
Abstract
Three FeCrAl alloys and two chromia-formers (a stainless steel, and a Ni-base alloy) have been exposed in four environments (dry air, air + 20% H2O, 20% H2 + 20% H2O + Ar and 95% N2 + 5% H2) for 168 h at 800 °C. The corroded samples were investigated by SEM/EDS, XRD and gravimetry, and the formation of CrO2(OH)2(g) was measured as a function of time using a denuder technique. The Fe-base alloy formed a Cr-rich protective oxide scale in dry air and wet air but suffered break-away oxidation in 20% H2 + 20% H2O + Ar. In contrast, the Ni-base alloy suffered extensive NiO formation and internal oxidation in dry air and wet air but formed a protective chromia scale in 20% H2 + 20% H2O. All three FeCrAl alloys formed protective alumina scales in dry air, wet air and 20% H2 + 20% H2O + Ar. The FeCrAl alloy Kanthal APMT was severely nitrided in the 95% N2 + 5% H2 environment due to defects in the oxide scale associated with RE-rich inclusions which allowed nitrogen to enter the alloy. In contrast, the two Cr-lean FeCrAl alloys Kanthal EF101 and Kanthal EF100 did not suffer nitridation at all.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High-temperature corrosion is often a lifetime-limiting factor for alloys used at temperatures > 500 °C, and it is essential that a slow-growing oxide scale is formed on the alloy surface which acts as a barrier between the environment and the alloy. Alumina and chromia are preeminent among the oxides forming such protective scales.
While chromium-rich oxide scales often perform excellently below 1000 °C, they can be subject to breakaway oxidation, triggering rapid growth of other transition metal oxides [1]. Such breakaway oxidation is often caused by chromium depletion of the alloy matrix. Thus, in environments containing both O2 and H2O, volatilization of CrO2(OH)2 can trigger rapid growth of Fe-oxide on stainless steel [2, 3]. Chromium volatilization can be mitigated by alloying with manganese, causing MnCr2O4 spinel oxide to form at the scale/gas interface [4, 5] or by the formation of a NiO “cap layer” provided that the alloy contains sufficient amounts of nickel [2]. The formation of alkali chromates by the presence of, e.g., KCl has similar corrosive effects as chromium volatilization [6]. Moreover, the preferential oxidation of chromium to form a protective scale can also be compromised by formation of chromium-rich precipitates, e.g. nitrides and carbides, in the alloy.
Stainless steels are known to suffer faster oxidation when water is the only oxidant, compared to when O2 is also present [7,8,9], the faster oxidation being accompanied by more inward-growing oxide [10, 11]. Chromium volatilization does not play a role here because CrO2(OH)2 cannot form in the absence of O2. Several explanations of this effect have been proposed, including the smaller ionic radius of OH− compared to O2− causing faster transport across the scale [12]; uptake of hydrogen by the alloy decreasing the availability of chromium [13]; and formation of smaller oxide grains in the presence of water [8].
At sufficiently low oxygen activity (below about 10–27 at 800 °C [14]), chromia cannot form. Under these conditions, the bare metal surface reacts rapidly with other oxidants present, which then enter the alloy and may form compounds. For example, nitrogen- or carbon-containing species cause ingress of nitrogen or carbon into the alloy, resulting in the formation of nitrides or carbides with, e.g. chromium, aluminium and titanium. This tends to make the alloy brittle, reducing component life. It is noted that the alloy can suffer nitridation or carburization, although at a reduced rate, even in the presence of a surface chromia layer, because chromia scales are somewhat permeable to both nitrogen and carbon [15, 16]. A previous paper showed that the presence of a chromia surface layer reduced the rate of nitrogen pick-up by 50–95% at 900 °C [16]. The resistance towards nitridation is reported to increase with the concentration of nickel, while lower concentrations of chromium in the alloy reduce nitrogen uptake [16, 17].
