
Abstract
Type 2 diabetes (T2DM) is a well known risk factor for Alzheimer’s disease. Mitochondria are the center of intracellular energy metabolism and the main source of reactive oxygen species. Mitochondrial dysfunction has been identified as a key factor in diabetes-associated brain alterations contributing to neurodegenerative events. Defective insulin signaling may act in concert with neurodegenerative mechanisms leading to abnormalities in mitochondrial structure and function. Mitochondrial dysfunction triggers neuronal energy exhaustion and oxidative stress, leading to brain neuronal damage and cognitive impairment. The normality of mitochondrial function is basically maintained by mitochondrial quality control mechanisms. In T2DM, defects in the mitochondrial quality control pathway in the brain have been found to lead to mitochondrial dysfunction and cognitive impairment. Here, we discuss the association of mitochondrial dysfunction with T2DM and cognitive impairment. We also review the molecular mechanisms of mitochondrial quality control and impacts of mitochondrial quality control on the progression of cognitive impairment in T2DM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is estimated that there are 537 million people living with diabetes worldwide and this number is expected to over 783 million in 2045, due to the aging population and increased longevity [1]. Type 2 diabetes (T2DM) is a major subtype of diabetes, accounting for more than 90% of all diabetes cases [2]. T2DM is characterized by relative insulin deficiency caused by pancreatic β-cell dysfunction and insulin resistance in target organs [3, 4]. Clinical studies have found that T2DM causes atrophy in frontal and temporal regions of the brain, especially the hippocampus, which is associated with poorer visuospatial construction, executive function, and memory function [5,6,7]. It has been found that T2DM increases the risk of multiple forms of cognitive impairment, including Alzheimer’s disease (AD) [8, 9]. A meta-analysis suggested that patients with diabetes had a 56% increased risk of AD [10].
The cognitive changes associated with T2DM appear to begin in the prediabetic phase of insulin resistance [11]. The brain is an organ susceptible to insulin. It is rich in insulin receptors in areas closely related to cognitive memory, such as the hippocampus [12]. Studies have shown that decreased brain sensitivity to insulin can lead to energy failure and ad like pathological changes, including amyloid beta peptide (Aβ) deposition and aberrant tau phosphorylation, leading to impaired nerve growth, synaptic plasticity, and cognitive function [13,14,15]. This suggests an association between AD and T2DM. In fact, AD is considered a brain-specific form of diabetes. AD patients have shown reduced brain insulin receptor sensitivity, hyperphosphorylation of insulin receptor and downstream second messenger such as insulin receptor substrate-1 (IRS-1) [16,17,18]. Cognitive function improved after treatment with insulin sensitizer or intranasal insulin in patients with AD [19,20,21].
The cognitive function of the brain depends on synaptic communication between neurons. This leads to high levels of energy demand, so synapses are rich in mitochondria [22, 23]. Mitochondria are also a major source and target of intracellular reactive oxygen species (ROS). Multiple studies have shown that mitochondrial dysfunction may be a key player in diabetes-associated brain alterations contributing to neurodegenerative events [24,25,26]. In the insulin resistant brain, the structural and functional damage of brain mitochondria was observed, including reduced mitochondrial electron transport chain (mETC) activity, decreased mitochondrial respiration and massive production of ROS [27,28,29,30]. This not only leads to energy exhaustion, but also oxidative stress. Uncontrolled oxidative stress can promote the accumulation of Aβ in synaptic mitochondria, induce neuronal apoptosis and lead to cognitive impairment [30,31,32,33].
Numerous quality control mechanisms have evolved within mitochondria to maintain proper function basally and in response to stress, including proteostasis, biogenesis, dynamics, and mitophagy [34]. Mitochondrial protein import is controlled and occurs in their unfolded form through various translocases, which is supported by mitochondrial membrane potential [35, 36]. Molecular chaperones and intramitochondrial proteases control the integrity and proper assembly of the imported proteins [37, 38]. In addition, parts of or even entire mitochondria can be removed via several mechanisms as discussed later [39, 40]. In this review, we focus on mitochondrial dysfunction, and summarize current knowledge of the role of mitochondrial quality control mechanisms in cognitive dysfunction in diabetes.
Mitochondrial Dysfunction as a Link Between T2DM and AD
Studies have shown that a high-fat diet results in an increased flux of free fatty acids into circulation, which are absorbed by the liver or skeletal muscle for beta-oxidation in the mitochondria or stored as triglycerides [41, 42]. When fatty acid flux exceeds the processing capacity of these pathways, fatty acid metabolic intermediates (particularly ceramide) will accumulate [43, 44]. Although the role of ceramide in the formation of insulin resistance is contentious, some evidence suggests that ceramide may contribute to brain insulin resistance [45,46,47,48,49,50,51]. Ceramide can enter the brain through the blood–brain barrier, and can also be produced in the brain through de novo synthesis or sphingomyelin hydrolysis [50,51,52,53]. In canonical insulin signaling, insulin binds to insulin receptor, inducing IR autophosphorylation, recruitment of insulin receptor substrate (IRS) adaptor proteins, and then activates phosphatidylinositol 3-kinase (PI3K)/AKT pathway, thereby exerting a variety of anabolic activities [54]. Ceramide can activate c-Jun N-terminal kinase (JNK) and IκB kinase to inactivate IRS-1 phosphorylation, and also inhibit the PI3-K/Akt pathway through protein phosphatase 2A dephosphorylation, leading to insulin signaling disruption and neuronal apoptosis [55, 56]. Interestingly, exposure to ceramide also leads to neuronal mitochondrial dysfunction. However, the role of mitochondrial dysfunction in the formation of brain insulin resistance is largely unknown, and few studies suggest that mitochondrial dysfunction contributes to brain insulin resistance [27, 57]. A study has shown that high glucose induces neuronal mitochondrial dysfunction, and subsequent mitochondrial dysfunction leads to impairment of the AMPK/Akt pathway, which is part of the insulin pathway and may lead to insulin resistance [57].
Insulin signaling has important implications for brain mitochondrial function [58, 59]. PI3-K/Akt can activate glucose transporter 3 to promote glucose uptake in neurons, and induce hexokinase II to bind to the mitochondrial outer membrane to promote glycolysis [60,61,62]. Pyruvate produced by glycolysis enters the mitochondria and is converted into acetyl-CoA as a substrate for the TCA Cycle. In addition, the interaction between Akt and hexokinase may inhibit the release of cytochrome c and maintain the structural and functional integrity of mitochondria, thus inhibiting neuronal apoptosis [63, 64]. In addition, Akt can also regulate mitochondrial biogenesis by regulating PGC-1α expression via mTOR to control mitochondrial oxidation [65, 66]. insulin signalling can also inhibit forkhead box O 1 (FOXO1) to inhibit the expression of heme oxygenase-1, which oxidizes heme to bilirubin and free Fe3+ to affect the activity of mitochondrial electron transport chain (mETC) and reduce NAD/NADH ratio and ATP production [59, 67]. Indeed, mitochondrial dysfunction, including reduced mitochondrial membrane potential, decreased mETC activity, reduced ATP production, and increased ROS, have been observed in T2DM and in insulin-resistant brains, while improved insulin signaling reversed these changes and improved synaptic plasticity and cognitive function [25, 68,69,70].
Neurodegenerative mechanism may work synergistically with T2DM to damage brain mitochondrial structure and function and cognition. During the development of brain insulin resistance, ceramide promote cleavage of amyloid beta precursor protein (APP) by β and γ-secretase to produce Aβ, which is the pathogenic molecule of AD [71, 72]. Meanwhile, high insulin levels in T2DM circulation can compete with Aβ for binding to insulin-degrading enzymes, reducing Aβ degradation [73]. Abnormal Aβ production and clearance will lead to its excessive accumulation in the brain. In turn, accumulated Aβ can compete with insulin for binding to the insulin receptor, reduce insulin receptor autophosphorylation, and decrease the affinity of insulin to its receptors, leading to disruption of insulin signaling [74]. Abeta oligomer also activates JNK leading to IRS-1 phosphorylation and degradation [75]. Therefore, a vicious circle may exist between insulin resistance and AD pathology. Elevated Aβ accumulates in synaptic mitochondria prior to extracellular accumulation, inhibiting mitochondrial respiration and biogenesis, resulting in overproduction of ROS, impaired mETC function, and altered calcium homeostasis [76,77,78,79].The increase of ROS in turn increases APP processing and Aβ production [80, 81].
