
Abstract
Central nervous system (CNS)-related conditions are currently the leading cause of disability worldwide, posing a significant burden to health systems, individuals and their families. Although the molecular mechanisms implicated in these disorders may be varied, neurological conditions have been increasingly associated with inflammation and/or impaired oxidative response leading to further neural cell damages. Therefore, therapeutic approaches targeting these defective molecular mechanisms have been vastly explored. Hydrogen sulphide (H2S) has emerged as a modulator of both inflammation and oxidative stress with a neuroprotective role, therefore, has gained interest in the treatment of neurological disorders. H2S, produced by endogenous sources, is maintained at low levels in the CNS. However, defects in the biosynthetic and catabolic routes for H2S metabolism have been identified in CNS-related disorders. Approaches to restore H2S availability using H2S-donating compounds have been recently explored in many models of neurological conditions. Nonetheless, we still need to elucidate the potential for these compounds not only to ameliorate defective biological routes, but also to better comprehend the implications on H2S delivery, dosage regimes and feasibility to successfully target CNS tissues. Here, we highlight the molecular mechanisms of H2S-dependent restoration of neurological functions in different models of CNS disease whilst summarising current administration approaches for these H2S-based compounds. We also address existing barriers in H2S donor delivery by showcasing current advances in mediating these constrains through novel biomaterial-based carriers for H2S donors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurological disorders may result from insult to the brain, spinal cord or peripheral nerves as well as congenital defects, degeneration or structural defects. These impairments are increasingly recognised as major burdens to the affected patient, families and health systems as they are listed as a major cause of disability and second leading cause of death [1]. Neurological disorders, especially those with a degenerative nature, including Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD), are incurable, debilitating conditions resulting in progressive damage of nerve cells. In 2006, the Global Burden of Disease (WHO) estimated that combined, the prevalence of PD, AD and other dementias was estimated to reach 5.25 (per 1000 individuals) in 2015, however, these figures are projected to increase to 6.47 (per 1000) by 2030 [2] with all neurological-related cases predicted to triple in the coming decades to 152 million worldwide [1]. Therefore, improving therapeutic management options is of critical importance.
Hydrogen sulphide (H2S) is an endogenous gas which, along with nitric oxide and carbon monoxide, belongs to a family of transmitters known as gaseous transmitters [3]. H2S has increasingly gained recognition as a protective agent. Studies have shown that H2S is involved in a variety of physiological processes in the body, able to exert cytoprotective effects owing to its antioxidant properties and ability to modulate oxygen consumption [4]. However, H2S was once considered a toxic molecule. It has been demonstrated that H2S has a biphasic effect; low concentrations are beneficial, allowing enhanced mitochondrial respiration whereas higher concentrations inhibit the respiratory chain at level of mitochondrial complex IV, resulting in cytotoxicity [5].
In neurodegenerative conditions, the use of H2S-releasing compounds have demonstrated neuroprotective roles [6,7,8] when administered at physiological levels (approximately 20 to 300 µM) [9]. Although many molecular mechanisms have been described, our understanding of the potential for H2S-based compounds in the treatment or prevention of neurological disorders is not yet clear. Many of the challenges yet to be addressed include the rapid release of H2S and fast metabolism in vivo, the ability of H2S-related compounds to cross the blood brain barrier (BBB) and the optimisation of laboratory techniques to measure H2S levels in tissues or cells.
This literature review aims to highlight current evidence demonstrating the role of H2S in the central nervous system (CNS) whilst exploring the neuroprotective potential of H2S-donating compounds in different models of neurological disease, from in vitro to in vivo settings. Additionally, this review will explore challenges in H2S delivery to the nervous system and showcase potential approaches to mitigate these challenges.
H2S in the Central Nervous System
Synthesis of H2S
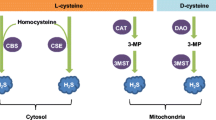
H2S is produced enzymatically by the action of three enzymes: cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate (3-MST) [10]. CBS catalyses a β-replacement reaction between homocysteine and cysteine, producing cystathionine and H2S [11]. While CSE employs a similar method, it can also catalyse a reaction that converts cystathionine into cysteine, which can then be used in further reactions to produce H2S [12]. Furthermore, 3-MST is a mitochondrial enzyme that produces H2S by transferring sulphur from 3-meracaptopyruvate (produced by cysteine aminotransferase) to sulphurous acid. This produces thiosulfate as one of the products, which is reduced into H2S [13].
In the CNS, CBS and 3-MST are the main enzymes involved in H2S production, with 3-MST being responsible for approximately 90% of the H2S produced in the brain [14]. While 3-MST is present mainly in neurons, CBS is present within astrocytes and microglia, suggesting that each enzyme may have a distinct role to promote H2S signalling [15]. Given that 3-MST is a located within the mitochondrial matrix [16], it highlights the potential role of mitochondrial H2S in regulating degenerative CNS processes whilst CBS has been found accountable for defective neurological functions such as neurogenesis, synaptogenesis and controlling BBB permeability [17]. Interestingly, CSE is hardly detected in brain tissue. Very low levels of CSE mRNA have been detected in the brain whilst CSE inhibitors have shown no significant suppression of H2S production [18], suggesting that CSE does not play a key role as a H2S producing enzyme in the brain (Fig. 1).
The importance of both CBS in generating H2S in brain cells (mainly astrocytes and microglia) suggest that their patterns of expression and/or activity may have an impact in the development of brain-related disorders. In neurodegenerative disorders such as PD and AD, hyperhomocysteinemia has been regarded as risk factor for the development of these conditions. Although accumulation of amino acid homocysteine leading to hyperhomocysteinemia may occur due to defects in different enzymes, evidence shows that CBS 844ins68 mutation and VNTR polymorphisms of the CBS gene are independent risk factors for AD development in subjects aged 75 years or more [19]. Moreover, recent evidence by Bjørke-Monsen, et al. 2022 showed that severe hyperhomocysteinemia in a patient with PD associated with reduced availability of CBS cofactor, pyridoxal 5-phosphate (PLP) [20]. These observations suggest that deficient CBS signalling, due to deficient availability of cofactor PLP and polymorphisms of CBS may be considered risk factors for the development of neurodegenerative diseases.

Synthesis of H2S in CNS-related cells. Cytoplasmatic cystathionine-β-synthase (CBS) expressed in astrocytes and microglia, and mitochondrial 3-mercaptopyruvate (3-MST) expressed in neurons, are mainly involved in the endogenous generation of H2S. CBS catalyse the reaction between homocysteine and cysteine, producing cystathionine and H2S whereas 3-MST produces H2S by transferring sulphur from 3-meracaptopyruvate (3-MP) (produced by cysteine aminotransferase, CAT) to sulphurous acid
There are also non-enzymatic routes for H2S synthesis. These routes are usually coupled to reducing reactions such as glycolysis, which produces glucose and reducing equivalents such as nicotinamide adenine dinucleotide. These reducing molecules can then participate in reducing numerous sulphur compounds into H2S [21, 22].