Alumina scales have excellent barrier properties at high temperature. Thus, alumina features low reactivity and very low vapour pressure [2, 18] and defect-free alumina scales are virtually impermeable to most oxidants, e.g. nitrogen [16]. Yet, alumina-forming alloys are seldom used at intermediate temperatures. Thus, for applications at, e.g. 800 °C, chromia-forming Ni-base alloys and stainless steels are predominant. The use of alumina-forming alloys at intermediate temperatures has traditionally been discouraged by the slow formation of protective alumina scales and by the tendency to form brittle intermetallic phases. This situation may now be about to change due to a new class of lean chromium FeCrAl´s which is being developed [19, 20], the low chromium content mitigating the formation of brittle phases. Also, the new class of alloys is showing some promise from a corrosion point-of-view. Thus, it was recently reported that a lean chromium FeCrAl alloy containing silicon forms protective alumina scales at 600 °C, being able to resist attack by, e.g. wet air [21, 22].
The main idea with this paper is to evaluate how two novel lean chromium FeCrAl alloys compete corrosion-wise with two well-known chromia-forming alloys (Alloy 600 and 310H stainless steel) at 800 °C. The powder-produced FeCrAl alloy Kanthal APMT is also included for comparison. The five alloys have been exposed in four environments, two featuring high pO2 (dry air and air + 20% H2O) and two with low pO2 (20% H2 + 20% H2O + Ar and 95% N2 + 5% H2). The wet air environment thus tests the ability of the alloys to cope with chromium volatilization. The H2 + H2O environment investigates the tendency for scale breakdown when the oxygen for oxide scale growth is supplied by water vapour. Lastly, the ability to resist nitridation is investigated in the N2 + H2 exposures.
Materials and Experimental
Materials
The chemical composition of the five studied alloys is presented in Table 1. The stainless steel 310H is an austenitic high-temperature alloy, Alloy 600 is an austenitic Ni-base alloy, and EF101, EF100 and APMT are ferritic FeCrAl-alloys. Alloys EF101 and EF100 are “lean FeCrAl alloys” and feature a low chromium content in order to avoid formation of secondary phases which causes brittleness. EF101 is alloyed with silicon in order to improve the ability to form a protective scale at intermediate temperature as previously reported [22]. APMT has higher aluminium and chromium concentrations and is designed to have excellent oxidation resistance and high creep strength at temperatures up to 1250 °C [23].
Sample coupons were prepared with the dimension 15 × 10 × 2 mm (alloys 310H and 600) and 15 × 15 × 2 mm (the three FeCrAl-alloys). The coupon faces were grinded and polished to a mirror-bright finish with 1 µm diamond paste as the final preparation step. Edges were grinded with SiC paper with a 1000# grit. After grinding and polishing, all samples were subject to cleaning, first in acetone and then in ethanol using an ultrasonic bath.
Experimental Setup
Exposures were performed in four different, carefully controlled gas environments, two at high oxygen activity and two at low oxygen activity, see Table 2 for details. The temperature was 800 °C ± 3 °C in all cases, and the furnaces were calibrated using thermocouples of type S. Dew points were calibrated using Michelle instruments high-precision chilled mirror hygrometers. The activity of oxygen in the different environments was calculated using Thermo-Calc with the database SSUB6.
Exposures were performed in horizontal tube furnaces equipped with fused silica reaction tube, except for environment 20% H2 + 20% H2O + Ar where a sintered alumina tube was used. In the exposure with air + 20% H2O, chromium(VI)-oxy-hydroxide which evaporated from the samples was collected by a “denuder technique” described in [2]. Gas flow rate is only considered to be important in the environment with air + 20% H2O where chromium(VI)-oxy-hydroxide is volatilized. In the exposures at high oxygen activity, the samples were inserted into a pre-heated furnace, and after 168 h of exposure, the samples were taken out and left to cool in ambient air. In the exposures at low oxygen activity, the samples were placed inside the cold furnace, and the furnace was purged for 24 h (either with N2 or Ar) before the furnace temperature was increased and the exposure started. In the case where the environment contained water, this was turned on after the furnaces reached about 150 °C. The time to reach exposure temperature was approximately one hour. After 168 h of exposure, the furnace was turned off, the samples in the furnace reaching ambient temperature after about 8 h. In the exposure with 20% H2O + 20% H2, the flow of water vapour was turned off at about 150 °C in order to avoid condensation. All exposures were performed using triplicate samples placed parallel to the gas flow standing in alumina samples holders with 2 mm slits.
