
Abstract
5-ethoxymethyl-8-hydroxyquinoline was synthesized and characterized using spectroscopic methods (1H, 13C NMR, IR). The crystal structure determined at room temperature (295 K) by means of X-ray powder diffraction is orthorhombic, with space group Pbca and eight molecules per unit cell (Z = 8, Z′ = 1). The lattice parameters are: a = 7.9551(12) Å, b = 17.981(3) Å, c = 15.125(2) Å and V = 2163.5(6) Å3. Geometric parameters and properties depending on the charge distribution around the different types of donors and acceptors bonds within the molecule are calculated by density functional theory (DFT/B3LYP) and Hartree–Fock methods. Atomic charges and dipole moment value permit qualitative predictions about high reactivity of this molecule. The 5-ethoxymethyl-8-hydroxyquinoline adopts a non-planar structure in the solid state and the molecule is stabilized by contact system as π–π stacking interactions, weak intra and intermolecular H-Bonding O–H···N and C–H···O types, this latter involving the rings of both adjacent molecules in plans with a gap from 0.638 Å.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
8-hydroxyquinoline derivatives are very attractive bioactive molecules especially in the field of drugs and pharmaceuticals (Di Varia et al. 2004; Salkovic-Petrisic et al. 2015; Cacciatore et al. 2013) and as chelating agents for a large number of metal ions (Pollak et al. 2013; La Deda et al. 2004). Derivatives such as zinc(II) bis (8-hydroxyquinoline) complex have been investigated as electroluminescent (EL) materials over the past decades (Yang et al. 2013; Yuan et al. 2012; Sokołowski et al. 2012; Yuan et al. 2013; Zawadzka et al. 2013; Yuan et al. 2015; Wang et al. 2013; D’Souza et al. 2008) including electro-transporting and emitting materials, which are applied in molecular-based OLEDs for their thermal stability, high fluorescence and excellent electron-transporting mobility (Shan et al. 2014; Huo et al. 2010; Zawadzka et al. 2002; Zawadzka et al. 2016; Zawadzka et al. 2013; Zhang et al. 2015; Yuan et al. 2012).
In this way, many studies have been published to understand the reactivity of this derivatives in different solvents (Himmi et al. 2008) and to determine the most stable conformation in acidic and alkaline aqueous solutions (Saylam et al. 2014). But up today only few structural data on 8-hydroxyquinoline derivatives have been published, showing, for example dependence of the molecule structure on the synthesis procedures of 8-hydroxyquinoline derivatives (Pearson et al. 1967; Gershon et al. 1969; Collis et al. 2003). In this study, we focus on a new one of 8-hydroxyquinoline derivatives named 5-(ethoxymethyl)quinolin-8-ol (5-EMQO) (Scheme 1). This molecule was synthesized and analyzed by different spectroscopic methods. The X-ray analysis, DFT and HF calculations permit us to study the geometric and charge transfer properties in the ground state of the compound.

Synthesis of 5-ethoxymethyl-8-hydroxyquinoline
2 Experimental section
2.1 Synthesis method
5-EMQ was synthesized according to the method described by Mishra et al. (2005). The obtained product (Scheme 1) was characterized by IR and 1H, 13C NMR methods, the spectral data are shown in Table 1.
Melting points were determined on an automatic electrothermal IA 9200 digital melting point apparatus in capillary Mp: 78–80 °C. 1H NMR spectra were recorded on a Bruker 300 WB spectrometer at 300 MHz for solutions in Me2SO-d6. Chemical shifts are given as δ values with reference to tetramethylsilane (TMS) as internal standard. Infrared spectra were recorded from 400 to 4000 cm−1 on a Bruker IFS 66v Fourier transform spectrometer using KBr pellets. Mass spectrum was recorded on THERMO Electron DSQ II.