Catabolism of H2S
H2S is mainly metabolised through oxidation reactions in the mitochondria known as the “sulphur oxidation unit” pathway [23]. A major enzyme in this pathway is sulphide quinone oxidoreductase (SQR), which oxidizes H2S to persulfide. SQR is found in many organs such as the liver and colon. SQR protein and mRNA are expressed in mouse primary microglia and astrocytes but not in primary neurons [24]. The patterns of SQR expression shows lower levels of SQR mRNA and protein in the brain when compared to other tissues, suggesting that SQR may have limited contribution to the breakdown of H2S in the brain [22, 25].
Nevertheless, due to the remarkable importance of SQR in the metabolism of H2S in other tissues, researchers have investigated the presence of homolog forms of SQR in neuronal tissue. Ackermann et al. demonstrated the presence of a SQR homolog known as SQRDL (sulphur quinone oxidoreductase-like protein), which metabolises sulphur in rat brain [26]. This study also revealed that the SQRDL mRNA increased in rats with age, which may result in excess H2S degradation, contributing to age-related neurodegenerative disorders [26]. More research is required to better understand the existence of other relevant SQR variants that may play a significant role in the degradation of H2S.
Other routes of H2S metabolism suggest the neuroglobin may have a role in H2S oxidation in the brain. Neuroglobin is a member of the haemoprotein family which uses its Fe2+ ion to bind to oxygen [27]. Neuroglobin binds to H2S and oxidises it to thiosulfate, however it has been suggested that this reaction is less efficient, compared to its haemoglobin and myoglobin counterparts [28]. In addition, as neuroglobin expression decreases with age in rodents, it has been suggested that it may play a role in age-related neurodegeneration [29] (Fig. 2). These association is still not yet well explored.

The oxidation (catabolism) of H2S in the brain is primarily mediated by the mitochondrial sulphide quinone oxidoreductase homolog, SQRDL (sulphur quinone oxidoreductase-like protein) generating sulphate (SO42−) and thiosulphate (S2O32−). Other routes of catabolism include the generation of thiosulphate (S2O32−) from the reaction of H2S with ferrous neuroglobin (FeIII–Ngb)2
Altogether, the interplay between SQR and its homolog SQRDL, along with neuroglobin in the metabolism of H2S in the brain is not yet well established. Because SQR mRNA and protein expression is low in the brain, pathways involving neuroglobin and/or alternative SQR variants, warrant further investigation to better understand their role in sustaining H2S oxidation in the brain.
Neuroprotective Mechanisms of H2S
New avenues of research have resulted in the development and refinement of several H2S donor compounds. These donors have been useful to elucidate key molecular mechanisms mediated by H2S in the context of physiological responses. Moreover, they have also allowed the exploration of their potential to modulate impaired responses and act as potential therapeutics in a myriad of human conditions. Table 1 summarises identified molecular mechanisms of H2S in diverse models of neurological conditions.
H2S as ROS Scavenger
H2S is involved in a variety of physiological processes in the nervous system [10]. Kimura et al. demonstrated the protective effect of H2S in the nervous system, showing H2S is able to protect neuronal cells against oxidative stress (imbalance between oxidative molecules production and antioxidant mechanisms) by enhancing the synthesis of the antioxidant substrate glutathione (GSH) which functions as a storage for cysteine [8]. Importantly, GSH is maintained at high levels in astrocytes [8], where also CBS and SQR have been identified. In environments mimicking oxidative stress, such as in the presence of hydrogen peroxide (H2O2), H2S has also demonstrated the ability to reduce oxidative damage in mouse brain neuroblastoma cells Neuro2a, by enhancing GSH levels reduced by H2O2 [30]. It is possible that the regulation of sulphur/GSH metabolism in astrocytes is a key regulatory mechanism for the protection against oxidative molecules in the CNS. In addition to the enhanced production of GSH resulting from H2S, other mechanisms involving the direct scavenging of reactive oxygen species (ROS) by H2S have been reported. In this regard, H2S donor NaHS, has been identified as a direct scavenger of peroxynitrate, a reactive molecule derived from nitrogen in neuroblastoma cells SHSY-5Y [31].
Anti-apoptotic Role of H2S
Apoptosis, or programmed cell death, may occur through the intrinsic (mediated by the mitochondria) or extrinsic (associated with death receptor) pathways [32]. H2S has been shown to exert anti-apoptotic effects through the intrinsic pathway linked to its ROS neutralising properties, thus preventing oxidative stress to neuronal cells [33]. H2S can exert its anti-apoptotic effect via modulating nuclear translocation of nuclear factor kappa B (NF- κB) which is a transcription factor that can activate anti-apoptotic genes [34]. Additionally, the administration of NaHS has been observed to abrogate the generation of H2O2, reducing hippocampal neuronal apoptosis in mouse [35]. Studies by Shan et al., showed pre-treatment with H2S donors in a model of intracerebral haemorrhage in mice, significantly reduced caspase 3 and Bcl-2 (regulators of apoptosis) [36]. The molecular mechanisms linking anti-apoptotic function and ROS neutralising effects might work in parallel in the presence of H2S and this evidence suggest that H2S therapies may be effective to ameliorate a broad spectrum of pathophysiological mechanisms underpinning neurological conditions.
Role of H2S in Neuroinflammation
H2S has been shown to play a vital role in preventing neuroinflammation. Damage to neuronal cells can result in neuroinflammation by activation of pro-inflammatory cytokines that can aggravate the inflammation leading to neuronal death [37]. Several reports have demonstrated that H2S is able to prevent neuroinflammation by modulating the inflammatory response. In this regard, NaHS, has been observed to modulate inflammation by inhibiting the release of tumour necrosis factor- α (TNF-α) and reducing the expression of toll-like receptor 4 (TLR4), and NF-κB in the hippocampus of Sprague-Dawley rats subject to subarachnoid haemorrhage [38]. Furthermore, in rat models of AD established by injections of Amyloid-β1–40 into the hippocampus, H2S significantly reduced the release of inflammatory cytokines interleukin IL-1β and TNF-α whilst ameliorating spatial learning and memory impairment [39]. These observations suggest an interesting link between neuroinflammation and cognitive impairments that might be ameliorated by H2S.
CNS-Related Disorders and H2S
Parkinson’s Disease (PD)
PD is one of the most prevalent neurodegenerative disorders worldwide. Motor symptoms of Parkinson’s include bradykinesia, resting tremor, and cogwheel rigidity whereas non-motor symptoms include sleep disturbance, cognitive decline, and depression [67]. The pathophysiology of PD involves the progressive destruction of the dopaminergic neurons in the nigrostriatal pathway in the midbrain [68]. Accumulation of ROS can damage the DNA leading to oxidative stress and cell death are associated with PD [69].
Several models of PD have been proposed in order to better understand this disorder. Examples include the use of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its active metabolite 1-methyl-4-phenylpyridinium (MPP+). MPP+ plays an active role in destroying the dopaminergic neurons that results in parkinsonism [70]. An in vitro study used the human neuroblastoma cells (SH-SY5Y) to investigate the role of H2S in attenuating the oxidative stress on the neurons in PD cell model [40]. This study used an MPP+-treated SH-SY5Y cell model showing that NaHS increased the cell viability, reduced cytotoxicity, and reduced oxidative stress-induced cell apoptosis in a dose dependent manner in the MPP+ treated cells [40].