After the exposures, the mass gain of the samples was measured using a Sartorius™ 6-decimal balance.
Analysis Methods
After exposure, samples were examined by scanning electron microscopy (SEM), energy-dispersive X-ray spectrometry (EDX) and X-ray diffraction (XRD). Cross-sectional samples were prepared by Broad-Ion-Beam (BIB) milling in a Leica TIC3X™. A Zeiss Ultra 55™ FEG-SEM was used for imaging in 50° plan view and cross-sectional view. For SEM–EDX examination, a FEI Quanta™ 200 Field Emission Gun was utilized. XRD measurements were performed by a Bruker D8 Advance (Cu Kɑ source) in Bragg–Brentano geometry.
Results
Gravimetry
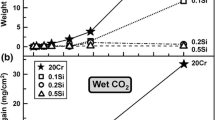
Figure 1 shows the mass gains of the five alloys after 168 h exposure in the four environments. For the exposures in air + 20% H2O, the mass gain of the two chromia-forming alloys 310H and 600 has been added with the mass of Cr2O3 corresponding to the amount of evaporated chromium-oxy-hydroxide (CrO2(OH)2). A comparison of the two chromia-formers shows that Alloy 600 exhibits higher mass gains than 310H in the two high-pO2 environments and lower mass gains in the low pO2 environments. The two “lean” FeCrAl alloys EF101 and EF100 show small mass gains in all four environments. The mass gains of APMT are also low, except for the nitriding, 95% N2 + 5% H2, environment.

Mass gains observed after exposure in the different environments at 800 °C for 168 h
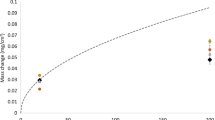
Figure 2 presents chromium evaporation rates for alloys 310H and 600. During the first 30 h of exposure, alloy 600 exhibits about 15% higher evaporation rate than 310H. For both alloys, the evaporation rate drops rapidly in the beginning of the experiment and levels out after about 80 h. At this stage, the evaporation rate of alloy 600 is about half that of 310H. CrO2(OH)2(g) was detected in the off-gas from the exposures of the three FeCrAl-alloys in air + 20% H2O. However, the evaporation rate was very low, being close to the detection limit (2 ng/cm2h).

Evaporation rate of alloy 310H and 600 in exposure in air + 20% H2O at 800 °C for 168 h
310H
Figure 3 shows SEM images of 310H after exposure in the four environments, while Table 3 shows the corrosion products identified by XRD. In dry air, the oxide scale consists of an inner chromia layer and an outer, chromium-rich spinel oxide layer forming large facetted crystals, the scale morphology showing little variation with alloy microstructure. This is in contrast to humid air where there is a marked difference between the scale formed on the grain interior and on grain boundaries. Thus, while the oxide scale on the interior of grains consists of an inner chromium- and iron-rich spinel (M3O4) layer and an outer hematite (Fe2O3) layer, the scale at the grain boundaries (not shown in Fig. 3(air + 20% H2O)) only consists of (Cr,Fe)-spinel, with no hematite present. While chromia was not observed in the cross-sectional examination, a weak chromia signal was detected by XRD. In 20% H2 + 20% H2O environment, spinel-type oxide was the only crystalline product detected. SEM-SE imaging (Fig. 3(20% H2 + 20% H2O) revealed two distinct types of scale morphologies. On the right-hand side of the image, the scale consists of a single layer of Cr-rich spinel oxide. The left-hand side image shows a complex scale consisting of an outer Fe-rich spinel and an inner Cr-rich spinel with Cr-depleted metal matrix sandwiched between the oxide layers. In the nitriding environment, no oxide scale formed on the alloy surface, while significant amounts of Cr-nitride precipitates are observed in the alloy bulk and at the surface.