Mitochondria are cellular energy factories. Cognitive function depends on the activity of neurons and synaptic connections, including the generation of action potentials, vesicle circulation, and neurotransmitter release [82, 83]. The high energy requirements generated by these processes and limited glycolysis capacity cause neurons to be extremely dependent on mitochondria [84]. Both insulin resistance and accumulation of Aβ lead to mitochondrial dysfunction, affecting energy supply to brain neurons, resulting in failure of neuronal metabolic control and promoting neurodegeneration [85,86,87]. In addition, the brain is highly vulnerable to oxidative stress due to its high rate of oxygen consumption and high levels of polyunsaturated fatty acids, coupled with low activity of antioxidant enzymes and high levels of pro-oxidative metal ions (such as Fe2+) [88, 89]. Mitochondria are also a major source and target of ROS, the initial form of ROS being superoxide (O2·−), which is later converted to hydrogen peroxide (H2O2). Mitochondrial dysfunction produces excessive ROS, reduces mETC activity and ATP synthesis [90]. Meanwhile, mitochondrial DNA (mtDNA) encoding respiratory chain complexes is susceptible to ROS, resulting in oxidative damage and mutation of mtDNA, which further damages the function of mETC and aggravates energy failure and oxidative stress [91]. In T2DM, enhanced mitochondrial ROS levels have also been observed to activate the apoptotic cascade by triggering the release of cytochrome c, leading to neuronal apoptosis and impaired cognition [92, 93]. In T2DM, oxidative stress also induces a novel form of iron-mediated cell death via phospholipid peroxidation, ferroptosis. In hippocampal neurons of db/db mice, upregulation of transferrin receptor 1 levels and decreased levels of ferroportin-1 and Ferritin heavy chain, decreased expression of mitochondrial ferritin and increased expression of mitoferrin were observed, suggesting hippocampal neuronal and mitochondrial iron overload [94, 95]. Excess Fe2+ can react with H2O2 generated to generate hydroxyl radicals (•OH) with stronger oxidative ability through Fenton reaction, and undergo lipid peroxidation reaction with unsaturated fatty acids [96]. Elevated mitochondrial ROS and decreased glutathione peroxidases activity lead to accumulation of lipid peroxides, which trigger ferroptosis and cognitive deficits in hippocampal neurons in T2DM and AD [94, 97].
Given the complex links between mitochondrial dysfunction and insulin resistance, impaired energy metabolism, accumulation of Aβ, and oxidative stress, mitochondrial dysfunction may be a bridge between T2DM and AD, leading to cognitive impairment.
Mitochondrial Protein Quality Control
In humans, only 13 proteins involving the subunit of respiratory chain complexes are encoded by the mitochondrial genome, while the remaining 1500 proteins are encoded by nuclear DNA [98]. Mitochondrial-encoded proteins can be inserted co-translationally into the inner membrane via the oxidase assembly protein complex [99, 100]. Precursor proteins encoded by nuclear DNA are produced on cytoplasmic ribosomes and subsequently imported into mitochondria in unfolded form with the help of molecular chaperones [101, 102]. With the support of mitochondrial membrane potential, precursor proteins are transported to the mitochondrial matrix through the translocase of the outer membrane complex on the outer mitochondrial membrane (OMM) and translocase of the inner membrane complex on the inner mitochondrial membrane (IMM) [103,104,105,106]. Precursor proteins entering the matrix are processed by mitochondrial processing peptidase, and then the molecular chaperones assists in folding imported proteins [107,108,109,110]. Misfolded or redundant proteins are degraded by ATP-dependent proteases [111,112,113,114]. When the accumulation of unfolded or misfolded proteins exceeds the cleaning capacity of mitochondria, the mitochondrial unfolded protein response (UPRmt) is induced [115]. In UPRmt, signals released from mitochondria trigger transcription of nuclear genes that encode mitochondrial chaperones and proteases to prevent harmful proteins from accumulating in the mitochondria [116,117,118]. Figure 1 illustrates the process of mitochondrial protein quality control.

Mitochondrial protein quality control. Mitochondrial proteins encoded by nuclear genes are produced in the cytoplasmic ribosome, and molecular chaperones keep the precursor proteins unfolded. Precursor proteins pass through and enter the mitochondria through the TOM and TIM complexs on the OMM. After the precursor protein that enters the matrix is processed by MPP, the mitochondrial chaperones folds it to maturity. Misfolded proteins can be hydrolyzed by proteases. Mitochondrial-encoded proteins can be co-translationally inserted into the IMM via OXA1. TOM translocases of the outer mitochondrial membrane, TIM translocase of the inner membrane, OMM outer mitochondrial membrane, IMS intermembrane space, IMM inner mitochondrial membrane, MPP mitochondrial processing peptidase, OXA1 oxidase assembly protein 1
Limited evidence shows defects in the quality control of mitochondrial proteins in diabetes. The levels of mitochondrial protease (Lon Peptidase 1) and mitochondrial chaperones (heat shock protein 60 and 70) were significantly reduced in the brain of T2DM mice, suggesting a deficiency of UPRmt [119, 120]. Metformin can induce UPRmt and significantly improve the function of brain mitochondria in T2DM mice [120]. In addition, the increased expression of heat shock protein 70 in the brain can improve insulin sensitivity and glycemic control [121]. Decreased mitochondrial chaperones heat shock protein 60 and 10 in the hypothalamus of T2DM lead to mitochondrial dysfunction and trigger neuronal insulin resistance, suggesting that defects in mitochondrial protein quality control may play an important role in the development of insulin resistance [122, 123]. However, the link between mitochondrial protein quality control system and cognitive impairment in T2DM remains largely unknown.
Mitochondrial Biogenesis
Mitochondrial biogenesis is a compensatory response secondary to the damaged respiratory apparatus and low ATP production, aiming to replenish mitochondrial components. In neurons, mitochondrial biogenesis mainly occurs in soma [124]. Peroxisome proliferator-activated receptor gamma coactivator-1-alpha (PGC-1α) is regarded as the core of transcriptional control of mitochondrial biogenesis, activated by sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK) induced deacetylation and phosphorylation, respectively [125]. It can augment the expression and activity of several critical transcription factors, including nuclear respiratory factor 1 (NRF1) and nuclear factor erythroid 2-related factor 2 (NRF2), peroxisome proliferator-activated receptor-α, oestrogen-related receptor-α and transcriptional repressor protein YY1 [126, 127]. Recent studies have found that NRF2 transcriptionally not only increases mitochondrial biogenesis, but also regulates mitochondrial genes in cooperation with PGC-1α. NRF2 can bind to PGC-1α to enhance gene induction of NRF1 and mitochondrial transcription factor A (TFAM), which is a key enhancer protein ensuring mtDNA replication by mtDNA polymerase γ [128,129,130]. These transcription factors can improve mitochondrial function and against oxidative stress and inflammation by regulating the expression of related proteins [131, 132]. Figure 2 illustrates the process of mitochondrial biogenesis.

Mitochondrial biogenesis. Mitochondrial biogenesis is mainly regulated by PGC1-α. PGC1-α activates the expression of NRF1/2, thereby enhancing the expression of TFAM and promoting mitochondrial DNA replication and transcription. PGC1-α peroxisome proliferator-activated receptor-γ co-activator 1α, NRF1 nuclear respiratory factor 1, NRF2 nuclear factor erythroid 2-related factor 2, TFAM transcription factor A
The study showed that the expression of SIRT1 and PGC-1α in the brain of T2DM mice decreased significantly [120]. Decreased PGC-1α expression and activation in hippocampal neurons of T2DM mice leads to blocked mitochondrial biogenesis and mitochondrial dysfunction, triggers neuronal loss, and promotes cognitive impairment in diabetes [133, 134]. Downregulation of PGC-1α in the hippocampus of T2DM may result from dipeptidyl peptidase-4 binding to protease-activated receptor 2 and triggering glycogen synthase kinase-3β (GSK3β) activation [133]. Insulin signaling has also been found to be involved in regulating PGC-1α expression, affecting mitochondrial biogenesis [135, 136]. It was found that hyperinsulinemia caused by T2DM activates the hyperactivation of the insulin signaling factor Akt in the anterior cortex and hippocampus, resulting in the phosphorylation inactivation of FOXOs and subsequent reduction of PGC-1α, and accumulation of Aβ [137]. Interestingly, another study in UCD-T2DM rats showed that with the progression of brain insulin resistance, AMPK phosphorylation and SIRT levels in hippocampal neurons decreased, and mitochondrial biogenesis-related PGC-1α and TFAM expressions were significantly decreased, leading to increased lipid peroxidation and decreased synaptic plasticity in hippocampal neurons [138]. A study using primary rat cortical neurons also showed that palmitate induces neuronal insulin resistance and suppresses PGC-1α expression, contributing to mitochondrial dysfunction and decreased cell viability [139]. These studies suggest that PGC-1α has beneficial effects on diabetic brain neurons, and impaired insulin signaling may induce and exacerbate neuronal damage through inhibition of PGC-1α.