Another study investigated the neuroprotective role of H2S in a mouse model of PD. In line with the study carried out by Liu et al. [40], this study also showed that H2S elicited neuroprotective properties. MPTP was used in this study to generate a mouse model of PD and NaHS was used to investigate the effect of H2S. This study demonstrated that H2S prevents MPTP-induced neuronal damage and promotes neurogenesis through Akt/glycogen synthase kinase-3β (GSK-3β)/β-catenin pathways in adult neuronal stem cells [42]. Consistently, in a similar model of MPTP-induced PD in mice established by Lu et al., it was observed that NaHS reduced primary mesencephalic neurons cytotoxicity whilst explored molecular mechanism associated highlighting that H2S was able to enhance mitochondrial uncoupling protein 2 (UCP2) antioxidation resulting in abrogated ROS generation and reduced caspase 12-induced apoptosis [43]. Mitochondria are the main site of ROS generation thus may play a key role in the development of PD whilst compounds modulating mitochondrial-ROS, such as H2S, may be potential therapeutic candidates in the treatment of PD.
Although the molecular mechanisms of H2S-induced neuroprotection in PD may be broad (Fig. 3), these new avenues in PD research position H2S as a potential mitochondrial protectant. Thus, H2S may be a suitable therapeutic candidate for clinical use in treating Parkinson’s in the future.

Protective molecular mechanisms of H2S in Parkinson’s disease (PD). The neuroprotective effects of H2S in PD are associated with neurogenesis mediated by GSK3β/β-catenin pathway. H2S has antioxidant properties observed by the induction of antioxidant defence mediated by upregulation of superoxide dismutase (SOD) and glutathione (GSH). H2S can also reduce levels of reactive oxygen species (ROS) acting as a direct scavenger or indirectly by restoring the mitochondrial membrane potential (ΔΨm). The reduction in accumulation of mitochondrial-ROS results in inhibition of apoptosis mechanisms via caspase 3. Moreover, H2S may also regulate parkin functions by mediating posttranslational modifications of active cysteine residues, a process known as S-sulfhydration or persulfidation
Alzheimer’s Disease (AD)
AD is the most common neurodegenerative disorder and is the leading cause of dementia in the elderly population. Oxidative stress, neuroinflammation and damage to cholinergic neurons are some of the pathophysiological changes that are involved in AD [71].Furthermore, the accumulation of the microtubule-associated protein Tau [72] and β-amyloid peptides in neuronal cells [73] have been proposed as key molecular mechanism leading to this devastating disorder.
A research study investigated the neuroprotective role of H2S in an animal model of AD by using the 3xTg-AD mouse model which includes Tau protein mutations: PS1M146V, APPSwe, and Tau P301L [46]. This study showed that H2S prevents hyperphosphorylation of the Tau protein by S-sulfhydration (post-translational modification of proteins mediated by H2S) of GSK3β. Additionally, it was observed that the CSE catalytic effect was augmented by binding wild type Tau which resulted in decreased levels of Tau protein in the cell. In contrast, CSE could not bind the mutated Tau P301L. Consequently, CSE levels were decreased in the cortex and hippocampus of the brain in the 3xTg-AD mouse model compared to wild type. This interesting report demonstrated a direct relationship between decreased endogenous levels of H2S and AD-like condition in vivo. Interestingly, it was demonstrated that administrating H2S donor, GYY4137, by daily intraperitoneal injection for 12 weeks (100 mg/kg) to 3xTg-AD mice improved their motor and cognitive function. The level of Tau protein S-sulfhydration was higher in the group treated with GYY4137 compared to the control group [46].
Aligned with the study carried out by Giovinazzo et al. [46], a study by Vandini et al. [47] using 3xTg-AD transgenic mouse model, previously showed that intraperitoneal injection of sulphur water and NaHS daily for three consecutive months improved memory and cognitive functions in both young and aged animals Additionally, treatment with NaHS and sulphur water in 3xTg-AD mouse decreased the size of the β-amyloid plaques in the cortex and hippocampus. The molecular mechanisms implicated suggest the inhibition of c-Jun N-terminal kinases, extracellular signal-regulated kinases, and p38 protect against neuroinflammation and Tau protein hyperphosphorylation, leading to a decrease in accumulation of β-amyloid plaques in the cortical and hippocampal regions of the brain [47].
Moreover, a recently synthesised H2S donor targeted to the mitochondria, AP39, was probed against a model of AD in mice (APP/PS1 double-transgenic mice) [49]. AP39 injected intraperitoneally for 6 months enhanced cellular bioenergetic function and had mitochondrial protective effect on AD neurons and mice. Neurons treated with AP39 showed reduced ROS levels and increased ATP production. Within the AD neurons, it was observed that mitochondrial DNA was damaged, however AP39 treatment prevented these alterations by increasing mitochondrial DNA integrity. Furthermore, APP/PS1 double-transgenic mice treated with AP39 observed an improvement in their learning and memory impairments [49] suggesting that H2S, targeted to the mitochondria, could be a potential therapeutic candidate for AD. Given the fact that accumulation of Tau protein disturbs the neuronal mitochondrial respiration, molecular mechanism implied may include the restoration of the flow of electrons in the oxidative phosphorylation and/or antioxidant effects abrogating neurotoxicity induced by Tau, however, these theories warrant further exploration.
Overall, H2S has proven to play a significant neuroprotective role in AD models (Fig. 4). However, the translation of these results to the clinical setting might be hindered by complex dosage regimen of the H2S donor, requiring multiple and extensive dose regimes (3 to 6 months) as observed in these studies [46, 47, 49]. This emphasises the necessity to further explore patient-friendly approaches including better delivery systems and/or more stable H2S donors to reduce frequency of administration.

Protective mechanisms of H2S in AD. The neuroprotective effects of H2S in AD are associated with inhibition of anti-inflammatory cascade via reduction of TNF-β and IL-6 levels. Moreover, H2S has been linked to reduction of size of β-amyloid plaques and reduction in hyperphosphorylation of Tau. H2S has been observed to reduce levels of mitochondrial reactive oxygen species (ROS) leading to inhibition of apoptosis via caspase 3. H2S may also maintain the integrity of mitochondrial DNA whilst it may also regulate GSK3β protein functions via S-sulfhydration (persulfidation)
Huntington’s Disease (HD)
HD is an autosomal dominant progressive neurodegenerative disorder resulting from the toxic accumulation of mutant huntingtin protein in neurons [74]. The pathophysiology of HD involves the selective destruction of the corpus striatum in the brain which controls motor activities leading to involuntary and irregular muscle movements, behavioural changes and cognitive decline [75].
Paul et al. [76] explored the neuroprotective role of H2S in a mouse model of HD showing a direct correlation between reduction in CSE levels and increased neurodegeneration in the striatum. This study used both in vivo and in vitro models to demonstrate the impact of CSE depletion in the pathophysiology of HD. CSE depleted (CSE−/−) mice model exhibited abnormal hindlimb clasping and clenching resembling HD. Additionally, this study demonstrated that CSE level was significantly decreased in the striatal cell line of HD cell model consisting of 111 glutamine repeats (STHdhQ111/Q111(Q111) compared to the control group [76]. These observations suggest the importance of H2S in HD and imply the potential application of H2S donors in halting the progression of the disorder.