2.2 X-ray diffraction
High quality powder X-ray diffraction measurements were performed with an Inel CPS120 powder diffractometer (Debye–Scherrer geometry, transmission mode). The sample was introduced into 0.5 mm diameter Lindemann glass capillary. The latter was rotated around its axis in order to minimize preferential orientations of the crystallites and to obtain reflections with exploitable intensities. The time of acquisition was set to 8 h approximately for a good signal-to-nose ratio. Monochromatic Cu-Kα1 radiation (λ = 1.54056 Å) was selected with asymmetric focusing incident-beam curved quartz monochromator, the generator power was set to 40 kV and 25 mA. The diffracted lines were collected on a 4096 channel detector over an arc of 120° and centered on the sample. External calibration using the Na2Ca2Al2F14 (NAC) cubic phase mixed with silver behenate was performed using cubic spline fittings.
2.3 Theoretical calculation
Geometrical data of the molecule in ground state was optimized by density functional theory (DFT) and Hartree–Fock (HF) methods with 6-311G(d,p) basis set, using Gaussian 03 program package at room temperature (Frisch et al. 2004). These methods are more used, actually, to study chemical and biochemical phenomena within a large variety of molecules (Prasad et al. 2013; Li et al. 2014). Because they can reach exactitude similar to other methods as ab initio in less time and less expensive from the computational point of view, on the other hand, they offers similar accuracy for middle sized and large systems.
3 Results and discussion
3.1 Crystal structure analysis
The chemical structure of the compound deduced from its IR, 1H, 13C and NMR spectra was confirmed by X-ray analysis. Thermal analysis was performed with Perkin-Elmer DSC-7 apparatus using heating rates of 10 K min−1 revealed no solid–solid phase transition between room temperature and the melting point. The melting point temperature (Mp = 351.8 K) was in good agreement with that measured by using digital melting point apparatus (Electrothermal IA 9200) and enthalpy of melting was equal to ∆H = 21.136 kJ/mol.
The crystal structure was determined with materials studio modeling 5.5 (http://accelrys.com/products/materials-studio). First, the pattern was indexed by means of the peak picking option of the software package and using the X-cell algorithm (Neumann 2003). Then, a Pawley profile-fitting procedure (Pawley 1981) was applied including refined cell parameters, experimental profile fitting with pseudo-Voigt function, zero shift and asymmetry Finger-Cox-Jephcoat function. Distances, angles and torsions in the molecule were obtained via energy-minimization calculations using the Dreiding force field (Mayo et al. 1990). Then, a Monte-Carlo approach was carried out in the direct space to solve the structure (Serbezov et al. 2013) moving the molecule as a rigid-body and allowing the change of the chain torsion angles. Rwp agreement factor of 4.78 % is reached after Rietveld refinement (Rietveld 1969). Describing a good agreement between the measured and calculated spectra for 5-EMQ molecule at 295 K (Fig. 1).

Experimental (red) and calculated (blue) diffraction patterns along with the difference profile (black) and calculated Bragg reflections (green) of 5-ethoxymethyl-8-hydroxyquinoline at 295 K. The insert is enlarged 15×
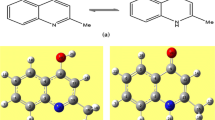
The molecular structure is represented in Fig. 2a. Relevant crystallographic data are summarized in Table 2. The parameters in the CIF form are available as Electronic Supplementary Information from the Cambridge Crystallographic Database Centre (1029560).

a Crystal molecular conformation of 5-ethoxymethyl-8-hydroxyquinoline, b geometric display showing the non planar conformation of the molecule
The molecule exibited non-planar structure, the dihedral angle calculated between the methylquinolin-8-ol group and other part of the molecule is 77.08° (Fig. 2b).
The stabilized crystal packing is mainly governed by hydrogen bonds (Fig. 3) and π–π stacking interactions (Fig. 4). The intramolecular hydrogen bonds observed in the structure involves the OH functional group, this latter presents strong interactions in many solvents, modify physical properties at room temperature (Kadiri et al. 1994; Cazeau-Dubroca 1991) and enhancing the biological activity of the molecule. Weak intra and intermolecular hydrogen bonds O–H···N and C–H···O types are detected in the structure involving adjacent molecules in different plans, with a gap from 0.638 Å (Fig. 3b). Selected distances (Å) and angles (°) describing the hydrogen bonds are reported in Table 3.