Mitochondrial Dynamics
Mitochondria are highly dynamic organelles that continually undergo fusion and fission [140, 141]. Mitochondrial fusion promotes mixing of membranes and contents between mitochondria to supplement oxidative damage components, safeguard mtDNA integrity and preserve mtDNA function in the face of mutations [142, 143]. Mitochondrial fission contributes to the even partitioning of mitochondria to daughter cells during mitosis, and separation of damaged mitochondria for autophagic degradation [144, 145]. Two classes of dynamin-like protein are involved in mitochondrial fusion, including mitofusin (MFN) and optic atrophy 1 (OPA1). MFN mediate IMM fusion. MFN1 and MFN2 on the OMM of two adjacent mitochondria form both homooligomeric and heterooligomeric complexes to promote mitochondrial fusion, which depends on GTP hydrolysis, and can be mediated by GTPase domain [146, 147]. The fusion of IMM is mediated by OPA1. OPA1 is encoded by nuclear genes and introduced into mitochondria through the mitochondria protein quality control system [148]. The OPA1 entering the mitochondria is hydrolyzed by protease in the matrix to form long isoform and short isoform OPA1 [149]. the long isoform OPA1 is anchored on the IMM, and the short isoform OPA1 regulates the fusion activity by forming a complex with the long isoform OPA1 [146, 150]. In addition, OPA1 is also involved in the formation of mitochondrial cristae [151].
The key to mitochondrial fission is dynamin-related protein 1 (Drp1). Most mitochondrial fission occurs where ER tubules crossing or wrapping around. During fission, endoplasmic reticulum (ER) tubules mark sites of mitochondrial division and mediated constriction [152]. Then, Drp1 is recruited from the cytosol to the OMM at ER tubules mark sites by its receptors fission protein 1 (Fis1), mitochondrial fission factor (MFF), mitochondrial elongation factor 2 and mitochondrial elongation factor 1 [153]. Researchs suggest that although they all have the ability to recruit Drp1 to mitochondria alone, MFF and MID seem to be more important that their interplay could regulate Drp1 to promote mitochondrial fission [154, 155]. The recruited Drp1 molecules assemble into a ring-like structure to constricts and cleaves mitochondria by GTP hydrolysis [156]. Dynamin 2 also found to be involved in mitochondrial fission, but not necessary [157]. Figure 3 illustrates the process of mitochondrial fusion and fission.

Mitochondrial fusion and fission. MFN1/2 interaction on adjacent mitochondria regulates OMM fusion. OPA1 mediates the fusion of IMM. Drp1 is recruited from the cytoplasm to the cleavage site of the OMM through its receptor, where it forms a ring-like structure to cleave mitochondria. MFN1/2 mitofusin ½, Drp1 dynamin-1-like protein
In the hippocampus of type 2 diabetic mice and high glucose cultured human SK cells, GSK3β was activated to promote the significant expression and mitochondrial translocation of Drp1, which exacerbated mitochondrial fission and subsequently damaged the morphology and function of mitochondria [30]. Interestingly, levels of Mfn1, Mfn2, OPA1 were not altered in diabetic hippocampus, which was consistent with some research results [30, 69, 158]. However, some studies report that Opa1 was reduced in the cortex of GK mice with T2DM [159]. Knockout or inhibition of Drp1 significantly improves mitochondrial mass and reduces diabetes-induced hippocampal synaptic damage [30, 133]. However, it has been found that hippocampal neurons in neuron-specific Drp1-deficient T2DM mice exhibited marked mitochondrial dysfunction and synaptic damage, and higher levels of oxidative stress and neuroinflammation, which may be due to the fact that Drp1 knockdown inhibits mitochondrial fission and impairs the autophagy process [160]. In the hippocampal neurons of T2DM mice and PC12 cells cultured with high glucose, it was further found that promoting FUNDC1-mediated mitophagy can eliminate mitochondrial fragmentation caused by overactivated Drp1, reduce mitochondria-derived apoptosis, and thus alleviate diabetic cognitive impairment [161]. In addition, in vitro studies have shown that high glucose increases nitric oxide in cortical and hippocampal neurons in an N-methyl-D-aspartate receptor-dependent manner, leading to S-nitrosylation of Drp1, which leads to excessive mitochondrial division, impairs neuronal energy generation, and leads to synaptic loss and reduced plasticity [162]. These studies suggest that mitochondrial fragmentation owing to a loss of mitochondrial dynamics has a key role in the progression of cognitive impairment in diabetes.
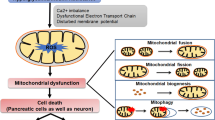
Mitophagy
Mitophagy is a selective form of autophagy, that mediates the removal of defective and superfluous mitochondria. Mitophagy can promote mitochondrial turnover, avoid the accumulation of damaged mitochondria that can lead to cell degeneration, and adjust mitochondrial numbers to meet the energy demand [163]. Defects in mitophagy have been implicated in a variety of neurodegenerative diseases [164]. The process of mitophagy includes the detection and separation of damaged mitochondria, the recruitment of phagosomes and subsequent autophagic degradation [165]. Figure 4 illustrates the process of mitophagy.

Mitophagy. ULK1 is activated by AMPK, induces nucleation of the phagophore by phosphorylating beclin 1 and activating Vps34 complex. Atg12-Atg5-Atg16 and LC3 further mediate autophagosome formation. LC3-I is processed into LC3-II and incorporated into the autophagosome membrane. Decreased mitochondrial membrane potential inhibits PINK1 entry into mitochondria, leading to PINK1 accumulation on OMM. Then, PINK1 recruits parkin from the cytoplasm to catalyze the formation of polyubiquitin chains on OMM proteins, which are then recognized by autophagy receptor proteins to form mitophagosome. Among other pathways of mitophagy, BNIP3, NIX and FUNDC1 in the OMM can directly bridge mitochondria to autophagosomes by interacting with LC3-II. ULK1 Unc-51 Like Autophagy Activating Kinase 1, AMPK AMP-activated protein kinase, Vps34 class III phosphatidylinositol 3-kinase, LC3 light chain 3, PINK1 PTEN- induced kinase 1, BNIP3 BCL2/Adenovirus E1B 19 kDa Interacting Protein 3, NIX Nip3-like protein X, FUNDC1 FUN14 domain containing 1
Like other forms of autophagy, mitophagy is activated by phosphorylation of the Unc-51 like autophagy activating kinase 1 (ULK1) complex, typically by AMPK [166]. ULK1 induces nucleation of the phagophore by phosphorylating beclin 1 and activating class III PI3K complex [167]. The phagophore membrane extends and completely engulfs the target mitochondria and matures into an autophagosome mediated by Atg12-Atg5-Atg16 and LC3/Atg8 systems. During this period, the microtubule-associated protein 1 light chain 3 (LC3-I) is processed to LC3-II that is incorporated into the double membranes of the autophagosomes [168]. Then, the autophagosomes fuses with the lysosomes, resulting in the degradation of its cargo by acid hydrolases [169]. Several pathways have been found to detect target mitochondria and recruit autophagosomes for degradation to achieve the selectivity and specificity of mitophagy.
PINK1/Parkin-mediated mitophagy is the most well-known pathway. PTEN- induced kinase 1 (PINK1) is regulated by mitochondrial protein quality control mechanisms [99]. Under normal circumstances, PINK1 is introduced into IMM through TOM and TIM complexes, where it is cleaved by IMM protease presenilin-associated rhomboid-like, and then it is released into cytoplasm and degraded by proteasome [170]. The decrease of the membrane potential during mitochondrial dysfunction inhibits the entry of PINK1 into IMM through TIM complexes, resulting in the accumulation of PINK1 on the OMM.
On the mitochondrial surface, PINK1 recruits and phosphorylates Parkin to relieve its auto inhibited state and increase its E3 ligase activity [171]. PINK1 also phosphorylates Ubiquitin. Activated Parkin ubiquitylates multiple OMM proteins builds poly-ubiquitin chains on the OMM proteins, which in turn recruit autophagy receptor proteins, including p62, Next to BRCA1 gene 1 (NBR1), Nuclear domain 10 protein 52, Optineurin and Tax 1 binding protein 1 [172]. Autophagy receptor proteins simultaneously bind poly-ubiquitin chains in mitochondria through their ubiquitin binding domains, and LC3 on autophagosome membranes, which promotes the target mitochondria to be engulfed by autophagosomes, then autophagosomes fuse with lysosomes and lysosomal hydrolases degrade polyubiquitinated mitochondria [172, 173].