In an attempt to explore the potential for NaHS in abrogating HD-like effects induced by 3-nitropropionic acid (3NP), a study in a rat HD model revealed that H2S improved overall cognitive and locomotor deficits whist provided antioxidant, anti-inflammatory and anti-apoptotic effects observed by reduced levels of oxidative stress marker malondialdehyde (MDA), TNF-α and caspase 3 activation [51]. Interestingly, this study also revealed that treatment with NaHS significantly enhanced CBS expression, suggesting that H2S availability may have a crucial effect in modulating endogenous H2S pathways. Nonetheless, these assumptions require further exploration to understand the potential molecular mechanisms implicated.
Cerebral Vascular Disease
Stroke is the second most prevalent cause of mortality worldwide. The two types of strokes include haemorrhagic and ischemic stroke [77]. Both lead to ischemia and consequent tissue damage/death.
Given the association between ischemic stroke and ROS, H2S as an antioxidant compound, may be a suitable candidate for management of these conditions. A study investigated the neuroprotective role of H2S in acute ischemic stroke using a model of cerebral ischemia/reperfusion injury in mice by administrating NaHS before reperfusion following an ischemic insult in the brain. By means of H-magnetic resonance spectroscopy/magnetic resonance spectroscopy (H-MRI/MRS) and immunohistochemistry, it was shown that the total infarct volume was reduced compared to sham, and administration of NaHS 1 min before perfusion exerted stronger effect that of 30 min [52]. Another study exploring the effect of NaHS on transient cerebral ischemia immediately after reperfusion, this, combined with mild hypothermia resulted in a decrease in ischemic-reperfusion injury via upregulation of N-methyl-D-aspartate receptor (NMDAR) [53].
To study mechanisms of haemorrhagic stroke in vivo, models such as the middle cerebral artery occlusion (MCAO) plus intravenous injection of tissue plasminogen activators (tPA), have been used [78]. Using this model, it was demonstrated that H2S reduces tPA-induced cerebral haemorrhage following MCAO. In this study, co-administration of two structurally distinct H2S donors, ADT-OH and NaHS reduced the tPA-induced cerebral haemorrhage through the inhibition of AKT-VEGF-MMP9 signalling cascade [55]. As one of the risk factors associated with tPA in the management of ischemic stroke, is the increased risk of cerebral haemorrhage, observations by Liu et al. [55] suggest that H2S donors may reduce the risk of haemorrhagic stroke in tPA-exposed subjects. Although more research is necessary to confirm this hypothesis, the co-administration of H2S and tPA may bring new avenues and may provide patients requiring treatment for ischemic stroke a better recovery profile.
Brain Tumours
Brain tumours carry a high morbidity and mortality rate which highlights their clinical significance and burden to health systems worldwide. The most malignant brain tumour is glioblastoma multiform (GBM) which can rapidly spread and invade brain parenchyma [79]. Despite the growing number of treatment options available for the patients, brain malignancies carry a poor prognosis [80].
An in vitro study investigated the anti-cancer properties of H2S in C6 glioma cells [58]. It was shown that NaHS induces C6 glioma cell apoptosis via upregulation of caspase 3 and Bax proteins and downregulation of Bcl-2 protein through p38 MAPK signalling pathway [58]. In contrast to this report, another in vitro study demonstrated that H2S promotes C6 glioma cell growth. However, when CBS activity was compromised, the enhanced cell proliferation was blunted [60]. This study showed that cell apoptosis was reduced resulting in increased proliferation and viability rates of C6 glioma cells when exposed to NaHS suggesting that p38 MAPK/ERK1/2-COX-2 pathways are involved in NaHS-induced cancer cell proliferation and anti-apoptosis in C6 glioma cells [60]. An in vivo study reported by Li et al. showed that the size of the tumour was significantly increased in the rats exposed to NaHS in GBM group compared to the GBM only group, and H2S promotes C6 glioma cell growth via augmenting neurovascular formation and increasing hypoxia-inducible factor-1alpha (HIF-1α) expression [61]. These contrasting results more investigation is required to better understand the role of H2S in promoting/mitigating the expansion of brain tumours.
Epilepsy
Epilepsy is one of the most common neurological disorders affecting around 50 million of the global population. This condition is characterised by recurrent seizures which is the clinical manifestation of alerted neuronal electrical activities [81]. Additionally, despite antiepileptic drugs offering many patients symptomatic relief, effective management is not achieved in almost one third of patients with current pharmacotherapies. Hence, new medical therapies for epilepsy are needed [82].
Pro- inflammatory cytokines such as IL-1β, IL-6, and TNF-α play an important role in the pathophysiology of the epileptic seizures and the level of these cytokines is raised in the serum and cerebrospinal fluid of epileptic patients [83]. Increased level of pro-inflammatory cytokines results in increased excitability of neurons which may be crucial in the pathophysiology epilepsy [84]. As H2S promotes the release of anti-inflammatory cytokines and is involved in preventing neuroinflammation [85], it might have a potential clinical application in the treatment of epilepsy.
The role of H2S in pilocarpine-induced status epilepticus (SE) in vivo has been explored. Adult male C57BL/6 mice were exposed to pilocarpine to induce SE whilst treatment consisted of exposure to a novel carbazole-based H2S donor. This study demonstrated that SE + H2S group had a shorter duration of seizure compared to the SE group. Additionally, this group showed delayed onset of seizure, reduced damage to the hippocampus and reduced pro-inflammatory state in microglia compared to the SE control group [62].
In contrast, a study performed by Luo et al. [63] showed that H2S delivered as NaHS exacerbates Pentylenetetrazole (PTZ)- and pilocarpine‐induced seizures in rats [63]. PTZ is a GABA-A receptor antagonist that when sequentially injected to animals, results in the development of chemical kindling, an epilepsy model [86]. Luo et al. [63] exposed rats to PTZ/pilocarpine and PTZ/pilocarpine + NaHS showing that the severity of seizures increased in the group treated with NaHS compared to the control group. Furthermore, the duration of seizure was longer in those rats exposed NaHS suggesting that H2S increased membrane excitability of entorhinal cortex neurones via facilitating the function of voltage‐gated sodium channel, AMPAR, and NMDAR [63]. These observations are not consistent about the effect of exogenous H2S on the manifestation of epilepsy. However, they suggest a potential role of H2S signalling in the modulation and severity of epilepsy. Further research is warranted to better understand the potential of the modulation of H2S signalling in the management of epilepsy.
Multiple Sclerosis (MS)
MS is an autoimmune disease affecting the myelin structure in the CNS. Currently, 2.8 million people are affected worldwide, however, this number is on the rise [87]. MS results in a wide range of signs and symptoms that are variable depending on the severity and location of the lesions with optic neuritis being the most common [88]. Currently, the available immune-based therapies do not significantly improve MS prognosis, therefore, new treatment strategies, such as H2S donors, warrant exploration in the pursuit of better patient outcomes.