Hydrogen Bonding contacts (dashed lines) in crystal lattice of molecule: a along the (bc) plan, b geometric display of the hydrogen bonds

View of crystal π–π stacking of molecule: a along the b axis, b π–π interactions in the crystal packing of molecule
In addition to the hydrogen bond, the crystal packing structure of 5-EMQO shows π–π stacking interactions between 8-quinolinol rings on adjacent molecules (Fig. 4a). Those contacts may stabilize the structure, with ring centroid to ring centroid distance of 3.473 Å (Fig. 4b). This value is similar to the values reported for other π–π interactions (Shi et al. 2007; Zawadzka et al. 2012).
3.2 Theoretical calculation
Figure 5a shows an optimized calculated structure of the compound. In Table 4 we report the most optimized structural parameters (bond distances, angles and torsions) obtained from crystal structure data (1), optimized by DFT (B3LYP) (2) and by HF (3), in accordance with the atom numbering scheme given in Fig. 2a. The global minimum energy obtained by DFT and HF structure optimization is calculated as −18244.67 and −18131.48 eV respectively. The dipole moment is equal to 3.0245 Debye.

a Optimized structure (B3LYP/6-311) of molecule; b Molecular orbital surfaces and energy levels for the HOMO, LUMO
The C–C bonds lengths optimized with both methods are in a fairly good agreement with the corresponding bond lengths obtained from X-ray analysis, however slight differences on C–C bond lengths may be noticed between DFT(B3LYP) and the HF calculations. The biggest gap between calculation and experience is observed for C(3)–C(4), C(7)–C(8) and C(9)–C(10) bonds at HF method, values are 0.045, 0.056 and 0.050 Å respectively. For the rest of bond lengths type, the highest difference between crystal structure and calculation is observed for the N(1)–C(2) bond with a value of 0.04 and 0.06 Å for DFT and HF methods respectively. Generally the HF method underestimate some bond lengths by Inclusion of the electron correlation which makes this bond shorter because of the increased resonance in the correlated wave function (Arici et al. 2005; Lakshmi et al. 2011; Zawadzka et al. 2015; Kosar and Albayrak 2011). As a result, the DFT method for bond lengths is much closer to experimental data.
The calculated bond angles values obtained with DFT and HF are very similar, but compared to crystal data some discrepancies appears for C(2)–N(1)–C(6) and C(9)C(10)–O(11) with a difference of 5.28° for DFT method and 5.34° for HF method, respectively. Concerning torsion angles, we presented in Table 3 only those with notable discrepancies between X-ray data and the two calculations (DFT and HF) methods. The Azine ring appears a little distorted with smaller C(6)–N(1)–C(2)–C(3), C(2)–N(1)–C(6)–C(5), N(1)–C(2)–C(3)–C(4) torsion angles and bigger C(2)–C(3)–C(4)–C(5), C(3)–C(4)–C(5)–C(6), C(4)–C(5)–C(6)–N(1) torsion angles (Table 4), comparing values from DFT and HF methods to the crystal data. The highest difference corresponds to the C(7)–C(12)–O(13)–C(14) torsion angle with the value of 77.1(13)° in the crystal structure and −178.6° and −178.4° respectively for DFT and HF calculations. The discrepancies between calculated and experimental structural parameters are related to the fact, that calculations refer to the isolated molecule, contrarily experimental results which correspond to interacting molecules in the crystal lattice where intra and intermolecular interactions with the neighboring molecules are present (Kavitha et al. 2010; Ghiasi et al. 2014; Pournamdari et al. 2012).
Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are been computed using B3LYP level 6-311(d,p) of DFT. Figure 5b shows the patterns of the HOMO and LUMO of 5-EMQO. The HOMO–LUMO gap is 4.463 eV. The HOMO energy shows the ability of giving an electron, whereas, the LUMO energy represents the ability of gaining an electron (Pournamdari et al. 2012). In many recent works, the energy gap between HOMO and LUMO has been used to prove the bioactivity from intra molecular charge transfer and to explain the chemical reactivity of the molecule (Fukui 1982; Gunasekaran et al. 2008).