In addition to the classic PINK1/Parkin-related mitophagy, other autophagy receptors have also been found to regulate mitophagy, including Nip3-like protein X (NIX), BCL2/Adenovirus E1B 19 kDa Interacting Protein 3 (BNIP3), FUN14 domain containing 1 (FUNDC1). Under hypoxia, the expression of BNIP3 can significantly up regulate through hypoxia-inducing factor-1α, the inactive monomer BNIP3 in the cytosol forms a stable homodimer and is anchored to the OMM via its C-terminal transmembrane domain [174, 175]. The homodimer of BNIP3 can interact with LC3, which is further regulated by Ser17 and Ser24 phosphorylation near the LIR motif [176]. BNIP3 and NIX are proteins with homology to BCL2 in the BH3 domain. Similar to BNIP3, NIX is integrated into the OMM through dimerization and then binds to LC3 [177]. Several studies have reported that BNIP3 and NIX are also involved in PINK1/Parkin mediated mitophagy. NIX is ubiquitylated by Parkin, which promotes the recruitment of NBR1 to mitochondria [178]. BNIP3 can inhibit PINK1 proteolysis and promote its accumulation on OMM [179]. Of note, BNIP3 and NIX can also induce cell death [180]. FUNDC1 is an OMM protein that mediates hypoxia-induced Parkin-independent mitophagy by directly binging to LC3 [175, 181]. Under basal conditions, the activity of DC1 binding to LC3 is inhibited by phosphorylation of casein kinase 2 at serine 13 and SRC kinase at tyrosine 18 [182]. When encountering hypoxia, FUNDC1 is dephosphorylated by phosphoglycerate mutase family member 5 (PGAM5) at serine 13 and phosphorylated by ULK1 at serine 17, which increases interaction with LC3 to promote mitophagy [183, 184]. In addition, FUNDC1 interacts with OPA1 under normal conditions, while under mitochondrial stress, this interaction is reduced and promotes Drp1 recruitment to mitochondria [185]. It further reveals the “coupling” mechanism between mitochondrial dynamics and mitophagy.
In vitro models of diabetes, mitophagy of SK-N-MC and SH-SY5Y cells is significantly triggered in response to mitochondrial dysfunction and apoptosis induced by high glucose. The mitophagy seems to depend on PINK1 rather than BNIP3 or NIX, and has the effect of protecting neuronal cells. Melatonin could enhance PINK1-dependent mitophagy via the MT2/Akt/NF-κB pathway, thereby preventing ROS accumulation and antiapoptosis in neuronal cells under high glucose conditions [186]. However, some research found that LC3-II and p62 increased and PINK1 decreased in the midbrain of diabetic mice, indicating that autophagic flux is blocked. This was also seen in neuron-like PC12 cells cultured in high glucose. High glucose significantly blocked autophagic flux and inhibited PINK1/Parkin mediated mitophagy to reduced the viability of PC12 cells [187]. Significantly, these studies suggest that the enhancement of PINK1/Parkin mediated mitophagy is an important way to rescue neuronal cells in diabetes [186, 187]. Recent studies have also found the role of FUNDC1 related mitophagy in diabetic cognitive impairments. In T2DM mice, the dephosphorylation of FUNDC1 was inhibited, which promoted oxidative stress and neuroinflammation, resulting in apoptosis of hippocampal neurons. Similarly, the destruction of autophagic flux and the inhibition of FUNDC1-dependent mitophagy induced by high glucose exacerbated the apoptosis of PC12 cells. Activation of the Rac1/ROS axis appears to be an effective approach to prevent hyperglycemia-induced neurotoxicity by modulating FUNDC1-dependent mitophagy [161].
Conclusion and Future Directions
Mitochondrial quality control is critical for the homeostasis of the mitochondrial network. The damage to multiple control mechanisms, such as imbalanced mitochondrial dynamics, impaired mitophagy and proteostasis disorder, was observed in diabetic cognitive impairment. However, the relative contribution of each dysregulated mechanism to cognitive impairment in diabetes is still unclear. In addition, because the mitochondrial quality control mechanism is a complex integrated hierarchical network of pathways, the changes of different quality control mechanisms can affect each other, and then alter the results of quality control. For example, proteins produced by mitochondrial biogenesis require the mitochondrial protein quality control system for import into mitochondria and proper assembly, and inhibition of mitochondrial fission might damage mitophagy and mitochondria biogenesis. Although current studies have noted that diabetes could affect interactions between mitochondrial quality control mechanisms, the functional consequences of these interactions are not fully understood and requires further experiments to determine the exact nature of their interplay. In conclusion, dissecting the mitochondrial quality control mechanisms and their interaction might be exploited to unveil new pathways for the prevention and treatment of diabetic cognitive impairment.
Data Availability
The datasets generated during or analysed during the current study are available from the corresponding author on reasonable request.
References
Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, Pavkov ME, Ramachandaran A, Wild SH, James S, Herman WH, Zhang P, Bommer C, Kuo S, Boyko EJ, Magliano DJ (2021) IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 183(10267):109119. https://doi.org/10.1016/j.diabres.2021.109119
Chatterjee S, Khunti K, Davies MJ (2017) Type 2 diabetes. Lancet 389(10085):2239–2251. https://doi.org/10.1016/S0140-6736(17)30058-2
Wang T, Lu J, Shi L, Chen G, Xu M, Xu Y, Su Q, Mu Y, Chen L, Hu R, Tang X, Yu X, Li M, Zhao Z, Chen Y, Yan L, Qin G, Wan Q, Dai M, Zhang D, Gao Z, Wang G, Shen F, Luo Z, Qin Y, Chen L, Huo Y, Li Q, Ye Z, Zhang Y, Liu C, Wang Y, Wu S, Yang T, Deng H, Zhao J, Lai S, Bi Y, DeFronzo RA, Wang W, Ning G, China Cardiometabolic Disease and Cancer Cohort Study Group (2020) Association of insulin resistance and β-cell dysfunction with incident diabetes among adults in China: a nationwide, population-based, prospective cohort study. Lancet Diabetes Endocrinol 8(2):115–124. https://doi.org/10.1016/S2213-8587(19)30425-5
Hudish LI, Reusch JE, Sussel L (2019) β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest 129(10):4001–4008. https://doi.org/10.1172/JCI129188
Moheet A, Mangia S, Seaquist ER (2015) Impact of diabetes on cognitive function and brain structure. Ann N Y Acad Sci 1353:60–71. https://doi.org/10.1111/nyas.12807
García-Casares N, Jorge RE, García-Arnés JA, Acion L, Berthier ML, Gonzalez-Alegre P, Nabrozidis A, Gutiérrez A, Ariza MJ, Rioja J, González-Santos P (2014) Cognitive dysfunctions in middle-aged type 2 diabetic patients and neuroimaging correlations: a cross-sectional study. J Alzheimers Dis 42(4):1337–1346. https://doi.org/10.3233/JAD-140702
Moran C, Phan TG, Chen J, Blizzard L, Beare R, Venn A, Münch G, Wood AG, Forbes J, Greenaway TM, Pearson S, Srikanth V (2013) Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care 36(12):4036–4042. https://doi.org/10.2337/dc13-0143
Ninomiya T (2019) Epidemiological evidence of the relationship between diabetes and dementia. Adv Exp Med Biol 1128:13–25. https://doi.org/10.1007/978-981-13-3540-2_2
Zhang J, Chen C, Hua S, Liao H, Wang M, Xiong Y, Cao F (2017) An updated meta-analysis of cohort studies: diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract 124:41–47. https://doi.org/10.1016/j.diabres.2016.10.024
Gudala K, Bansal D, Schifano F, Bhansali A (2013) Diabetes mellitus and risk of dementia: a meta-analysis of prospective observational studies. J Diabetes Investig 4(6):640–650. https://doi.org/10.1111/jdi.12087
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S (2011) Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol 68(1):51–57. https://doi.org/10.1001/archneurol.2010.225
Pomytkin I, Costa-Nunes JP, Kasatkin V, Veniaminova E, Demchenko A, Lyundup A, Lesch KP, Ponomarev ED, Strekalova T (2018) Insulin receptor in the brain: mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci Ther 24(9):763–774. https://doi.org/10.1111/cns.12866