In physiological settings, the BBB protects the CNS by making it inaccessible to immune cells, while in MS, the BBB gets damaged due to endothelial cells’ impairment [89]. This results in an exacerbated influx of inflammatory mediated cells into the brain tissue, which induces demyelination and axonal dysfunction. Recurrent inflammatory responses in MS irreversibly damage the CNS nerve cells with the involvement of ROS [90]. The pathophysiology of MS also involves chronic platelet activation that promotes differentiation of autoreactive T cells resulting in initiation and progression of autoimmune neuroinflammation [91]. Therefore, two potential molecular mechanisms by which H2S may show potential beneficial effects in MS include its antioxidant role as well as its ability to inhibit platelet activation and adhesion molecule-mediated aggregation [92].
An in vitro study by Talaei et al. [65] examined the effect of NaHS on peripheral blood mononuclear cells (PBMNCs) obtained from healthy individuals to access its transmigration across an endothelial cells’ barrier representing the BBB. This study showed that pre-treatment of human endothelial cells with NaHS decreased PBMNC transmigration of both control and serotonin-treated cells. Additionally, treatment with NaHS resulted in upregulation of IL-10 and downregulation of adhesion molecules: lymphocyte function-associated antigen 1 (LFA-1) and vascular cell adhesion protein 1 (VCAM-1). This suggests that whilst NaHS resulted in relaxation/expansion of endothelial cells it may also cause morphological changes to the BBB leading to increased BBB permeability during MS attacks [65].
Lazarević et al. [66] studied the in vitro effect of H2S donor GYY4137 on differentiated dendritic cells and T cells which are both involved in the pathogenesis of MS. GYY4137 enhanced transforming growth factor β (TGF-β) expression in dendritic cells, suggesting an anti-inflammatory effect of GYY4137 in an MS-like environment. Additionally, isolated lymph node and spinal cord T cells were obtained from mice and rats immunised with CNS antigens and treated with GYY4137. GYY4137 significantly reduced interferon gamma (IFN-γ) and IL-17 production and reduced the proportion of FoxP3 + regulatory CD4 + T cells in the lymph node and spinal cord T cells. Interestingly, when the expression of H2S-producing enzymes (CBS, CSE, and 3-MST) was assessed in immune cells from healthy donors and drug-naïve relapse-remitting MS patients, the authors reported no differences in the CSE and CBS expression but lower expression of 3-MST [66].
Given the relevance of 3-MST in the generation and availability of H2S within the mitochondria, this study further suggests a potential link between H2S signalling and mitochondria in the modulation of anti-inflammatory response in MS. However, this theory awaits more exploration.
Challenges in H2S Delivery to the Nervous System
Several limitations associated with the gaseous nature and rapid release of H2S from its various donor molecules hinder the translation of H2S-based therapeutics to the clinic. As shown throughout this review, it appears that a systemic increase in H2S availability may improve neurological function in certain neurological disorders such as PD and AD.
In conditions where H2S may have a therapeutic effect, desirable H2S-based compounds should bear long-circulating times with slow-releasing patterns. As Table 2 shows, current research evidence displaying the potential therapeutic effect of H2S donors in CNS related conditions, usually require extensive dosage regimes, ranging from weeks to months with daily administration of the donor, in order to observe therapeutic effect. This is consistent with previous observations that bolus administrations of H2S-donating compounds may result in spiked concentrations of H2S [93], leading to complex administration regimes. Understanding the burden that neurological conditions put on patients, families and healthcare professionals, patient-friendly approaches are necessary. This suggest that strategies to reduce administration of the compounds is of key importance. In an attempt to overcome this issue, several compounds with H2S-releasing properties have been developed which differ in their patterns of H2S release, including mechanisms triggered by hydrolysis, enzymatic reactions, pH variation, novel formulation strategies amongst others [94].
Another important limitation arises from the fact that to reach the brain, therapeutic compounds require to be able to cross the BBB. The BBB is a structure separating the blood and brain compartments. Its inner layer is composed of endothelial cells with tight junctions and its outer layer consists of pericytes and astrocytes [95]. The BBB allows hydrophobic substances (e.g., water and oxygen) to access the brain whilst preventing pathogens and large and hydrophilic substances (e.g., microorganisms and drugs) from entering the brain. For this reason, most drugs targeting the CNS cannot enter the brain, bringing difficulties in developing new efficient treatments for neurological diseases.
In terms of sulphide donating salts, ions diffuse passively across the BBB and their flow can be accelerated by partial association between anions and cations to form neutral ion-pair species in solution [96]. Thiosulfate does not permeate across cell membranes freely as it is an anion, therefore it must be transported by a transport mechanism to get into cells. The transport mechanism for thiosulfate is an SLC13 protein called sodium sulphate cotransporter 2 (SLC13A4, NaS-2), which is found within the brain. The transport mechanism helps the transportation of thiosulfate across the cell membrane [97], therefore, approaches targeting these transport mechanisms may offer a feasible method to enhance H2S delivery, when thiosulphate is employed. Therefore, along with developing targeted formulations, understating the chemistry of H2S-donating compounds is key in the successful delivery of these drugs to the CNS.
Biochemical structures of ACS84, ADT-OH, AP39, Carbazole-based H2S donor (C15H14N2S), Diallyl disulphide (DAD) and diallyl trisulfide (DAT), GYY4137, Sodium hydrosulphide (NaHS), S-ASP (sulphur-aspirin hybrid) and S-DI (sulphur-diclofenac) are depicted in Fig. 5.

Biochemical structures of ACS84, ADT-OH, AP39, Carbazole-based H2S donor (C15H14N2S), Diallyl disulphide (DAD) and diallyl trisulfide (DAT), GYY4137, Sodium hydrosulphide (NaHS), S-ASP (sulphur-aspirin hybrid) and S-DI (sulphur-diclofenac)
Perspectives: Overcoming Limitations in H2S Delivery
Thus far, H2S donors have not been specifically targeted to the brain even when indicated in neurological dysfunction. Drugs with molecular weight below 500 Da and high lipophilic content usually diffuse the BBB easily [104] thus forth, H2S donors targeting the brain must possess such qualities. Carrier-mediated transport and receptor-mediated endocytosis/transcytosis have been explored and, more recently, biomaterial-mediated strategies have been envisaged, including polymeric-based systems, already approved by the Food and Drug Administration Agency. These polymeric systems include polylactide, polycaprolactum and polyglycolide that can be employed as hydrogels and nanoparticle formulations. Another approach has been the use of liposomes. These structures, due to their similarity with the lipid bilayer in cellular membranes along with CNS targeting potential offers an interesting approach when exploring CNS drug delivery. In particular, approaches embedding antibodies within liposome structure, also referred as immunoliposomes, have been explored in the treatment of AD [105].