The charge distribution on different atoms (C, N and O) for molecule obtained by using X-ray analysis data B3LYP methods and HF methodes is plotted in Fig. 6. Results obtained by these two methods are similar. This distribution depending on the electronegativity of atoms; the most negative are those present at nitrogen and oxygen atoms. The values obtained from DFT calculations are in fairly agreement with crystal structure. The most negative values are those that are present at nitrogen and oxygen: N1, O11 and O13. It has to be noticed that the carbon atom attached to oxygen and nitrogen atoms: C2, C6, C10, C12 and C14 in both X-ray and B3LYP are electron deficient atoms possessing positive electronic charge due to the oxygen and nitrogen electronegativity . Remaining carbon atoms possess negative electronic charge. Except some discrepancies about C4 and C5 which possess a different electronic charge in DFT calculation, this deviation is probably due to the fact that the intermolecular hydrogen bonding interaction C3–H3–O13 in the crystalline state are dominant influencing the charge distribution in C4 and C5. Furthermore, the DFT calculation refers to the isolated molecule where the intermolecular interactions with the neighboring molecules are absent. This distribution within the molecule and the high dipole moment explain the notable polarization given by the small HOMO-LUMO energy calculated. The molecule will exhibit a significant degree of intramolecular charge transfer from the electron donor section to the electron acceptor section.

The atomic charges for 5-ethoxymethyl-8-hydroxyquinoline
4 Conclusion
In this work, we have synthesized 5-ethoxymethyl-8-hydroxyquinoline and determined its crystal structure by X-ray powder diffraction pattern. The crystal system is orthorhombic with Pbca space group and eight molecules per unit cell. The lattice parameters are: a = 7.9551(12) Å, b = 17.981(3) Å, c = 15.125(2) Å. The crystal structure is mainly governed by intra and intermolecular O–H···N and C–H···O hydrogen-bonding type and π–π stacking interactions. Density functional theory DFT(B3LYP) and Hartree–Fock (HF) calculations were compared with the measured data, some discrepancies appears, the big one occurs in the C(7)–C(12)–O(13)–C(14) torsion angle, showing the orientation of the O–C2H5 group in the opposite direction to that given by X-ray analysis. Furthermore, HOMO–LUMO patterns and Mulliken atomic charges have been analyzed by DFT to explain the possibility of charge transfer within the molecule. The small obtained gap and atomic charges distribution reveled possibility of the charge transfer within the molecule and a high reactivity of 5-ethoxymethyl-8-hydroxyquinoline, who permits to this type of molecule to be used as bioactive molecule.