SEM-SE images showing 310H exposed in the different environments for 168 h. Top row shows 50° tilted top view images and bottom row shows cross-sectional images
Alloy 600
Figure 4 shows SEM images of Alloy 600 after exposure in the four environments. Table 4 shows the corrosion products identified by XRD. The alloy suffered extensive oxidation in the two high-pO2 environments, and the oxidation morphology depended on the alloy microstructure. In dry air, the oxide scale on the interior of the alloy grains consists of an inward-growing part and an outward-growing part. The inward-growing scale consists of (Cr,Ni) spinel which is separated from the alloy substrate by a thin chromia layer. The outward growing scale has a bottom NiO layer and a top layer consisting of a mixture of NiO and Fe-rich spinel. In contrast, the scale at the alloy grain boundaries is much thinner, consisting of a single chromia layer. While the oxide scale formed in humid air is similar to that observed in dry air, the oxide layers tend to be thicker and an outward-growing oxide appears at the grain boundaries, on top of the chromia layer. In 20% H2 + 20% H2O environment, the oxide scale is thin and consists of a single chromia layer, no spinel oxides being detected. The chromia layer is slightly thicker at alloy grain boundaries and also features a higher concentration of dissolved iron (not shown in Figure). Similar to 310H, no surface oxide formed in 95% N2 + 5% H2 environment. Small (Ti,Cr)-nitride precipitates have formed throughout the alloy matrix, while extended Ti,Cr-nitride precipitates appear at alloy grain boundaries.

SEM-SE images showing 600 exposed in the different environments for 168 h. Top row shows 50° tilted top view images and bottom row shows cross-sectional images
APMT
Figure 5 shows SEM images of the FeCrAl alloy APMT after 168 h exposure. Al-rich surface layers formed in all four environments. However, the limited oxide layer thickness barred phase identification by the analysis instruments used. Table 5 shows the measured oxide scale thickness and the average scale thickness calculated from the mass gain. In air, humid air and 20% H2 + 20% H2O + Ar, there is good agreement between the measured and calculated scale thickness, implying that the entire mass gain is due to oxide scale growth. The thickest Al-rich surface scale formed in the H2 + H2O environment.

SEM-SE images showing APMT exposed in the different environments for 168 h. Top row shows 50° tilted top view images and bottom row shows cross-sectional images
The top view images in Fig. 5 show that the APMT samples exposed in dry and wet air have formed a large number of small oxide nodules and a smaller number of large oxide agglomerations, the latter sometimes having irregular shape. The large oxide agglomerations also appear in 20% H2 + 20% H2O + Ar environment, while the numerous small oxide nodules are missing. The plan view image of the sample exposed in N2 + H2 is dominated by a large bright feature corresponding to a RE-rich inclusion in the alloy. Smaller RE-rich inclusions (bright contrast) are also present. SEM–EDX analysis showed that the large oxide agglomerations formed in dry air and wet air and in H2 + H2O environment were overlying RE-rich inclusions in the alloy. Some scale spallation was observed around the RE-rich particles and the associated oxide agglomerations. In N2 + H2 environment, APMT suffered severe local nitridation, see the large AlN precipitates in Fig. 5 (cross section), corresponding to a significant mass gain (see Fig. 1).
EF101
Figure 6 shows SEM images of EF101 after 168 h exposure. All four environments resulted in thin Al-rich surface layers with similar morphologies. Table 6 shows the measured oxide scale thickness and the average oxide scale thickness calculated based on mass gains. The thickness of the oxide layers observed in SEM corresponded well to the mass gains recorded. The top view images in Fig. 6 show similar features as Fig. 5, oxide agglomerations having formed at RE-rich inclusions. It is noted that the very large RE-rich inclusions (and the associated oxide agglomerations) seen in the case of APMT were absent for EF101. Also, no scale spallation was observed in the vicinity of the RE-particles. In accordance with the mass gain results (i.e., the good agreement between measured and calculated alumina layer thickness, see Table 6), there was no evidence for AlN precipitates in the cross section of EF101 exposed to N2 + H2. The numerous small oxide nodules seen after exposure of APMT in dry and wet air are absent on EF101. The oxide microstructure of EF100 after exposure was essentially identical to EF101 and is therefore not presented.