Nanoparticle formulations based on polymer or liposomal structures may also assist drug movement across the BBB when CNS-related drugs cannot cross the BBB on their own. In other scenarios of human disease, preclinical studies of nanoparticle delivery of drugs have reported that nanoparticles could not only increase the compounds half-life time but also decrease the amount of drug being eliminated by the reticuloendothelial system, thus, improving stability and increasing drug exposure [106, 107]. Recently, in our laboratory, we have explored nanoparticle approaches to encapsulate H2S donors, protecting from rapid degradation and release of H2S whilst exploring different routes of administration aiming to reduce administration regimes [108,109,110]. Whilst observations must be confirmed in an in vivo setting, it was noted that liposomes employed to carry ADT-OH across murine skin observed a delayed flux across the skin compared to formulations without liposomes suggesting liposomes may be beneficial in giving a controlled release of drug [108]. Furthermore, cationic liposomes used in the controlled release of H2S from sodium thiosulphate were able to retain its biological properties in hypoxia-like environment [110]. Finally, PLGA nanoparticles carrying sodium thiosulphate observed a sustained cellular drug uptake whilst maintaining the pro-angiogenic potential of H2S [109]. These new approaches are supported by other researchers exploring the use of mesoporous iron oxide nanoparticle (MION) with polyethylene glycol (PEG) and lactoferrin as carriers for the H2S donor DATS [111]. Another relevant aspect of this novel method of H2S delivery is the fact that MION, due to their excellent superparamagnetism, enable non-invasive in vivo tracing through MRI techniques, that can be further applied as a means to measure pharmacokinetic parameters such as drug distribution [111].
Nonetheless, emerging pharmaceutical technology to successfully deliver H2S at consistent rates, limiting its degradation and successfully targeting the CNS is in its infancy. It is therefore paramount to encourage interdisciplinary collaborations bringing together the biochemistry and pharmacological aspect of H2S metabolism within the CNS field to offer better approaches and methods in the investigation of H2S-based therapeutics for neurological health.
Conclusion
H2S is an important mediator in nervous system physiology playing a protective role in most neurological disorders. H2S elicits anti-oxidative, anti-apoptotic, and anti-inflammatory properties in the CNS which may ameliorate a range of neurological disorders. This review highlights the potential therapeutic application of H2S-based compounds in treating neurological disease. Although many H2S releasing drugs have been tested in scenarios resembling neurological impairments (in vitro and in vivo), many limitations in the translation to the clinic are present, including the gaseous nature of H2S compounds, the rapid release of H2S from these molecules and lack of targeting to the CNS, limiting the successful crossing of the BBB. Recent approaches including chemical modifications to the H2S donor and nanoparticle formulations (both polymer- and liposome-based) to encapsulate H2S-based compounds may offer a solution to protect these compounds from rapid degradation, control their rates of release whilst providing targeting opportunities to reach and cross the BBB. Furthermore, we believe exploring additional routes of drug delivery such as buccal or transdermal drug delivery would aid patient self care and reduce the need for medical intervention. Understanding these new avenues in pharmacological formulations and enhancing interdisciplinary research may result in the generation of a suitable H2S donor treatments to address neurological conditions.
Data Availability
Enquiries about data availability should be directed to the authors.
References
Feigin VL, Nichols E, Alam T, Bannick MS, Beghi E, Blake N, Culpepper WJ, Dorsey ER, Elbaz A, Ellenbogen RG, Fisher JL (2019) Global, regional, and national burden of neurological disorders, 1990–2016:a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18(5):459–480
World Health O (2006) Neurological disorders: public health challenges. World Health Organization, Geneva
Ufnal M, Sikora M (2011) The role of brain gaseous transmitters in the regulation of the circulatory system. Curr Pharm Biotechnol 12(9):1322–1333
Stein A, Bailey SM (2013) Redox biology of hydrogen sulfide: implications for physiology, pathophysiology, and pharmacology. Redox Biol 1(1):32–39
Szabo C, Papapetropoulos A (2017) International union of basic and clinical pharmacology. CII: pharmacological modulation of H(2)S levels: H(2)S donors and H(2)S biosynthesis inhibitors. Pharmacol Rev 69(4):497–564
Panthi S et al (2016) Physiological importance of hydrogen sulfide: emerging potent neuroprotector and neuromodulator. Oxid Med Cell Longev 2016:9049782
Nagai Y et al (2004) Hydrogen sulfide induces calcium waves in astrocytes. FASEB J 18(3):557–559
Kimura Y, Kimura H (2004) Hydrogen sulfide protects neurons from oxidative stress. FASEB J 18(10):1165–1167
Kashfi K, Olson KR (2013) Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem Pharmacol 85(5):689–703
Kimura H (2011) Hydrogen sulfide: its production and functions. 96:833–8359
Chen X, Jhee KH, Kruger WD (2004) Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J Biol Chem 279(50):52082–52086
Kolluru GK et al (2013) Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide 35:5–20
Tanizawa K (2011) Production of H2S by 3-mercaptopyruvate sulphurtransferase. J Biochem 149(4):357–359
Polhemus DJ, Lefer DJ (2014) Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circul Res 114(4):730–737
Nicholson CK, Calvert JW (2010) Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol Res 62(4):289–297
Zhang F et al (2020) 3-Mercaptopyruvate sulfurtransferase/hydrogen sulfide protects cerebral endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury via mitoprotection and inhibition of the RhoA/ROCK pathway. Am J Physiol Cell Physiol 319(4):C720–C733. https://doi.org/10.1152/ajpcell.00014.2020. Epub 2020 Aug 19. PMID: 32813542.
Siracusa R, Fusco R, Cuzzocrea S (2019) Astrocytes: role and functions in brain pathologies. Front Pharmacol 10:1114. https://doi.org/10.3389/fphar.2019.01114. PMID: 31611796; PMCID: PMC6777416.
Abe K, Kimura H (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16(3):1066–1071
Beyer K et al (2004) Cystathionine beta synthase as a risk factor for Alzheimer disease. Curr Alzheimer Res 1(2):127–133
Bjørke-Monsen A-L et al (2022) Severe hyperhomocysteinemia in a patient with Parkinson disease. Clin Chem 68(3):396–401
Cao X et al (2019) A review of hydrogen sulfide synthesis, metabolism, and measurement: is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid Redox Signal 31(1):1–38
Lagoutte E et al (2010) Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta 1797(8):1500–1511
Hildebrandt TM, Grieshaber MK (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 275(13):3352–3361
Jia J et al (2020) SQR mediates therapeutic effects of H(2)S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci Adv 6(35):eaaz5752
Linden DR et al (2012) Sulphide quinone reductase contributes to hydrogen sulphide metabolism in murine peripheral tissues but not in the CNS. Br J Pharmacol 165(7):2178–2190
Ackermann M et al (2011) The vertebrate homolog of sulfide-quinone reductase is expressed in mitochondria of neuronal tissues. Neuroscience 199:1–12
Dewilde S et al (2001) Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J Biol Chem 276(42):38949–38955
Ruetz M et al (2017) A distal ligand mutes the interaction of hydrogen sulfide with human neuroglobin. J Biol Chem 292(16):6512–6528
Sun Y et al (2005) Effect of aging on neuroglobin expression in rodent brain. Neurobiol Aging 26(2):275–278
Yang W et al (2005) Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J Physiol 569(Pt 2):519–531
Whiteman M et al (2004) The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem 90(3):765–768
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516
Zhang JY et al (2017) Hydrogen sulfide therapy in brain diseases: from bench to bedside. Med Gas Res 7(2):113–119
Sen N et al (2012) Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol Cell 45(1):13–24
Luo Y et al (2012) Hydrogen sulfide prevents hypoxia-induced apoptosis via inhibition of an H2O2-activated calcium signaling pathway in mouse hippocampal neurons. Biochem Biophys Res Commun 425(2):473–477
Shan H et al (2019) Exogenous hydrogen sulfide offers neuroprotection on intracerebral hemorrhage injury through modulating endogenous H2S metabolism in mice. Front Cell Neurosci 13:349. https://doi.org/10.3389/fncel.2019.00349. PMID: 31440142; PMCID: PMC6693577.