References
Arici, K., Yurdakul, M., Yurdakul, S.: HF and DFT studies of the structure and vibrational spectra of 8-hydroxyquinoline and its mercury(II) halide complexes. Spectrochimica Acta Part A 61, 37–43 (2005)
Cacciatore, I., Fornasari, E., Baldassarre, L., Cornacchia, C., Fulle, S., Di Filippo, E.S., Pietrangelo, T., Pinnen, F.: A potent (R)-alpha-bis-lipoyl derivative containing 8-hydroxyquinoline scaffold: synthesis and biological evaluation of its neuroprotective capabilities in SH-SY5Y human neuroblastoma cells. Pharmaceuticals 6(1), 54–69 (2013)
Cazeau-Dubroca, C.: Wist of “TICT” molecules in the ground state by hydrogen bonding. Trends Phys. Chem. 2, 233–258 (1991)
Collis, G.E., Burrell, A.K., John, K.D., Plieger, P.G.: 7-Bromoquinolin-8-ol. Acta Cryst. C 59, o443–o444 (2003)
Di Varia, M., Bazzicalupi, C., Orioli, P., Messori, L., Bruni, B., Zatta, P.: Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg. Chem. 43(13), 3795–3797 (2004)
D’Souza, F., Maligaspe, E., Zandler, M.E., Subbaiyan, N.K., Ohkubo, K., Fukuzumi, S.: Metal quinolinolate-fullerene(s) donor-acceptor complexes: evidence for organic LED molecules acting as electron donors in photoinduced electron-transfer reactions. J. Am. Chem. Soc. 130(50), 16959–16967 (2008)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr, J.A., Vreven, T., Kudin, K.N., Burant, J.C., et al.: Gaussian 03 (Revision D.02). Gaussian, Inc, Wallingford (2004)
Fukui, K.: Role of frontier orbitals in chemical reactions. Science 218, 747–754 (1982)
Gershon, H., McNeil, M.W., Grefig, A.T.: Reinvestigation of the action of N-halosuccinimides on bis(8-quinolinolato)copper(II). J. Org. Chem. 34, 3268–3270 (1969)
Ghiasi, R., Bharifar, H., Hosseinzade, S., Ail Zarinfard, M., Hakimyoun, A.H.: The stability and properties of Mn+@ C26-2NBNNN (M = alkaline and earth alkaline metals; n = 0, 3) complexes for synthesis application. J. Appl. Chem. Res. 8, 29–36 (2014)
Gunasekaran, S., Balaji, R.A., Kumeresan, S., Anand, G., Srinivasan, S.: Experimental and theoretical investigations of spectroscopic properties of N-acetyl-5-methoxytryptamine. Can. J. Anal. Sci. Spectrosc. 53, 149–160 (2008)
Himmi, B., Messnaoui, B., Kitane, S., Eddaif, A., Alaoui, A., Bouklouz, A., Soufiaoui, M.: Study of Zn (II) extraction by 5-azidomethyl-8-hydroxyquinoline: experiment and modelling. Hydrometallurgy 93, 39–44 (2008)
Huo, Y.P., Zhu, S.Z., Hu, S.: Synthesis and luminescent properties of Zn complex based on 8-hydroxyquinoline group containing 3,5-bis(trifluoromethyl) benzene unit with unique crystal structure. Tetrahedron 66(45), 8635–8640 (2010)
Kadiri, A., Kabouchi, B., Benali, B., Cazeau-Dubroca, C., Nouchi, G.: Study of the fluorescence of 9,9′-bianthryl in polyvinylalcohol and in polyvinylchloride at 300 and 80 K Spectrochim. Acta Part A: Mol. Spectrosc. 50, 1–4 (1994)
Kavitha, E., Sundaraganesan, N., Sebastian, S., Kurt, M.: Molecular structure, Anharmonic vibrational frequencies and NBO analysis of naphthalene acetic acid by density functional theory calculations. Spectrochim. Acta Part A 77, 612–619 (2010)
Kosar, B., Albayrak, C.: Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta A 78, 160–167 (2011)
La Deda, M., Grisolia, A., Aiello, I., Crispini, A., Ghedini, M., Belviso, S., Amati, M., Lelj, F.: Investigations on the electronic effects of the peripheral 4′-group on 5-(4′-substituted)phenylazo-8-hydroxyquinoline ligands: zinc and aluminium complexes. Dalton Trans. 16, 2424–2431 (2004)