Cai HY, Wang ZJ, Hölscher C, Yuan L, Zhang J, Sun P, Li J, Yang W, Wu MN, Qi JS (2017) Lixisenatide attenuates the detrimental effects of amyloid β protein on spatial working memory and hippocampal neurons in rats. Behav Brain Res 318:28–35. https://doi.org/10.1016/j.bbr.2016.10.033
Bedse G, Di Domenico F, Serviddio G, Cassano T (2015) Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci 9:204. https://doi.org/10.3389/fnins.2015.00204
Ng RC, Cheng OY, Jian M, Kwan JS, Ho PW, Cheng KK, Yeung PK, Zhou LL, Hoo RL, Chung SK, Xu A, Lam KS, Chan KH (2016) Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol Neurodegener 11(1):71. https://doi.org/10.1186/s13024-016-0136-x
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis 7(1):63–80. https://doi.org/10.3233/jad-2005-7107
de la Monte SM (2012) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 9(1):35–66. https://doi.org/10.2174/156720512799015037
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM (2005) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis 8(3):247–268. https://doi.org/10.3233/jad-2005-8304
Rajasekar N, Nath C, Hanif K, Shukla R (2017) Intranasal insulin administration ameliorates streptozotocin (ICV)-induced insulin receptor dysfunction, neuroinflammation, amyloidogenesis, and memory impairment in rats. Mol Neurobiol 54(8):6507–6522. https://doi.org/10.1007/s12035-016-0169-8
Guo Z, Chen Y, Mao YF, Zheng T, Jiang Y, Yan Y, Yin X, Zhang B (2017) Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Sci Rep 7:45971. https://doi.org/10.1038/srep45971
Avgerinos KI, Kalaitzidis G, Malli A, Kalaitzoglou D, Myserlis PG, Lioutas VA (2018) Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: a systematic review. J Neurol 265(7):1497–1510. https://doi.org/10.1007/s00415-018-8768-0
Gundelfinger ED, tomDieck S (2000) Molecular organization of excitatory chemical synapses in the mammalian brain. Naturwissenschaften 87(12):513–523. https://doi.org/10.1007/s001140050770
Rangaraju V, Lauterbach M, Schuman EM (2019) Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 176(1–2):73-84.e15. https://doi.org/10.1016/j.cell.2018.12.013
Carvalho C, Cardoso S (2021) Diabetes-Alzheimer’s disease link: targeting mitochondrial dysfunction and redox imbalance. Antioxid Redox Signal 34(8):631–649. https://doi.org/10.1089/ars.2020.8056
Ruegsegger GN, Manjunatha S, Summer P, Gopala S, Zabeilski P, Dasari S, Vanderboom PM, Lanza IR, Klaus KA, Nair KS (2019) Insulin deficiency and intranasal insulin alter brain mitochondrial function: a potential factor for dementia in diabetes. FASEB J 33(3):4458–4472. https://doi.org/10.1096/fj.201802043R
Lee HJ, Jung YH, Choi GE, Kim JS, Chae CW, Lim JR, Kim SY, Yoon JH, Cho JH, Lee SJ, Han HJ (2021) Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ 28(1):184–202. https://doi.org/10.1038/s41418-020-0593-1
Sripetchwandee J, Chattipakorn N, Chattipakorn SC (2018) Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia. Front Endocrinol 9:496. https://doi.org/10.3389/fendo.2018.00496
Pipatpiboon N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC (2012) PPARγ agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology 153(1):329–338. https://doi.org/10.1210/en.2011-1502
Raza H, John A, Howarth FC (2015) Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell Physiol Biochem 35(3):1241–1251. https://doi.org/10.1159/000373947
Huang S, Wang Y, Gan X, Fang D, Zhong C, Wu L, Hu G, Sosunov AA, McKhann GM, Yu H, Yan SS (2015) Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes 64(5):1728–1742. https://doi.org/10.2337/db14-0758
Wang D, Yan J, Chen J, Wu W, Zhu X, Wang Y (2015) Naringin improves neuronal insulin signaling, brain mitochondrial function, and cognitive function in high-fat diet-induced obese mice. Cell Mol Neurobiol 35(7):1061–1071. https://doi.org/10.1007/s10571-015-0201-y
Choi J, Ravipati A, Nimmagadda V, Schubert M, Castellani RJ, Russell JW (2014) Potential roles of PINK1 for increased PGC-1α-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 18:41–48. https://doi.org/10.1016/j.mito.2014.09.005
Song M, Zhao X, Song F (2021) Aging-dependent mitophagy dysfunction in Alzheimer’s disease. Mol Neurobiol 58(5):2362–2378. https://doi.org/10.1007/s12035-020-02248-y
Ng MYW, Wai T, Simonsen A (2021) Quality control of the mitochondrion. Dev Cell 56(7):881–905. https://doi.org/10.1016/j.devcel.2021.02.009
Maity S, Chakrabarti O (2021) Mitochondrial protein import as a quality control sensor. Biol Cell 113(9):375–400. https://doi.org/10.1111/boc.202100002
Lenkiewicz AM, Krakowczyk M, Bragoszewski P (2021) Cytosolic quality control of mitochondrial protein precursors—the early stages of the organelle biogenesis. Int J Mol Sci 23(1):7. https://doi.org/10.3390/ijms23010007
Shin CS, Meng S, Garbis SD, Moradian A, Taylor RW, Sweredoski MJ, Lomenick B, Chan DC (2021) LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat Commun 12(1):265. https://doi.org/10.1038/s41467-020-20597-z
Matsushima Y, Takahashi K, Yue S, Fujiyoshi Y, Yoshioka H, Aihara M, Setoyama D, Uchiumi T, Fukuchi S, Kang D (2021) Mitochondrial Lon protease is a gatekeeper for proteins newly imported into the matrix. Commun Biol 4(1):974. https://doi.org/10.1038/s42003-021-02498-z
Pickles S, Vigié P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28(4):R170–R185. https://doi.org/10.1016/j.cub.2018.01.004
Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnauné-Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P (2016) Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis 90:3–19. https://doi.org/10.1016/j.nbd.2015.10.011
Khaleel EF, Abdel-Aleem GA, Mostafa DG (2018) Resveratrol improves high-fat diet induced fatty liver and insulin resistance by concomitantly inhibiting proteolytic cleavage of sterol regulatory element-binding proteins, free fatty acid oxidation, and intestinal triglyceride absorption. Can J Physiol Pharmacol 96(2):145–157. https://doi.org/10.1139/cjpp-2017-0001
Zheng F, Cai Y (2019) Concurrent exercise improves insulin resistance and nonalcoholic fatty liver disease by upregulating PPAR-γ and genes involved in the beta-oxidation of fatty acids in ApoE-KO mice fed a high-fat diet. Lipids Health Dis 18(1):6. https://doi.org/10.1186/s12944-018-0933-z
Rico JE, Giesy SL, Haughey NJ, Boisclair YR, McFadden JW (2018) Intravenous triacylglycerol infusion promotes ceramide accumulation and hepatic steatosis in dairy cows. J Nutr 148(10):1529–1535. https://doi.org/10.1093/jn/nxy155
Rico JE, Mathews AT, Lovett J, Haughey NJ, McFadden JW (2016) Palmitic acid feeding increases ceramide supply in association with increased milk yield, circulating nonesterified fatty acids, and adipose tissue responsiveness to a glucose challenge. J Dairy Sci 99(11):8817–8830. https://doi.org/10.3168/jds.2016-11296
Appriou Z, Nay K, Pierre N, Saligaut D, Lefeuvre-Orfila L, Martin B, Cavey T, Ropert M, Loréal O, Rannou-Bekono F, Derbré F (2019) Skeletal muscle ceramides do not contribute to physical-inactivity-induced insulin resistance. Appl Physiol Nutr Metab 44(11):1180–1188. https://doi.org/10.1139/apnm-2018-0850
Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, Wilkerson JL, Sweeney CR, Pereira RF, Sumida DH, Maschek JA, Cox JE, Kaddai V, Lancaster GI, Siddique MM, Poss A, Pearson M, Satapati S, Zhou H, McLaren DG, Previs SF, Chen Y, Qian Y, Petrov A, Wu M, Shen X, Yao J, Nunes CN, Howard AD, Wang L, Erion MD, Rutter J, Holland WL, Kelley DE, Summers SA (2019) Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365(6451):386–392. https://doi.org/10.1126/science.aav3722
Yazıcı D, Sezer H (2017) Insulin resistance, obesity and lipotoxicity. Adv Exp Med Biol 960:277–304. https://doi.org/10.1007/978-3-319-48382-5_12
de la Monte SM (2012) Triangulated mal-signaling in Alzheimer’s disease: roles of neurotoxic ceramides, ER stress, and insulin resistance reviewed. J Alzheimers Dis 30(Suppl 2):S231–S249. https://doi.org/10.3233/JAD-2012-111727