McGeer PL, Rogers J, McGeer EG (2016) Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimers Dis 54(3):853–857
Duan H et al (2020) Hydrogen sulfide reduces cognitive impairment in rats after subarachnoid hemorrhage by ameliorating neuroinflammation mediated by the TLR4/NF-kappaB pathway in microglia. Front Cell Neurosci 14:210
Xuan A et al (2012) Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in beta-amyloid rat model of Alzheimer’s disease. J Neuroinflamm 9(1):202
Liu L, Wang J, Wang H (2020) Hydrogen sulfide alleviates oxidative stress injury and reduces apoptosis induced by MPP(+) in Parkinson’s disease cell model. Mol Cell Biochem 472(1–2):231–240
Xie L et al (2013) Therapeutic effect of hydrogen sulfide-releasing L-dopa derivative ACS84 on 6-OHDA-induced Parkinson’s disease rat model. PLoS ONE 8(4):e60200
Wang M et al (2021) Hydrogen sulfide enhances adult neurogenesis in a mouse model of Parkinson’s disease. Neural Regen Res 16(7):1353–1358
Lu M et al (2012) The neuroprotection of hydrogen sulfide against MPTP-induced dopaminergic neuron degeneration involves uncoupling protein 2 rather than ATP-sensitive potassium channels. Antioxid Redox Signal 17(6):849–859
Vandiver MS et al (2013) Sulfhydration mediates neuroprotective actions of parkin. Nat Commun 4:1626
Giuliani D et al (2013) Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol Learn Mem 104:82–91
Giovinazzo D et al (2021) Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3beta and inhibiting tau hyperphosphorylation. Proc Natl Acad Sci USA 118:4
Vandini E et al (2019) Mechanisms of hydrogen sulfide against the progression of severe Alzheimer’s disease in transgenic mice at different ages. Pharmacology 103(1–2):50–60
Rosario-Alomar MF et al (2015) Hydrogen sulfide inhibits amyloid formation. J Phys Chem 119(4):1265–1274
Zhao F-L et al (2016) AP39, a mitochondria-targeted hydrogen sulfide donor, supports cellular bioenergetics and protects against Alzheimer’s disease by preserving mitochondrial function in APP/PS1 mice and neurons. Oxid Med Cell Longev 2016:8360738–8360738
Gong Q-H et al (2011) S-Propargyl-cysteine, a novel hydrogen sulfide-modulated agent, attenuates lipopolysaccharide-induced spatial learning and memory impairment: involvement of TNF signaling and NF-κB pathway in rats. Behav Immun 25(1):110–119Brain
Mohammed RA, Mansour SM (2021) Sodium hydrogen sulfide upregulates cystathionine β-synthase and protects striatum against 3-nitropropionic acid-induced neurotoxicity in rats. J Pharm Pharmacol 73(3):310–321
Woo CW et al (2017) The administration of hydrogen sulphide prior to ischemic reperfusion has neuroprotective effects in an acute stroke model. PLoS ONE 12(11):e0187910
Dai H-b et al (2015) Hydrogen sulphide and mild hypothermia activate the CREB signaling pathway and prevent ischemia-reperfusion injury. BMC Anesthesiol 15(1):119
Wang Y et al (2014) Hydrogen sulfide protects blood–brain barrier integrity following cerebral ischemia. J Neurochem 129(5):827–838. https://doi.org/10.1111/jnc.12695. Epub 2014 Mar 27. PMID: 24673410.
Liu H et al (2016) Hydrogen sulfide attenuates tissue plasminogen activator-induced cerebral hemorrhage following experimental stroke. Transl Stroke Res 7(3):209–219
Liu H et al (2016) Hydrogen sulfide attenuates tissue plasminogen activator-induced cerebral hemorrhage following experimental stroke. Transl Stroke Res 7(3):209–219
Xiao AY et al (2019) Sodium sulfide selectively induces oxidative stress, DNA damage, and mitochondrial dysfunction and radiosensitizes glioblastoma (GBM) cells. Redox Biol 26:101220
Zhao L et al (2015) Exogenous hydrogen sulfide exhibits anti-cancer effects though p38 MAPK signaling pathway in C6 glioma cells. Biol Chem 396(11):1247–1253
Das A, Banik NL, Ray SK (2007) Garlic compounds generate reactive oxygen species leading to activation of stress kinases and cysteine proteases for apoptosis in human glioblastoma T98G and U87MG cells. Cancer 110(5):1083–1095. https://doi.org/10.1002/cncr.22888. PMID: 17647244.