Lakshmi, A., Balachandran, V., Janaki, A.: Comparative vibrational spectroscopic studies, HOMO–LUMO and NBO analysis of 5,7-dibromo-8-hydroxyquinoline and 5,7-dichloro-8-hydroxyquinoline based on density functional theory. J. Mol. Struct. 1004, 51–66 (2011)
Li, Y., Liu, Y.Y., Chen, X.J., Xiong, X.H., Li, F.S.: Synthesis, spectroscopic characterization, X-Ray structure, and DFT calculations of some new 1,4-Dihydro-2,6-Dimethyl-3,5—pyridinedicarboxamides. PLoS ONE 9(3), e91361 (2014). doi:10.1371/journal.pone.0091361
Materials Studio Modeling 5.5. http://accelrys.com/products/materials-studio
Mayo, S.L., Olafson, B.D., Goddard III, W.A.: Dreiding: a generic force field for molecular. J. Phys. Chem. 94, 8897–8909 (1990)
Mishra, A., Periasamy, N., Patankar, M.P., Narasimhan, K.L.: Synthesis and characterisation of soluble aluminium complex dyes based on 5-substituted-8-hydroxyquinoline derivatives for OLED applications. Dyes Pigm. 66(2), 89–97 (2005)
Neumann, M.A.: X-Cell: a novel indexing algorithm for routine tasks and difficult cases. J. Appl. Cryst. 36, 356–365 (2003)
Pawley, G.S.: Unit-cell refinement from powder diffraction scans. J. Appl. Cryst. 14, 357–361 (1981)
Pearson, D.E., Wysong, R.D., Breder, C.V.: Ortho bromination of phenols. J. Org. Chem. 32, 2358–2360 (1967)
Pollak, Y., Mechlovich, D., Amit, T., Bar-Am, O., Manov, I., Mandel, S.A., Weinreb, O., Meyron-Holtz, E.G., Iancu, T.C., Youdim, M.B.H.: Effects of novel neuroprotective and neurorestorative multifunctional drugs on iron chelation and glucose metabolism. Neural Transm. 120(1), 37–48 (2013)
Pournamdari, E., Monajjemi, M., Zare, K.: Ab initio study of thermodynamic properties, IR spectra and electrical properties of Cu4O3H2 nanosemiconductor. Life Sci. J. 9(4), 1729–1738 (2012)
Prasad, M.V.S., Sri, N.U., Veeraiah, A., Veeraiah, V., Chaitanya, K.: Molecular structure, vibrational spectroscopic (FT-IR, FT-Raman), UV–Vis spectra, first order hyperpo-larizability, NBO analysis, HOMO and LUMO analysis, thermodynamic properties of 2, 6-dichloropyrazine by ab inito HF and density functional method. J. At. Mol. Sci. 4(1), 1–17 (2013)
Rietveld, H.M.: A profile refinement method for nuclear and magnetic structures. J. Acta. Crystallogr. 2(2), 65–71 (1969)
Salkovic-Petrisic, M., Knezovic, A., Osmanovic-Barilar, J., Smailovic, U., Trkulja, V., Riederer, P., Amit, T., Mandel, S., Youdim, M.B.H.: Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer’s disease. Life Sci. 136, 108–119 (2015)
Saylam, A., Seferoğlu, Z., Ertan, N.: Azo-8-hydroxyquinoline dyes: the synthesis, characterizations and determination of tautomeric properties of some new phenyl- and heteroarylazo-8-hydroxyquinolines. J. Mol. Liq. 195, 267–276 (2014)
Serbezov, V., Sotirov, S., Khalil, B., Zawadzka, A., Sahraoui, B.: Investigation of superfast deposition of metal oxide and diamond-like carbon thin films by nanosecond ytterbium (Yb+) Fiber laser. Opt. Mater. 36, 53–59 (2013)
Shan, W., Shan, X., Li, X., Liu, J., Zhang, L., Yuan, G.: Self-assembly and luminescent properties of one novel tetranuclear Cd(II) complex based on 8-hydroxyquinolinate ligand. Inorg. Chem. Commun. 48, 131–135 (2014)
Shi, W.J., Hou, L., Li, D., Yin, Y.G.: Supramolecular assembly driven by hydrogen-bonding and π–π stacking interactions based on copper (II)-terpyridyl complexes Inorg. Chim. Acta 360(2), 588–598 (2007)