Maciejczyk M, Żebrowska E, Chabowski A (2019) Insulin resistance and oxidative stress in the brain: what’s new? Int J Mol Sci 20(4):874. https://doi.org/10.3390/ijms20040874
Lyn-Cook LE Jr, Lawton M, Tong M, Silbermann E, Longato L, Jiao P, Mark P, Wands JR, Xu H, de la Monte SM (2009) Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J Alzheimers Dis 16(4):715–729. https://doi.org/10.3233/JAD-2009-0984
de la Monte SM, Tong M, Nguyen V, Setshedi M, Longato L, Wands JR (2010) Ceramide-mediated insulin resistance and impairment of cognitive-motor functions. J Alzheimers Dis 21(3):967–984. https://doi.org/10.3233/JAD-2010-091726
Signorelli P, Conte C, Albi E (2021) The multiple roles of sphingomyelin in Parkinson’s disease. Biomolecules 11(9):1311. https://doi.org/10.3390/biom11091311
Campana M, Bellini L, Rouch C, Rachdi L, Coant N, Butin N, Bandet CL, Philippe E, Meneyrol K, Kassis N, Dairou J, Hajduch E, Colsch B, Magnan C, Le Stunff H (2018) Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances β-cell function of obese Zucker rats. Mol Metab 8:23–36. https://doi.org/10.1016/j.molmet.2017.10.013
Akhtar A, Sah SP (2020) Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem Int 135:104707. https://doi.org/10.1016/j.neuint.2020.104707
Milstein JL, Ferris HA (2021) The brain as an insulin-sensitive metabolic organ. Mol Metab 52:101234. https://doi.org/10.1016/j.molmet.2021.101234
Arboleda G, Huang TJ, Waters C, Verkhratsky A, Fernyhough P, Gibson RM (2007) Insulin-like growth factor-1-dependent maintenance of neuronal metabolism through the phosphatidylinositol 3-kinase-Akt pathway is inhibited by C2-ceramide in CAD cells. Eur J Neurosci 25(10):3030–3038. https://doi.org/10.1111/j.1460-9568.2007.05557.x
Peng Y, Liu J, Shi L, Tang Y, Gao D, Long J, Liu J (2016) Mitochondrial dysfunction precedes depression of AMPK/AKT signaling in insulin resistance induced by high glucose in primary cortical neurons. J Neurochem 137(5):701–713. https://doi.org/10.1111/jnc.13563
Ruegsegger GN, Creo AL, Cortes TM, Dasari S, Nair KS (2018) Altered mitochondrial function in insulin-deficient and insulin-resistant states. J Clin Invest 128(9):3671–3681. https://doi.org/10.1172/JCI120843
Cheng Z, Tseng Y, White MF (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 21(10):589–598. https://doi.org/10.1016/j.tem.2010.06.005
Ni Y, Zhou Y, Zhou M, Zhang L (2015) Akt and cAMP response element binding protein mediate 17β-estradiol regulation of glucose transporter 3 expression in human SH-SY5Y neuroblastoma cell line. Neurosci Lett 604:58–63. https://doi.org/10.1016/j.neulet.2015.07.041
Fernie AR, Carrari F, Sweetlove LJ (2004) Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol 7(3):254–261. https://doi.org/10.1016/j.pbi.2004.03.007
Wu B, Luo H, Zhou X, Cheng CY, Lin L, Liu BL, Liu K, Li P, Yang H (2017) Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: therapeutical effects of kaempferol. Biochim Biophys Acta Mol Basis Dis 1863(9):2307–2318. https://doi.org/10.1016/j.bbadis.2017.06.011
Roberts DJ, Miyamoto S (2015) Hexokinase II integrates energy metabolism and cellular protection: akting on mitochondria and TORCing to autophagy. Cell Death Differ 22(2):248–257. https://doi.org/10.1038/cdd.2014.173
Li Y, Yang Y, Zhao Y, Zhang J, Liu B, Jiao S, Zhang X (2019) Astragaloside IV reduces neuronal apoptosis and parthanatos in ischemic injury by preserving mitochondrial hexokinase-II. Free Radic Biol Med 131:251–263. https://doi.org/10.1016/j.freeradbiomed.2018.11.033
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P (2007) mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450(7170):736–740. https://doi.org/10.1038/nature06322
Fu X, Li K, Niu Y, Lin Q, Liang H, Luo X, Liu L, Li N (2022) The mTOR/PGC-1α/SIRT3 pathway drives reductive glutamine metabolism to reduce oxidative stress caused by ISKNV in CPB cells. Microbiol Spectr 10(1):e0231021. https://doi.org/10.1128/spectrum.02310-21
Lin CC, Yang CC, Hsiao LD, Yang CM (2021) Carbon monoxide releasing molecule-3 enhances heme oxygenase-1 induction via ROS-dependent FoxO1 and Nrf2 in brain astrocytes. Oxid Med Cell Longev 8:1–22. https://doi.org/10.1155/2021/5521196
Beirami E, Oryan S, Seyedhosseini Tamijani SM, Ahmadiani A, Dargahi L (2018) Intranasal insulin treatment restores cognitive deficits and insulin signaling impairment induced by repeated methamphetamine exposure. J Cell Biochem 119(2):2345–2355. https://doi.org/10.1002/jcb.26398
Ruegsegger GN, Vanderboom PM, Dasari S, Klaus KA, Kabiraj P, McCarthy CB, Lucchinetti CF, Nair KS (2019) Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 4(18):e130681. https://doi.org/10.1172/jci.insight.130681
Wang D, Liu L, Li S, Wang C (2018) Effects of paeoniflorin on neurobehavior, oxidative stress, brain insulin signaling, and synaptic alterations in intracerebroventricular streptozotocin-induced cognitive impairment in mice. Physiol Behav 191:12–20. https://doi.org/10.1016/j.physbeh.2018.03.016
Takasugi N, Sasaki T, Shinohara M, Iwatsubo T, Tomita T (2015) Synthetic ceramide analogues increase amyloid-β 42 production by modulating γ-secretase activity. Biochem Biophys Res Commun 457(2):194–199. https://doi.org/10.1016/j.bbrc.2014.12.087
Jazvinšćak Jembrek M, Hof PR, Šimić G (2015) Ceramides in Alzheimer’s disease: key mediators of neuronal apoptosis induced by oxidative stress and Aβ accumulation. Oxid Med Cell Longev 2015:346783. https://doi.org/10.1155/2015/346783
Kurochkin IV, Guarnera E, Berezovsky IN (2018) Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends Pharmacol Sci 39(1):49–58. https://doi.org/10.1016/j.tips.2017.10.008
Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R (2002) Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci 22(10):RC221. https://doi.org/10.1523/JNEUROSCI.22-10-j0001.2002
Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, Chen PP, Hudspeth B, Chen C, Zhao Y, Vinters HV, Frautschy SA, Cole GM (2009) Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci 29(28):9078–9089. https://doi.org/10.1523/JNEUROSCI.1071-09.2009
Chen Z, Tao S, Li X, Zeng X, Zhang M, Yao Q (2019) Anagliptin protects neuronal cells against endogenous amyloid β (Aβ)-induced cytotoxicity and apoptosis. Artif Cells Nanomed Biotechnol 47(1):2213–2220. https://doi.org/10.1080/21691401.2019.1609979
Akhter F, Chen D, Yan SF, Yan SS (2017) Mitochondrial perturbation in Alzheimer’s disease and diabetes. Prog Mol Biol Transl Sci 146:341–361. https://doi.org/10.1016/bs.pmbts.2016.12.019
Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA (2002) Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80(1):91–100. https://doi.org/10.1046/j.0022-3042.2001.00681.x
Tong BC, Wu AJ, Li M, Cheung KH (2018) Calcium signaling in Alzheimer’s disease & therapies. Biochim Biophys Acta Mol Cell Res 11:1745–1760. https://doi.org/10.1016/j.bbamcr.2018.07.018
Pedrós I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, Auladell C, Pallàs M, Vázquez-Carrera M, Casadesús G, Folch J, Camins A (2014) Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta 1842(9):1556–1566. https://doi.org/10.1016/j.bbadis.2014.05.025
Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440(7083):528–534. https://doi.org/10.1038/nature04543
Gao Z, Wu R, Chen C, Wen B, Liu Y, Lu W, Chen N, Feng J, Fan R, Wang D, Cui S, Wang JH (2019) Coactivations of barrel and piriform cortices induce their mutual synapse innervations and recruit associative memory cells. Brain Res 1721:146333. https://doi.org/10.1016/j.brainres.2019.146333
Wu R, Cui S, Wang JH (2020) miRNA-324/-133a essential for recruiting new synapse innervations and associative memory cells in coactivated sensory cortices. Neurobiol Learn Mem 172:107246. https://doi.org/10.1016/j.nlm.2020.107246
Potenza MA, Sgarra L, Desantis V, Nacci C, Montagnani M (2021) Diabetes and Alzheimer’s disease: might mitochondrial dysfunction help deciphering the common path? Antioxidants 10(8):1257. https://doi.org/10.3390/antiox10081257
Sharma VK, Singh TG (2020) Insulin resistance and bioenergetic manifestations: targets and approaches in Alzheimer’s disease. Life Sci 262:118401. https://doi.org/10.1016/j.lfs.2020.118401