SEM-SE images showing EF101 exposed in the different environments for 168 h. Top row shows 50° tilted top view images and bottom row shows cross-sectional images
Discussion
The Chromia-Forming Alloys
A comparison of the corrosion behaviour of the two chromia-forming alloys in dry air shows that while 310H forms a continuous chromia layer covered by Cr-rich spinel oxide, the Ni-base Alloy 600 is unable to form a protective chromia layer early in the exposure (see Figs. 3 and 4). After 168 h, the latter has formed a continuous chromia layer at the scale/alloy interface. According to Wagner [24], a basic requirement which must be fulfilled for, e.g. chromia and alumina to form protective layers on an alloy surface, rather than precipitating within the metal matrix, is that the outward transport of the oxide-forming element is faster than the inward transport of oxygen into the alloy. The capacity for chromium transport towards the alloy surface is the product of the concentration (properly the activity) and diffusivity of chromium. The higher concentration of chromium is therefore expected to promote the formation of an external chromia scale on 310H in comparison to Alloy 600. (The two austenites are considered to feature similar Cr diffusivities.) A comparison of the two alloys with respect to oxygen inward transport is less clear-cut. On one hand, both the solubility and the diffusivity of oxygen in austenitic FeCrNi alloys are reported to diminish with increasing Ni content [25], favouring the formation of an external chromia scale on Alloy 600. On the other hand, the high Ni- and low Fe- content of Alloy 600 may promote NiO formation during early stages of oxidation. If that is the case, it would interfere with the formation of an external chromia layer. This is because NiO has a greater tendency to decompose into metal plus O(ss) compared to thermodynamically more stable oxides such as iron or chromium oxides [10, 26].
As expected, exposure of the two alloys to a combination of air and water vapour resulted in significant evaporation of CrO2(OH)2, see Figs. 1 and 2. As noted above, in the case of 310H the presence of water vapour also produces a scale morphology which reflects the alloy microstructure, e.g. hematite forming on the interior of grains but not at grain boundaries. It is argued that the evaporation of CrO2(OH)2 increases the chromium supply needed to suppress iron oxidation and that hematite formation is still hindered in the vicinity of grain boundaries because of relatively fast grain-boundary diffusion of chromium. Yet, corrosion-wise 310H is relatively unaffected by water vapour and the associated chromium loss (Fig. 3). While chromium volatilization is known to trigger breakaway oxidation on a variety of stainless steels, including 310H at 600 °C [27], the ability of 310H to resist breakaway in the present case is attributed to the much higher diffusivity of chromium in the steel at 800 °C. Chromium volatilization at the end of the exposure was about half for Alloy 600 compared to 310H, see Fig. 2. A recent paper [2] on the oxidation properties of a related Ni-base chromia former (Alloy 690) in a similar environment reported that a NiO “cap layer” tended to form on the scale surface, resulting in a significant decrease in chromium volatilization. In accordance with [2], the rapid decrease in chromium volatilization from Alloy 600 (see Fig. 2) is attributed to the NiO-rich oxide scale, acting as a barrier to Cr3+ diffusion towards the scale/gas surface. Still, the faster corrosion of Alloy 600 in wet air compared to dry air (see Figs. 1 and 4) is attributed to the loss of chromium by volatilization which decreases the availability of chromium in the alloy near-surface region in the substrate, making it more difficult to form a “healing” chromia layer at the bottom of the scale.