Sokołowski, K., Justyniak, I., Siliwinski, W., Sołtys, K., Tulewicz, A., Kornowicz, A., Moszynski, R., Lipkowski, J., Lewinski, J.: Towards a new family of photoluminescent organozinc 8-hydroxyquinolinates with a high propensity to form noncovalent porous materials. Chem. A Eur. J. 18, 5637–5645 (2012)
Wang, R., Cao, Y., Jia, D., Liu, L., Li, F.: New approach to synthesize 8-hydroxyquinoline-based complexes with Zn2+ and their luminescent properties. Opt. Mater. 36, 232–237 (2013)
Yang, F., Kubota, F., Kamiya, N., Goto, M.: Extraction of rare-earth ions with an 8-hydroxyquinoline derivative in an ionic liquid. Solvent Extr. Res. Dev. 20, 123–129 (2013)
Yuan, G.Z., Huo, Y.P., Nie, X.L., Fang, X.M., Zhu, S.Z.: Structure and photophysical properties of a dimeric Zn(II) complex based on 8-hydroxyquinoline group containing 2,6-dichlorobenzene unit. Tetrahedron 68, 8018–8023 (2012a)
Yuan, G.Z., Huo, Y.P., Rong, L.L., Nie, X.L., Fang, X.M.: Photoluminescences and 3D supramolecular structure with unique dimeric Zn (II) units featuring 2-substituted 8-hydroxyquinoline. Inorg. Chem. Commun. 23, 90–94 (2012b)
Yuan, G., Rong, L., Qiao, X., Ma, L., Wei, X.: Anion-controlled structures and luminescent properties of three Cd(II)complexes assembled by a2-substituted 8-hydroxyquinoline ligand. Cryst. Eng. Comm. 15, 7307–7314 (2013)
Yuan, G., Shan, W., Chen, J., Tian, Y., Wang, H.: Synthesis, structure and photophysical properties of a binuclear Zn(II) complex based on 8-hydroxyquinoline ligand with naphthyl unit. J. Lumin. 160, 16–21 (2015)
Zawadzka, A., Dygdała, R.S., Raczyński, A., Zaremba, J., Kobus, J.: Three-photon resonances due to autoionizing states in calcium. J. Phys. B: At. Mol. Phys. 35, 1801–1817 (2002)
Zawadzka, A., Płóciennik, P., Czarnecka, I., Sztupecka, J., Łukasiak, Z.: The effects of annealing process influence on optical properties and the molecular orientation of selected organometallic compounds thin films. Opt. Mater. 34, 1686–1691 (2012)
Zawadzka, A., Płóciennik, P., Strzelecki, J., Łukasiak, Z., Sahraoui, B.: Photophysical properties of Alq3 thin films. Opt. Mater. 36(1), 91–97 (2013a)
Zawadzka, A., Płóciennik, P., Strzelecki, J., Pranaitis, M., Dabos-Seignon, S., Sahraoui, B.: Structural and nonlinear optical properties of as-grown and annealed metallophthalocyanine thin films. Thin Solid Films 545, 429–437 (2013b)
Zawadzka, A., Karakas, A., Płóciennik, P., Szatkowski, J., Łukasiak, Z., Kapceoglu, A., Ceylan, Y., Sahraoui, B.: Optical and structural characterization of thin films containing metallophthalocyanine chlorides. Dyes Pigm. 112, 116–126 (2015)
Zawadzka, A., Płóciennik, P., El Kouari, Y., Bougharraf, H., Sahraoui, B.: Linear and nonlinear optical properties of ZnO thin films deposited by pulsed laser deposition. J. Lumin. 169, 483–491 (2016)
Zhang, L., Sun, L., Li, X., Tian, Y., Yuan, G.: Five 8-hydroxyquinolinate-based coordination polymers with tunable structures and photoluminescent properties for sensing nitroaromatics. Dalton Trans. 44, 401–410 (2015)
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is part of the Topical Collection on Advanced Materials for photonics and electronics.
Guest Edited by Bouchta Sahraoui, Yahia Boughaleb, Kariem Arof, Anna Zawadzka.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bougharraf, H., Benallal, R., El faydy, M. et al. Synthesis, spectroscopic characterization, X-Ray analysis, and DFT-HF calculations of 5-ethoxymethyl-8-hydroxyquinoline. Opt Quant Electron 48, 141 (2016). https://doi.org/10.1007/s11082-016-0393-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-016-0393-5