the band. Most likely, the molten phase present had contributed to the formation of the dense layer.
The oxide scale on a heat-transfer material grows either by outward diffusion of metal cations (outward-growing scale) or by inward diffusion of anions (inward-growing scale) [38]. Typically, oxide scales grow simultaneously by both mechanisms so that the outward-growing scale builds up at the gas/scale interface and the inward-growing scale at the scale/metal interface. The inward-growing scales in Figs. 4d–h and 7 suggest internal oxidation in the presence of O2 in the atmosphere. The chemical composition was studied from the elemental maps to study the formation of oxide formation. The oxide scales formed on the low-alloyed materials all have a duplex structure, consisting of an outward-grown iron oxide layer and an inward-grown oxide layer containing iron and chromium (Fig. 7 illustrates a typical scale structure).

SEM elemental maps of the 10CrMo9-10 samples exposed to CO2/H2O + 1% O2 atmosphere at 450 °C for 168 h. O (red), Fe (yellow), and Cr (green). The scale bar is shown in the image. The original alloy surface is marked with a dotted line (Color figure online)
The elemental maps show that mainly iron (Fe) has diffused to form an oxide scale with some porosity above the original sample surface marked with a dotted line in the figure. The internal oxidation front forming by outward diffusing Fe and inward diffusing oxygen is clearly seen in the elemental maps. In contrast, the chromium content is constant in the surface-section as suggested by the even green coloring below the original surface. The locally higher chromium contents at the upper part of the Cr map are due to chromium detached during the polishing step.
According to Jonsson et al. [39], the formation of hematite and magnetite for iron at 500 °C depends on the atmosphere. In dry and wet air, a two-layered magnetite forms beneath a thin, fine-grained hematite layer. In contrast, a duplex magnetite forms in H2O. In this work, the straight interface in the layer structure in Figs. 4 and 7 was assumed to indicate duplex magnetite layer. However, this could not be verified using SEM-EDXA.
Iron Chloride Formation and Migration
Although temperature and especially the prevailing atmosphere affect the formed oxide scale thickness, neither alters the chemical composition of the scale. The line scan in Fig. 8 presents a typical chemical composition of the formed scale. The iron concentration remains constant through the corrosion oxide scale from the metal surface up to the KCl layer (areas 2 and 3). Chromium on the other hand is constant in the bulk material and internally oxidized zone but has not diffused into the oxide layer found above the original sample (areas 1 and 2). In area 4, manganese is found enriched in a layer between KCl and the outermost iron oxide. This enrichment was found in other places of the sample as well. This Mn-rich layer on top of the Fe oxide layer has also been reported in other studies [37]. The Mn-layer has been attributed to chlorination and subsequent oxidation of manganese in the outer corrosion layer due to where the oxygen partial pressure is high enough to convert manganese chloride to manganese oxide [40]. However, the diffusion mechanisms of Mn in the present conditions remain unclear. A peak in the amounts of Fe and O can be seen between areas 5 and 6 indicating the possible oxidation of outward diffused FeClx. At low oxygen partial pressures between the intact metal and oxide layer (areas 1 and 2), traces of chlorine are found, indicating that chlorine has penetrated through the oxide layer to the metal surface. This provides further proof for possible iron chloride formation and through that, to the decreased melting point of pure KCl.

An SEM image of the 10CrMo9-10 sample exposed in O2 atmosphere at 450 °C for 168 h (a) and the line scan (b) along the yellow arrow in a. (Area 1: bulk material, 2: inward-grown oxide, 3: outward-grown oxide, 4: manganese enriched layer, and 5 and 6: KCl) (Color figure online)
Regardless of the temperature, samples exposed to conditions with added O2 or O2 + HCl show the presence of iron oxide in the dense KCl layer, which indicates the presence of gaseous FeCl2 followed by its diffusion and oxidation both on and within KCl. This formation of iron oxide on the KCl can be seen in Fig. 9 below. The data from the elemental maps were further processed with The X-Ray Map Analyzer software which allows a more precise presentation of chosen data and pinpointing certain elemental combinations and/or concentrations. Chlorine, potassium, and iron are shown in concentrations between 1 and 5 at% to exclude most of the signal originating from KCl or iron oxide.

Processed elemental maps of 10CrMo9-10 samples exposed CO2/H2O + O2 + HCl at 550 °C for 168 h. The upper left corner shows the original backscattering image. Other images indicate where Cl, K, and iron, respectively, are found in concentrations between 1 and 5 at%. The scale bar is shown in the image
The processed images in Fig. 9 clearly indicate the presence of chlorine without potassium below the formed oxide which supports the idea of inward diffusion of chlorine, thus enabling the formation of iron chlorides. The presence of a continuous KCl layer on top of the iron oxide scale suggests that the salt has been at least partly molten during the exposure. This provides further evidence for iron chloride formation, where the chlorine originates mostly likely from both chlorine sources, KCl and HCl.