Bosco D, Fava A, Plastino M, Montalcini T, Pujia A (2011) Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med 15(9):1807–1821. https://doi.org/10.1111/j.1582-4934.2011.01318.x
Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S (2019) Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J Clin Invest 129(1):34–45. https://doi.org/10.1172/JCI120848
Wang X, Michaelis EK (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2:12. https://doi.org/10.3389/fnagi.2010.00012
Shanmugam KR, Mallikarjuna K, Kesireddy N, Sathyavelu Reddy K (2011) Neuroprotective effect of ginger on anti-oxidant enzymes in streptozotocin-induced diabetic rats. Food Chem Toxicol 49(4):893–897. https://doi.org/10.1016/j.fct.2010.12.013
Subramaniam SR, Chesselet MF (2013) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 106–107:17–32. https://doi.org/10.1016/j.pneurobio.2013.04.004
Kowluru RA, Kowluru A, Mishra M, Kumar B (2015) Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res 48:40–61. https://doi.org/10.1016/j.preteyeres.2015.05.001
Yang L, Han W, Luo Y, Hu X, Xu Y, Li H, Hu C, Huang D, Ma J, Yang Y, Chen Q, Li Y, Zhang J, Xia H, Chen Z, Wang H, Ran D, Yang J (2018) Adapentpronitrile, a new dipeptidyl peptidase-iv inhibitor, ameliorates diabetic neuronal injury through inhibiting mitochondria-related oxidative stress and apoptosis. Front Cell Neurosci 12:214. https://doi.org/10.3389/fncel.2018.00214
Wang G, Zhao Z, Ren B, Yu W, Zhang X, Liu J, Wang L, Si D, Yang M (2022) Exenatide exerts a neuroprotective effect against diabetic cognitive impairment in rats by inhibiting apoptosis: role of the JNK/c-JUN signaling pathway. Mol Med Rep 25(4):111. https://doi.org/10.3892/mmr.2022.12627
An JR, Su JN, Sun GY, Wang QF, Fan YD, Jiang N, Yang YF, Shi Y (2022) Liraglutide alleviates cognitive deficit in db/db mice: involvement in oxidative stress, iron overload, and ferroptosis. Neurochem Res 47(2):279–294. https://doi.org/10.1007/s11064-021-03442-7
Sha W, Hu F, Xi Y, Chu Y, Bu S (2021) Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res 2:1–10. https://doi.org/10.1155/2021/9999612
Zhang S, Xin W, Anderson GJ, Li R, Gao L, Chen S, Zhao J, Liu S (2022) Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis 13(1):40. https://doi.org/10.1038/s41419-021-04490-1
Gao Y, Li J, Wu Q, Wang S, Yang S, Li X, Chen N, Li L, Zhang L (2021) Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int Immunopharmacol 99:108002. https://doi.org/10.1016/j.intimp.2021.108002
Schon EA, DiMauro S, Hirano M (2012) Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 13(12):878–890. https://doi.org/10.1038/nrg3275
Song J, Herrmann JM, Becker T (2021) Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol 22(1):54–70. https://doi.org/10.1038/s41580-020-00300-2
Pfanner N, Warscheid B, Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20(5):267–284. https://doi.org/10.1038/s41580-018-0092-0
Artigues A, Iriarte A, Martinez-Carrion M (2002) Binding to chaperones allows import of a purified mitochondrial precursor into mitochondria. J Biol Chem 277(28):25047–25055. https://doi.org/10.1074/jbc.M203474200
Vazquez-Calvo C, Suhm T, Büttner S, Ott M (2020) The basic machineries for mitochondrial protein quality control. Mitochondrion 50:121–131. https://doi.org/10.1016/j.mito.2019.10.003
Drwesh L, Rapaport D (2020) Biogenesis pathways of α-helical mitochondrial outer membrane proteins. Biol Chem 401(6–7):677–686. https://doi.org/10.1515/hsz-2019-0440
Model K, Prinz T, Ruiz T, Radermacher M, Krimmer T, Kühlbrandt W, Pfanner N, Meisinger C (2002) Protein translocase of the outer mitochondrial membrane: role of import receptors in the structural organization of the TOM complex. J Mol Biol 316(3):657–666. https://doi.org/10.1006/jmbi.2001.5365
Gebert N, Chacinska A, Wagner K, Guiard B, Koehler CM, Rehling P, Pfanner N, Wiedemann N (2008) Assembly of the three small Tim proteins precedes docking to the mitochondrial carrier translocase. EMBO Rep 9(6):548–554. https://doi.org/10.1038/embor.2008.49
Davis AJ, Alder NN, Jensen RE, Johnson AE (2007) The Tim9p/10p and Tim8p/13p complexes bind to specific sites on Tim23p during mitochondrial protein import. Mol Biol Cell 18(2):475–486. https://doi.org/10.1091/mbc.e06-06-0546
Tamura Y, Harada Y, Shiota T, Yamano K, Watanabe K, Yokota M, Yamamoto H, Sesaki H, Endo T (2009) Tim23-Tim50 pair coordinates functions of translocators and motor proteins in mitochondrial protein import. J Cell Biol 184(1):129–141. https://doi.org/10.1083/jcb.200808068
Kawai A, Nishikawa S, Hirata A, Endo T (2001) Loss of the mitochondrial Hsp70 functions causes aggregation of mitochondria in yeast cells. J Cell Sci 114(Pt 19):3565–3574. https://doi.org/10.1242/jcs.114.19.3565
Kunová N, Havalová H, Ondrovičová G, Stojkovičová B, Bauer JA, Bauerová-Hlinková V, Pevala V, Kutejová E (2022) Mitochondrial processing peptidases-structure, function and the role in human diseases. Int J Mol Sci 23(3):1297. https://doi.org/10.3390/ijms23031297
Wang J, Enriquez AS, Li J, Rodriguez A, Holguin B, Von Salzen D, Bhatt JM, Bernal RA (2019) MitCHAP-60 and hereditary spastic paraplegia SPG-13 arise from an Inactive hsp60 chaperonin that fails to fold the ATP synthase β-subunit. Sci Rep 9(1):12300. https://doi.org/10.1038/s41598-019-48762-5
Nahar S, Chowdhury A, Ogura T, Esaki M (2020) A AAA ATPase Cdc48 with a cofactor Ubx2 facilitates ubiquitylation of a mitochondrial fusion-promoting factor Fzo1 for proteasomal degradation. J Biochem 167(3):279–286. https://doi.org/10.1093/jb/mvz104
Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, Becker T (2019) Mitochondrial protein translocation-associated degradation. Nature 569(7758):679–683. https://doi.org/10.1038/s41586-019-1227-y
Pryde KR, Taanman JW, Schapira AH (2016) A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Rep 17(10):2522–2531. https://doi.org/10.1016/j.celrep.2016.11.027
Fischer F, Langer JD, Osiewacz HD (2015) Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci Rep 5:18375. https://doi.org/10.1038/srep18375
Shpilka T, Haynes CM (2018) The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 19(2):109–120. https://doi.org/10.1038/nrm.2017.110
Weidberg H, Amon A (2018) MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 360(6385):eaan4146. https://doi.org/10.1126/science.aan4146
Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM (2016) The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 26(15):2037–2043. https://doi.org/10.1016/j.cub.2016.06.002
Deng P, Haynes CM (2017) Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol 47:43–49. https://doi.org/10.1016/j.semcancer.2017.05.002
Fernandes V, Choudhary M, Kumar A, Singh SB (2020) Proteotoxicity and mitochondrial dynamics in aging diabetic brain. Pharmacol Res 159:104948. https://doi.org/10.1016/j.phrs.2020.104948
Docrat TF, Nagiah S, Naicker N, Baijnath S, Singh S, Chuturgoon AA (2020) The protective effect of metformin on mitochondrial dysfunction and endoplasmic reticulum stress in diabetic mice brain. Eur J Pharmacol 875:173059. https://doi.org/10.1016/j.ejphar.2020.173059
Mulyani WRW, Sanjiwani MID, Sandra PIPY, Lestari AAW, Wihandani DM, Suastika K, Saraswati MR, Bhargah A, Manuaba IBAP (2020) Chaperone-based therapeutic target innovation: heat shock protein 70 (HSP70) for type 2 diabetes mellitus. Diabetes Metab Syndr Obes 13:559–568. https://doi.org/10.2147/DMSO.S232133
Kleinridders A, Lauritzen HP, Ussar S, Christensen JH, Mori MA, Bross P, Kahn CR (2013) Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J Clin Invest 123(11):4667–4680. https://doi.org/10.1172/JCI67615
Wardelmann K, Rath M, Castro JP, Blümel S, Schell M, Hauffe R, Schumacher F, Flore T, Ritter K, Wernitz A, Hosoi T, Ozawa K, Kleuser B, Weiß J, Schürmann A, Kleinridders A (2021) Central acting Hsp10 regulates mitochondrial function, fatty acid metabolism, and insulin sensitivity in the hypothalamus. Antioxidants 10(5):711. https://doi.org/10.3390/antiox10050711