Zhen Y et al (2015) Exogenous hydrogen sulfide promotes C6 glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2 pathways. Oncol Rep 34(5):2413–2422
Li ZY et al (2012) Hydrogen sulfide stimulates the development of rat glioblastoma. Zhonghua Zhong Liu Za Zhi 34(4):254–258
Liu Z et al (2021) A novel hydrogen sulfide donor reduces pilocarpine-induced status epilepticus and regulates microglial inflammatory profile. Front Cell Neurosci 15:780447
Luo Y et al (2014) Aggravation of seizure-like events by hydrogen sulfide: involvement of multiple targets that control neuronal excitability. CNS Neurosci Ther 20(5):411–419
Zhu Z et al (2021) A hydrogen sulfide donor suppresses pentylenetetrazol-induced seizures in rats via PKC signaling. Eur J Pharmacol 898:173959
Talaei F (2016) Pathophysiological concepts in multiple sclerosis and the therapeutic effects of hydrogen sulfide. Basic Clin Neurosci 7(2):121–136
Lazarevic M et al (2020) Upregulation of tolerogenic pathways by the hydrogen sulfide donor GYY4137 and impaired expression of H2S-producing enzymes in multiple sclerosis. Antioxidants 9(7):608
Sveinbjornsdottir S (2016) The clinical symptoms of Parkinson’s disease. J Neurochem 139(Suppl 1):318–324
Struzyna LA et al (2018) Tissue engineered nigrostriatal pathway for treatment of Parkinson’s disease. J Tissue Eng Regen Med 12(7):1702–1716
Zhou C, Huang Y, Przedborski S (2008) Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci 1147:93–104
Sun Y et al (2018) MPP(+) decreases store-operated calcium entry and TRPC1 expression in mesenchymal stem cell derived dopaminergic neurons. Sci Rep 8(1):11715
Breijyeh Z, Karaman R (2020) Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25(24):5789
Huang HC, Jiang ZF (2009) Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: relationship and links in Alzheimer’s disease. J Alzheimers Dis 16(1):15–27
Murphy MP, LeVine H(2010) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimer’s Dis 19(1):311–323
McColgan P, Tabrizi SJ (2018) Huntington’s disease: a clinical review. Eur J Neurol 25(1):24–34
Ross CA et al (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10(4):204–216
Paul BD et al (2014) Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 509(7498):96–100
Katan M, Luft A (2018) Global burden of stroke. Semin Neurol 38(2):208–211
García-Yébenes I et al (2011) A mouse model of hemorrhagic transformation by delayed tissue plasminogen activator administration after in situ thromboembolic stroke. Stroke 42(1):196–203
Porcu E et al (2018) BMP9 counteracts the tumorigenic and pro-angiogenic potential of glioblastoma. Cell Death Differ 25(10):1808–1822
Blumenthal DT et al (2017) Is more better? The impact of extended adjuvant temozolomide in newly diagnosed glioblastoma: a secondary analysis of EORTC and NRG Oncology/RTOG. Neuro Oncol 19(8):1119–1126
Sander JW (1993) Some aspects of prognosis in the epilepsies: a review. Epilepsia 34(6):1007–1016
Schmidt D, Rogawski MA (2002) New strategies for the identification of drugs to prevent the development or progression of epilepsy. Epilepsy Res 50(1–2):71–78
Webster KM et al (2017) Inflammation in epileptogenesis after traumatic brain injury. J Neuroinflamm 14(1):10
Vezzani A et al (2013) Epilepsy and brain inflammation. Exp Neurol 244:11–21
Seifert HA, Pennypacker KR (2014) Molecular and cellular immune responses to ischemic brain injury. Transl Stroke Res 5(5):543–553
Shimada T, Yamagata K (2018) Pentylenetetrazole-induced kindling mouse model. J Vis Exp 136:e56573
Walton C et al (2020) Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult Scler 26(14):1816–1821
Dobson R, Giovannoni G (2019) Multiple sclerosis—a review. Eur J Neurol 26(1):27–40
Waubant E (2006) Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Dis Markers 22(4):235–244
Miljkovic D, Spasojevic I (2013) Multiple sclerosis: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal 19(18):2286–2334
Starossom SC et al (2015) Platelets play differential role during the initiation and progression of autoimmune neuroinflammation. Circ Res 117(9):779–792
Grambow E et al (2014) Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets 25(3):166–174
Zheng Y et al (2018) Toward hydrogen sulfide based therapeutics: critical drug delivery and developability issues. Med Res Rev 38(1):57–100
Corvino A et al (2021) Trends in H(2)S-donors chemistry and their effects in cardiovascular diseases. Antioxidants 10(3):429
Thangudu S, Cheng F-Y, Su C-H (2020) Advancements in the blood-brain barrier penetrating nanoplatforms for brain related disease diagnostics and therapeutic applications. Polymers 12(12):3055
Breschi GL et al (2013) Different permeability of potassium salts across the blood-brain barrier follows the hofmeister series. PLoS ONE 8(10):e78553
Marutani E et al (2015) Thiosulfate mediates cytoprotective effects of hydrogen sulfide against neuronal ischemia. J Am Heart Assoc 4(11):e002125
Pomierny B et al (2021) The slow-releasing and mitochondria-targeted hydrogen sulfide (H(2)S) delivery molecule AP39 induces brain tolerance to Ischemia. Int J Mol Sci 22(15):7816
Wallace GCt et al (2013) Multi-targeted DATS prevents tumor progression and promotes apoptosis in ectopic glioblastoma xenografts in SCID mice via HDAC inhibition. J Neurooncol 114(1):43–50
Han X et al (2020) GYY4137 protects against MCAO via p38 MAPK mediated anti-apoptotic signaling pathways in rats. Brain Res Bull 158:59–65
Lee M et al (2010) Hydrogen sulfide-releasing NSAIDs attenuate neuroinflammation induced by microglial and astrocytic activation. Glia 58:103–1131
Shi H-Q et al (2016) Sodium sulfide, a hydrogen sulfide-releasing molecule, attenuates acute cerebral ischemia in rats. CNS Neurosci Ther 22:625–632
Cheng X-j et al (2019) Tacrine–hydrogen sulfide donor hybrid ameliorates cognitive impairment in the aluminum chloride mouse model of Alzheimer’s disease. ACS Chem Neurosci 10(8):3500–3509
Kamphorst W, de Boer AG, Gaillard PJ (2001) Brain drug targeting: the future of brain drug development: pardridge W M. Cambridge University Press, 2001. J Clin Pathol 55(2):158–158
Ordóñez-Gutiérrez L et al (2017) ImmunoPEGliposome-mediated reduction of blood and brain amyloid levels in a mouse model of Alzheimer’s disease is restricted to aged animals. Biomaterials 112:141–152
Kyle S, Saha SJAHM (2014) Nanotechnology for the detection and therapy of stroke. Adv Healthc Mater 3(11):1703–1720. https://doi.org/10.1002/adhm.201400009. Epub 2014 Apr 1. PMID: 24692428.
Goyal K et al (2014) Targeted drug delivery to central nervous system (CNS) for the treatment of neurodegenerative disorders: trends and advances. Cent Nerv Syst Agents Med Chem 14(1):43–59
Marwah MK et al (2022) Transdermal delivery of a hydrogen sulphide donor, ADT-OH using aqueous gel formulations for the treatment of impaired vascular function: an ex vivo study. Pharm Res 39(2):341–352
Marwah MK et al (2022) Novel controlled-release polylactic-co-glycolic acid (PLGA) nanoparticles for sodium thiosulphate, a hydrogen sulphide donor, retains pro-angiogenic potential of hydrogen sulphide. J Exp Nanosci 17(1):197–213
Sanchez-Aranguren L et al (2022) Sodium thiosulphate-loaded liposomes control hydrogen sulphide release and retain its biological properties in hypoxia-like environment. Antioxidants 11(11):2092
Sun X et al (2021) Novel controlled and targeted releasing hydrogen sulfide system exerts combinational cerebral and myocardial protection after cardiac arrest. J Nanobiotechnol 19(1):40
Funding
This work was supported by the Royal Society Grant - Round 1 2021 (RGS\R1\221169) and by the Sir Halley Stewart Trust (Ref number 2728). The views expressed within this report are those of the authors and not necessarily those of the Trust, both awarded to LSA.
Author information
Authors and Affiliations
Contributions
LS-A conceptualised the manuscript and revised the intellectual concept of the work. AHS, MI and NG participated in the writing of the original draft. LS-A, BM, MM and DM edited the manuscript, revised critically the intellectual nature of the final draft and approved final version for submission. LS-A secured funding for this work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharif, A.H., Iqbal, M., Manhoosh, B. et al. Hydrogen Sulphide-Based Therapeutics for Neurological Conditions: Perspectives and Challenges. Neurochem Res 48, 1981–1996 (2023). https://doi.org/10.1007/s11064-023-03887-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03887-y