The rate of the chlorine cycle may be substantially accelerated if molten chlorides are formed which may cause fluxing of the oxide scale, making the scale lose its integrity and become more vulnerable to further attack [28]. This fluxing may also happen if HCl is present in the atmosphere. Mayer et al. [41] reported that at 0.1 vol% HCl in the flue gas, blisters in the Fe2O3 (hematite) layer started to occur, and at 0.2 vol% the layer was porous and non-continuous.
In very low partial pressure of O2, the partial pressure of HCl was probably not high enough to cause severe gas-phase corrosion attack alone, but it may have still played a role in the scale failure and thus improved the possibilities of Cl penetration [32].
The experiments in this work aimed to clarify whether a small oxygen slip from the air reactor or the surrounding atmosphere would induce boiler material corrosion in the CLC fuel reactor. Adding 1% O2 in the atmosphere doubled the corrosion thickness compared to water vapor + CO2. When 500 ppm HCl was added to the very corrosive 1% O2 atmosphere, the corrosion thickness increased by a factor of two. The presence of oxygen may affect the oxidation in many ways, for example, Ihara et al. [42] have found that adding oxygen to the HCl gas greatly accelerated the corrosion of iron due to the formation of low-melting FeCl3 by oxychlorination (Reaction 6). Folkeson et al. [43] studied austenitic stainless steel at 500 °C in an atmosphere containing 5% O2; the weight gain was 0.01 mg/cm2 for a week. Adding 500 ppm HCl increased the weight gain to 0.06 mg/cm2, while 40% of H2O resulted in a further gain of 0.09 mg/cm2. Larsson et al. [29] studied Fe–2.25Cr–1Mo at 400 °C for 24 h in different atmospheres. With 20% H2O, 5% O2, and 500 ppm HCl, the weight gain was 0.2 mg/cm2. A deposit of KCl in the same atmosphere increased the weight gain to 0.5 mg/cm2. Interestingly, when HCl was removed from the atmosphere the gain was 0.9 mg/cm2. However, these tests are not totally comparable to our setup. Larsson et al. [29] used test pieces that were not pre-oxidized before the exposure, the total test time was 24 h, and they sprayed the salt as separate particles instead of covering the whole surface with KCl, as in the present work. However, the experiments in typical CLC conditions suggest that oxygen and KCl increase the corrosion of low-chromium steels. Further, the presence of HCl increased the corrosion at higher temperatures.
Under all studied conditions simulating the fuel reactor, the low-Cr steels 10CrMo9-19 and X10 showed clear indications of corrosion at 450 °C. As expected, the thickest oxide scale, 45 µm, was detected for steels 10CrMo9-10 and X10 under conditions where both oxygen and HCl were present in the atmosphere.
At 550 °C, all low-Cr steel samples showed clear corrosion. The thickest oxide scale of 97 µm induced again in the presence of oxygen and HCl. For high-Cr steels, corrosion was negligible, and the mean thickness of the corrosion scales was markedly thinner than reported for the same type of materials exposed to pure KCl at 600 °C in the air [44, 45].
Conclusions
The influence of the simultaneous presence of potassium chloride, hydrochloric acid, and oxygen on the corrosion of five heat-exchanger materials was studied on a laboratory scale under conditions simulating the fuel reactor side in the chemical-looping combustion of biomass.
Under reducing conditions (50/50 vol% CO2/H2O), the low-Cr steels 10CrMo9-19 and X10 showed clear indications of corrosion already at 450 °C, and at 550 °C, the corrosion was more pronounced. At both temperatures, the oxide thickness grew in the series CO2/H2O → CO2/H2O/HCl → CO2/H2O/O2 → CO2/H2O/HCl/O2. The combined effect of O2, HCl, and KCl was significant, resulting in a notable corrosion of both studied low-Cr steels. The oxide scales were ~ 45 µm at 450 °C and ~ 90 µm at 550 °C. The presence of HCl in the atmosphere enables the formation of iron chloride(s), which degrades the low-Cr alloys. At the same time, the presence of only 1% of O2 in the atmosphere enhances the oxidation of formed intermediate FeClx, thus releasing chlorine, which subsequently can react with the alloy. In some of the tests, signs of molten KCl were seen near the metal surface at significantly lower temperatures than the melting point of pure KCl, 770 °C. Evidence that KCl had been partly molten during the experiment was found only with the low-Cr steel samples together with notable corrosion. It is believed to originate from the formation of FeClx as an intermediate in the corrosion reaction. When mixing with KCl, FeClx lowers the first melting point of the mixture significantly when compared to that of pure KCl. The formation of such a partly molten salt phase may accelerate the corrosion, suggesting that attention should be paid to the intermediates and reaction products when estimating the corrosivity of certain conditions.