The present study shows that the 20% H2 + 20% H2O environment triggers partial breakdown of the protective chromia/spinel oxide layer on 310H, the resulting scale including large chunks of Cr-depleted metal (see Fig. 3). In contrast, Alloy 600 forms a protective chromia scale in the same environment (see Figs. 1 and 4). Several reports show that chromia-forming alloys and stainless steels tend to suffer more severe corrosion when H2O is the oxidant rather than O2 [8, 10]. Recently, work on the so-called “dual atmosphere effect” has shown that ferritic stainless steels become more prone to suffer breakaway oxidation in air when the steel contains dissolved hydrogen [28,29,30]. The deleterious effect of hydrogen is reportedly present in the 600–800 °C range and was attributed to a decrease in chromium diffusivity caused by the association of hydrogen with chromium at alloy grain boundaries. It is proposed that in this case, hydrogen is picked up by the metal and that, in line with [30], hydrogen interacts with chromium at alloy grain boundaries, retarding the supply of chromium to the growing scale, thus causing the 310H stainless steel to suffer breakaway in the 20% H2 + 20% H2O environment. While 800 °C is at the high end of the temperature range reported for the dual atmosphere effect in ferrites [28], the argument is still considered to be valid because of the lower bulk diffusivity of chromium in austenites compared to ferrites. Hence, the dominance of grain boundary transport in the supply of chromium to the growing chromia scale extends to higher temperature for austenites and that, consequently, the hydrogen effect on chromium diffusion also extends to higher temperature.
The results show that the relative ability of the two austenitic alloys to resist corrosion in H2 + H2O is reversed compared to the situation in dry and wet air. Thus, Alloy 600 forms a protective surface chromia layer in the H2 + H2O environment in contrast to 310H which suffers breakaway oxidation (see Figs. 3 and 4). It is argued that this reversal is caused by changes in the ability of the two alloys to fulfil the condition for the formation of a protective surface chromia, i.e. that the outward chromium transport in the alloy must be faster than the inward transport of oxygen. In this respect, the situation is essentially the same as in dry air and wet air, with two exceptions. Firstly, we have the appearance of the hydrogen effect just mentioned. Secondly, and as discussed above, the absence of Ni oxide in the oxide scale is expected to decrease the flux of oxygen into the alloy, in comparison to the situation in dry air and wet air. It is suggested that this effect is greater for Alloy 600 because of the higher Ni content (73% compared to 20% in 310H). In contrast, there is no obvious reason for the adverse effect of alloy hydrogen on the oxidation properties to differ between the two alloys.
In the 95% N2 + 5% H2 environment, which only contains traces of H2O, chromia is not thermodynamically stable, while the formation of chromium nitride (both Cr2N and CrN) is spontaneous. Nitridation involves adsorption and dissociation of N2 molecules on the metal surface, followed by dissolution of nitrogen in the metal matrix. Chromium nitride particles then nucleate as the solid solution becomes supersaturated. As expected, both alloys exhibit nitride precipitates both at the surface and in the alloy bulk, no surface chromia layer being present. Because Alloy 600 contains titanium (Table 1), the nitride precipitates contain titanium in addition to chromium. The greater volume fraction of chromium nitride in 310H compared to Alloy 600 (see Figs. 3 and 4) is in agreement with previous studies [16] and is attributed to the higher concentration of chromium and lower nickel in 310H. It is well-known that the solubility of nitrogen in austenitic alloys decreases as the nickel content increases [17], resulting in a slower permeation of nitrogen into the alloy. This effect may explain why large nitride particles primarily occur at alloy grain boundaries in alloy 600, while large nitride precipitates are not restricted to grain boundaries in the case of 310H.
The Alumina-Forming Alloys
In this study, the oxidation behaviour of the alumina-forming alloys is strikingly different from that of the chromia-formers. Thus, all three alumina-formers grow protective Al-rich scales in air, wet air and H2 + H2O + Ar environment, showing no sign of internal oxidation. Also, there is no competing oxide formation by chromium and iron, implying that there is a sufficient supply of aluminium to the growing scale by diffusion in the alloy. Moreover, in these three environments, the Al-rich layer morphology appears unaffected by the presence of alloy grain boundaries (see Figs. 5 & 6). The H2 + N2 environment apparently presents a much greater challenge to the alumina formers as evidenced by the extensive formation of aluminium nitride (AlN) in alloy APMT (see Fig. 1). The greater tendency for nitridation in H2 + N2 environment is attributed to be due to the low availability of oxygen, the oxygen needed to grow the protective Al-rich layer being supplied by traces of water vapour (about 10 ppm by volume) in the gas. Even though the formation of alumina remains spontaneous in this gas, the low concentration of water means that imperfections in the Al-rich scale, such as cracks, are difficult to heal, allowing N2 to penetrate to the metal.