The results indicate that low-Cr steels are not suitable as heat-exchanger materials under the reducing conditions in the fuel reactor if humidity, HCl, and/or oxygen are present in the atmosphere above 450 °C. Furthermore, in the second-generation CLC, KCl may deposit on heat-exchanger surfaces in the fuel reactor and cause rapid corrosion. Based on the results, KCl in the CLC fuel reactor might induce corrosion, especially if even small amounts of O2 and/or HCl are also present. The high-Cr steels, again, withstand corrosion induced by potassium-containing deposits in all the tested atmospheres.
References
V. Masson-Delmotte, P. Zhai, H. O. Pörtner, D. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J. B. Matthews, Y. Chen, X. Zhou, M. I. Gomis, E. Lonnoy, T., Maycock, M. Tignor and T. Waterfield, Global Warming of 1.5°C. An IPCC Special Report on the Impacts of Global Warming of 1.5°C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty; IPCC, 2018.
C. Azar, K. Lindgren, Obersteiner, et al., Climatic Change 100, 2010 (195).
A. Lyngfelt, B. Leckner, and T. Mattisson, Chemical Engineering Science 56, 2001 (3101).
W. K. Lewis and E. R. Gilliland production of pure carbon dioxide. US Patent US Patent 2,665,972, 1954.
M. Ishida and H. Jin, Energy 19, 1994 (415).
A. Lyngfelt, Applied Energy 113, 2014 (1869).
A. Lyngfelt, Energy and Fuels 34, 2020 (9077).
H. Leion, T. Mattison, and A. Lyngfelt, International Journal of Greenhouse Gas Control 2, 2008 (180).
T. Mattisson, A. Lyngfelt, and H. Leion, International Journal of Greenhouse Gas Control 3, 2009 (11).
A. Lyngfelt and C. Linderholm, Energy Procedia 63, 2014 (98). https://doi.org/10.1016/j.egypro.2014.11.011.
A. Lyngfelt and B. Leckner, Applied Energy 157, 2015 (475). https://doi.org/10.1016/j.apenergy.2015.04.057.
A. Lyngfelt, A. Brink, Ø. Langørgen, T. Mattisson, M. Rydén, and C. Linderholm, International Journal of Greenhouse Gas Control 88, 2019 (38).
M. Ryden, A. Lyngfelt, O. Langoergen, Y. Larring, A. Brink, S. Teir, H. Havaag, and P. Karmhagen, Energy Procedia 114, 2017 (6074).
F. I. Kluger, I. Abdulally, H. Andrus, A. Levasseur, B. Beal, and J. Marion, Overview of Alstom’s Chemical Looping Programs, 5Th Meeting of the IEAGHG International Oxyfuel Combustion Research Network Wuhan, China—October 28 (2015).
M. Zevenhoven, P. Yrjas, B.-J. Skrifvars, and M. Hupa, Energy Fuels 26, 2012 (6366). https://doi.org/10.1021/ef300621j.
M. Zevenhoven, P. Yrjas, and M. Hupa, Ash-forming matter and ash-related problems. in Handbook of combustion; solid fuels, vol. 4, eds. M. Lackner, F. Winter and A. K. Agarwal (Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010), pp. 493–531.
B.-J. Skrifvars, R. Backman, M. Hupa, K. Salmenoja, and E. Vakkilainen, Corrosion Science 50, 2008 (1274).
S. Enestam, D. P. Bankiewicz, J. Tuiremo, K. Makela, and M. Hupa, Fuel 104, 2013 (294). https://doi.org/10.1016/j.fuel.2012.07.020.
H. Wu, D. Bankiewicz, P. Yrjas, and M. Hupa, Energy Fuels 29, 2015 (2709).
A. Zahs, M. Spiegel, and H. J. Grabke, Corrosion Science 42, 2000 (1093).
H. J. Grabke, E. Reese, and M. Spiegel, Corrosion Science 37, 1995 (1023). https://doi.org/10.1016/0010-938X(95)00011-841.44.
M. Spiegel, A. Zahs, and H. J. Grabke, Materials at High Temperatures. 20, 2003 (153).
V. Cantatore, M. A. Olivas Ogaz, J. Liske, T. Jonsson, J. E. Svensson, L. G. Johansson, and I. Panas, The Journal of Physical Chemistry 123, 2019 (25957).
N. Folkeson, T. Jonsson, M. Halvarsson, L.-G. Johansson, and J.-E. Svensson, Materials and Corrosion. 62, 2011 (606). https://doi.org/10.1002/maco.201005942.