Considering the oxide agglomerations which correspond to underlying RE inclusions, a comparison of the plan view images in Figs. 5 and 6 shows that APMT features a population of very large RE-rich inclusions in the alloy which is lacking in EF101. Working at 900 °C, Mortazavi et al. [31] showed that nitridation of alumina-forming alloys in N2 + H2 environment is associated with imperfections in the alumina layer, the layer itself being practically impermeable to nitrogen. Also, it was reported that FeCrAl(RE) alloys tend to develop such scale imperfections at RE-rich inclusions in the alloy. Moreover, it has been shown that the tendency to act as points-of-entry for nitrogen increases steeply with inclusion size, RE inclusions smaller than a critical size not causing nitridation [31]. In accordance to [31] and [32], it is argued that the relative vulnerability of alloy APMT towards nitridation in comparison to EF101 and EF100, as evidenced in this study, can be attributed to the occurrence of a small number of large RE-inclusions in the former alloy. These large inclusions disturb the formation of the alumina layer, causing cracks that do not easily heal in the very dry N2 + H2 exposure environment. The differences between the size and distribution of RE-rich inclusions are related to alloy production, APMT being produced by powder processing, while EF101 and EF100 are produced by conventional methods.
The two Cr-lean alumina formers in this study are very similar except that EF101 is alloyed with silicon, whereas EF100 is not (c.f. Table 1). It was recently reported [21] that silicon-containing FeCrAls resist corrosion in wet air at 600 °C better than several other alumina-forming alloys, the superior properties being attributed to the silicon content. In the present investigation, the two alloys were indistinguishable regarding both microstructure and oxidation behaviour, implying that this level of silicon alloying has little influence on the corrosion properties of FeCrAl´s at 800 °C.
The thinnest Al-rich oxide scales formed in dry air. Exposure in wet air resulted in thicker Al-rich oxide scales compared to dry air and exposure to the low pO2-environment, 20% H2 + 20% H2O, produced an even thicker oxide scale c.f. the cross section images in Figs. 5 and 6. This is in line with previous investigations of alumina scale formation on a FeCrAl alloy at 900 °C [33] [34]. The effect was attributed to a stabilization of metastable aluminas in the scale by water, resulting in delayed transformation to the slower-growing α-alumina. The higher oxidation rate when water is present has been observed before, and a proposed mechanism in this environment is the possible inward diffusion of hydroxide ions along alumina grain boundaries [32].
Conclusions
-
The lean chromium FeCrAl alloys EF101 and EF100 withstand oxidation and nitridation best among the alloys investigated, forming Al-rich oxide scales which resisted water vapour at both low and high pO2 as well as coping with the H2 + N2 environment without suffering nitridation.
-
The silicon alloying of EF101 had no apparent effect, EF101 and EF100 showing essentially the same corrosion behaviour.
-
While the powder metallurgical FeCrAl alloy APMT formed thin and protective alumina scales in dry and wet air and in H2 + H2O environment, it suffered severe nitridation in H2 + N2 environment. The poor resistance to nitridation is attributed to the presence of a small number of relatively large RE-rich inclusions in the alloy, giving rise to defects in the alumina scale. The low concentration of water vapour makes it difficult for such defects to heal, allowing them to act as points-of-entry for nitrogen into the metal.
-
The two chromia-forming austenites studied were both nitrided in H2 + N2 environment, 310H suffering much more extensive nitridation than alloy 600. The better resistance towards nitridation of the latter is attributed to its higher nickel content and lower chromium content.
-
In H2 + H2O environment, alloy 310H suffers partial breakaway oxidation while alloy 600 forms a thin and protective chromia film. The vulnerability of 310H is attributed to hydrogen which dissolves into the alloy and interacts with chromium at alloy grain boundaries, retarding the supply of chromium to the growing scale. The superior behaviour of alloy 600 is attributed to the low pO2 which disallows Ni2+ from entering the scale, causing less oxygen to dissolve into the alloy.
-
Alloy 600 and 310H form relatively thick oxide layers in dry air and wet air but do not suffer breakaway oxidation.
-
In wet air, the two alloys form CrO2(OH)2(g), the rate of chromium volatilization decreasing with time. The rate of chromium volatilization from the alumina formers was < 0.1% of the chromia-formers.
-
The faster corrosion of alloy 600 in wet air compared to dry air is attributed chromium volatilization which makes the formation of a chromia healing layer at the scale/alloy interface more difficult.
References
Asteman, H., Water Vapour induced active oxidation of stainless steel. (2002).
Sand, T., et al., Oxidation of Metals 1–21. (2019).
H. Asteman, et al., Oxidation of Metals 52, 1999 (95).
H. Falk-Windisch, J. E. Svensson, and J. Froitzheim, Journal of Power Sources 287, 2015 (25).
M. Stanislowski, E. Wessel, K. Hilpert, T. Markus, and L. Singheiser, Journal of The Electrochemical Society 154, 2007 (A295).
J. Pettersson, et al., Oxidation of metals 76, 2011 (93).
Y. P. Jacob, et al., Corrosion Science 44, 2002 (2027).
J. Zurek, et al., Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing 477, 2008 (259).
H. Hooshyar, et al., Oxidation of Metals 85, 2016 (321).
E. Essuman, et al., Oxidation of Metals 69, 2008 (143).
M. Michalik, et al., Materials at High Temperatures 22, 2005 (213).
S. Henry, et al., Materials at High Temperatures 17, 2000 (231).
G. Hultquist, B. Tveten, and E. Hörnlund, Oxidation of Metals 54, 2000 (1).
Gleeson, B., 1.09 - Thermodynamics and Theory of External and Internal Oxidation of Alloys, in Shreir's Corrosion, B. Cottis, et al., Editors. (Elsevier: Oxford. p. 180–194, 2010).
D. J. Young, et al., Scripta Materialia 77, 2014 (29).
Sand, T., et al., Corrosion Science, 110050. (2021).
K. Tjokro and D. J. Young, Oxidation of Metals 44, 1995 (453).
Reddy, M.J., J.E. Svensson, and J. Froitzheim, Corrosion Science, 190. (2021).
J. Ejenstam, B. Jonsson, and P. Szakalos, Oxidation of Metals 88, 2017 (361).
J. Ejenstam, et al., Journal of Nuclear Materials 457, 2015 (291).
J. Eklund, et al., Corrosion Science 144, 2018 (266).
T. Sand, et al., Oxidation of Metals 95, 2021 (221).
B. Jonsson, et al., OXIDATION OF METALS 79, 2013 (29).
C. Wagner, Zeitschrift für physikalische Chemie 21, 1933 (25).
D. Jullian, et al., Journal of Alloys and Compounds 732, 2018 (646).
F. Gesmundo and F. Viani, Oxidation of metals 25, 1986 (269).
H. Asteman, J.-E. Svensson, and L.-G. Johansson, Corrosion Science 44, 2002 (2635).
P. Alnegren, et al., Journal of Power Sources 392, 2018 (129).
C. Goebel, et al., International Journal of Hydrogen Energy 43, 2018 (14665).
K. O. Gunduz, et al., Corrosion Science 179, 2021 (109112).
A. N. Mortazavi, et al., Acta Materialia 201, 2020 (131).
N. Mortazavi, et al., Nature Materials 17, 2018 (610).
H. Götlind, et al., Oxidation of Metals 67, 2007 (251).
F. Liu, et al., Materials at High Temperatures 22, 2005 (521).
Funding
Open access funding provided by Chalmers University of Technology. Funding was provided by the Swedish Energy Agency (Energimyndigheten).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sand, T., Liske, J., Helander, T. et al. A Comparison of the Oxidation and Nitridation Properties of Selected Chromia- and Alumina-Forming Alloys at 800 °C. Oxid Met 98, 163–178 (2022). https://doi.org/10.1007/s11085-022-10117-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-022-10117-3