J. Pettersson, H. Asteman, J. E. Svensson, et al., Oxidation of Metals 64, 2005 (23). https://doi.org/10.1007/s11085-005-5704-3.
C. Pettersson, J. Pettersson, H. Asteman, J. E. Svensson, and L. G. Johansson, Corrosion Science 48, 2006 (1368).
J. Lehmusto, P. Yrjas, B. J. Skrifvars, and M. Hupa, Materials Science Forum 696, 2011 (218).
A. Ruh and M. Spiegel, Corrosion science 48, 2006 (679).
E. Larsson, J. Liske, A. Persdotter, et al., Oxidation of Metals 93, 2020 (29).
T. Jonsson, N. Folkeson, M. Halvarsson, J. E. Svensson, and L. G. Johansson, Oxidation of Metals 81, 2014 (575).
T. Dudziak, K. Jura, and J. Rutkowska, Oxidation of Metals 85, 2016 (647–664). https://doi.org/10.1007/s11085-016-9617-0.
J.-E. Eriksson, M. Zevenhoven, P. Yrjas, A. Brink, and L. Hupa, Energies 15, 2022 (2740). https://doi.org/10.3390/en15082740.
D. Bankiewicz, P. Yrjas, and M. Hupa, Energy & Fuels 23, 2009 (3469).
S. C. Okoro, S. Kiamehr, M. Montgomery, F. J. Frandsen, and K. Pantleon, Materials and Corrosion 68, 2017 (499). https://doi.org/10.1002/maco.201609173.
H. P. Nielsen, F. J. Frandsen, K. Dam-Johansen, and L. L. Baxter, Progress in Energy and Combustion Science. 26, 2000 (283). https://doi.org/10.1016/S0360-1285(00)00003-4.
G. J. Janz, C. B. Allen, Jr. J. R. Downey, RPT. Tamkins Eutectic data; safety, hazard, corrosion, melting points, compositions and bibliography. Troy: Molten Salts Data Center, Rensselaer Polytechnic Institute, 1976.
A. Zahs, M. Spiegel, and H. Grabke, Materials and Corrosion 50, 1999 (561). https://doi.org/10.1002/(SICI)1521-4176(199910)50:10%3c561::AID-MACO561%3e3.0.CO;2-L.
C. Wagner, Corrosion Science 5, 1965 (751).
T. Jonsson, B. Pujilaksono, S. Hallström, J. Ågren, J.-E. Svensson, L.-G. Johansson, and M. Halvarsson, Corrosion Science 51, 2009 (1914). https://doi.org/10.1016/j.corsci.2009.05.016.
S. C. Okoro, M. Montgomery, F. J. Frandsen, and K. Pantleon, Energy & Fuels. 28, 2014 (6447). https://doi.org/10.1021/ef5017335.
P. Mayer and A. V. Manolescu, Corrosion 36, 1980 (369). https://doi.org/10.5006/0010-9312-36.7.369.
Y. Ihara, H. Ohgame, K. Sakiyama, and K. Hashimoto, Corrosion Science 21, 1981 (805).
N. Folkeson, T. Jonsson, J-E. Svensson, The combined effect of hcl and water vapour on the high temperature corrosion of an austenitic stainless steel. 8th International Symposium on High-Temperature Corrosion and Protection of Materials, Les Embiez, 2012.
H. Wu, P. Yrjas, and M. Hupa, Energy Fuels. 29, 2015 (1186).
J. Lehmusto, P. Yrjas, B.-J. Skrifvars, and M. Hupa, Fuel Processing Technology. 104, 2012 (253).
Funding
Open access funding provided by Åbo Akademi University. This work has been carried out partly within the Research Council of Finland project “Initiation and propagation of high-temperature corrosion reactions in complex oxygen-containing environments” (Decision no. 348963). This work is part of the Nordic Flagship project, Enabling negative CO2 emissions in the Nordic energy system through the use of Chemical-Looping Combustion of Biomass (Bio-CLC)” financed by the Nordic Energy Research. The financial support by Åbo Akademi University is acknowledged.
Author information
Authors and Affiliations
Contributions
J-EE, MZ, and PY contributed to the conceptualization, J-EE contributed to the methodology, J-EE, LS, DL, and JL were involved in the analyses, J-EE assisted in the writing—original draft, JL, LH, MH, AB, and MZ contributed to the writing—review and editing, AB and MZ were involved in the project administration, AB assisted in the funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eriksson, JE., Lehmusto, J., Silvander, L. et al. Corrosion of Heat-Transfer Materials Induced by KCl, HCl, and O2 Under Chemical-Looping Conditions. High Temperature Corrosion of mater. (2024). https://doi.org/10.1007/s11085-024-10250-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11085-024-10250-1