Zheng YR, Zhang XN, Chen Z (2019) Mitochondrial transport serves as a mitochondrial quality control strategy in axons: implications for central nervous system disorders. CNS Neurosci Ther 25(7):876–886. https://doi.org/10.1111/cns.13122
Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T (2010) Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464(7293):1313–1319. https://doi.org/10.1038/nature08991
Ventura-Clapier R, Garnier A, Veksler V (2008) Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79(2):208–217. https://doi.org/10.1093/cvr/cvn098
Dumont S, Le Pennec S, Donnart A, Teusan R, Steenman M, Chevalier C, Houlgatte R, Savagner F (2018) Transcriptional orchestration of mitochondrial homeostasis in a cellular model of PGC-1-related coactivator-dependent thyroid tumor. Oncotarget 9(22):15883–15894. https://doi.org/10.18632/oncotarget.24633
Zhang X, Jing S, Lin H, Sun W, Jiang W, Yu C, Sun J, Wang C, Chen J, Li H (2019) Anti-fatigue effect of anwulignan via the NRF2 and PGC-1α signaling pathway in mice. Food Funct 10(12):7755–7766. https://doi.org/10.1039/c9fo01182j
Huang DD, Fan SD, Chen XY, Yan XL, Zhang XZ, Ma BW, Yu DY, Xiao WY, Zhuang CL, Yu Z (2019) Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp Gerontol 119:61–73. https://doi.org/10.1016/j.exger.2019.01.022
Li Y, Feng YF, Liu XT, Li YC, Zhu HM, Sun MR, Li P, Liu B, Yang H (2021) Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol 38:101771. https://doi.org/10.1016/j.redox.2020.101771
Eteghadi MR, Nasehi M, Vaseghi S, Hesami-Tackallou S (2021) The effect of Crocin on TFAM and PGC-1α expression and Catalase and Superoxide dismutase activities following cholestasis-induced neuroinflammation in the striatum of male Wistar rats. Metab Brain Dis 36(7):1791–1801. https://doi.org/10.1007/s11011-021-00748-x
Yang Q, Wang C, Jin Y, Ma X, Xie T, Wang J, Liu K, Sun H (2019) Disocin prevents postmenopausal atherosclerosis in ovariectomized LDLR-/- mice through a PGC-1α/ERα pathway leading to promotion of autophagy and inhibition of oxidative stress, inflammation and apoptosis. Pharmacol Res 148:104414. https://doi.org/10.1016/j.phrs.2019.104414
Sun C, Xiao Y, Li J, Ge B, Chen X, Liu H, Zheng T (2021) Nonenzymatic function of DPP4 in diabetes-associated mitochondrial dysfunction and cognitive impairment. Alzheimers Dement. https://doi.org/10.1002/alz.12437
Chandrasekaran K, Choi J, Arvas MI, Salimian M, Singh S, Xu S, Gullapalli RP, Kristian T, Russell JW (2020) Nicotinamide mononucleotide administration prevents experimental diabetes-induced cognitive impairment and loss of hippocampal neurons. Int J Mol Sci 21(11):3756. https://doi.org/10.3390/ijms21113756
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100(14):8466–8471. https://doi.org/10.1073/pnas.1032913100
Tiefenböck SK, Baltzer C, Egli NA, Frei C (2010) The Drosophila PGC-1 homologue Spargel coordinates mitochondrial activity to insulin signalling. EMBO J 29(1):171–183. https://doi.org/10.1038/emboj.2009.330
Sajan M, Hansen B, Ivey R 3rd, Sajan J, Ari C, Song S, Braun U, Leitges M, Farese-Higgs M, Farese RV (2016) Brain insulin signaling is increased in insulin-resistant states and decreases in FOXOs and PGC-1α and increases in Aβ1-40/42 and phospho-tau may abet Alzheimer development. Diabetes 65(7):1892–1903. https://doi.org/10.2337/db15-1428
Agrawal R, Zhuang Y, Cummings BP, Stanhope KL, Graham JL, Havel PJ, Gomez-Pinilla F (2014) Deterioration of plasticity and metabolic homeostasis in the brain of the UCD-T2DM rat model of naturally occurring type-2 diabetes. Biochim Biophys Acta 1842(9):1313–1323. https://doi.org/10.1016/j.bbadis.2014.05.007
Kwon B, Lee HK, Querfurth HW (2014) Oleate prevents palmitate-induced mitochondrial dysfunction, insulin resistance and inflammatory signaling in neuronal cells. Biochim Biophys Acta 1843(7):1402–1413. https://doi.org/10.1016/j.bbamcr.2014.04.004
Rovira-Llopis S, Bañuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM (2017) Mitochondrial dynamics in type 2 diabetes: pathophysiological implications. Redox Biol 11:637–645. https://doi.org/10.1016/j.redox.2017.01.013
Schulman JJ, Szczesniak LM, Bunker EN, Nelson HA, Roe MW, Wagner LE 2nd, Yule DI, Wojcikiewicz RJH (2019) Bok regulates mitochondrial fusion and morphology. Cell Death Differ 26(12):2682–2694. https://doi.org/10.1038/s41418-019-0327-4
Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141(2):280–289. https://doi.org/10.1016/j.cell.2010.02.026
Yan C, Duanmu X, Zeng L, Liu B, Song Z (2019) Mitochondrial DNA: distribution, mutations, and elimination. Cells 8(4):379. https://doi.org/10.3390/cells8040379
Moore AS, Coscia SM, Simpson CL, Ortega FE, Wait EC, Heddleston JM, Nirschl JJ, Obara CJ, Guedes-Dias P, Boecker CA, Chew TL, Theriot JA, Lippincott-Schwartz J, Holzbaur ELF (2021) Actin cables and comet tails organize mitochondrial networks in mitosis. Nature 591(7851):659–664. https://doi.org/10.1038/s41586-021-03309-5
Yamashita SI, Kanki T (2017) How autophagy eats large mitochondria: autophagosome formation coupled with mitochondrial fragmentation. Autophagy 13(5):980–981. https://doi.org/10.1080/15548627.2017.1291113
Gao S, Hu J (2021) Mitochondrial fusion: the machineries in and out. Trends Cell Biol 31(1):62–74. https://doi.org/10.1016/j.tcb.2020.09.008
Detmer SA, Chan DC (2007) Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol 176(4):405–414. https://doi.org/10.1083/jcb.200611080
El-Hattab AW, Craigen WJ, Scaglia F (2017) Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol Basis Dis 1863(6):1539–1555. https://doi.org/10.1016/j.bbadis.2017.02.017
Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204(6):919–929. https://doi.org/10.1083/jcb.201308006
Wang R, Mishra P, Garbis SD, Moradian A, Sweredoski MJ, Chan DC (2021) Identification of new OPA1 cleavage site reveals that short isoforms regulate mitochondrial fusion. Mol Biol Cell 32(2):157–168. https://doi.org/10.1091/mbc.E20-09-0605
Hu C, Shu L, Huang X, Yu J, Li L, Gong L, Yang M, Wu Z, Gao Z, Zhao Y, Chen L, Song Z (2020) OPA1 and MICOS regulate mitochondrial crista dynamics and formation. Cell Death Dis 11(10):940. https://doi.org/10.1038/s41419-020-03152-y
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362. https://doi.org/10.1126/science.1207385
Losón OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24(5):659–667. https://doi.org/10.1091/mbc.E12-10-0721
Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT (2013) Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288(38):27584–27593. https://doi.org/10.1074/jbc.M113.479873
Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT (2016) Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci 129(11):2170–2181. https://doi.org/10.1242/jcs.185165
Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, Frost A (2018) Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558(7710):401–405. https://doi.org/10.1038/s41586-018-0211-2
Fonseca TB, Sánchez-Guerrero Á, Milosevic I, Raimundo N (2019) Mitochondrial fission requires DRP1 but not dynamins. Nature 570(7761):E34–E42. https://doi.org/10.1038/s41586-019-1296-y
Ding Y, Liu H, Cen M, Tao Y, Lai C, Tang Z (2021) Rapamycin ameliorates cognitive impairments and Alzheimer’s disease-like pathology with restoring mitochondrial abnormality in the hippocampus of streptozotocin-induced diabetic mice. Neurochem Res 46(2):265–275. https://doi.org/10.1007/s11064-020-03160-6
Santos RX, Correia SC, Alves MG, Oliveira PF, Cardoso S, Carvalho C, Seiça R, Santos MS, Moreira PI (2014) Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes. Mol Cell Biochem 394(1–2):13–22. https://doi.org/10.1007/s11010-014-2076-5
Park G, Lee JY, Han HM, An HS, Jin Z, Jeong EA, Kim KE, Shin HJ, Lee J, Kang D, Kim HJ, Bae YC, Roh GS (2021) Ablation of dynamin-related protein 1 promotes diabetes-induced synaptic injury in the hippocampus. Cell Death Dis 12(5):445. https://doi.org/10.1038/s41419-021-03723-7
Wang Z, Xia P, Hu J, Huang Y, Zhang F, Li L, Wang E, Guo Q, Ye Z (2021) LncRNA MEG3 alleviates diabetic cognitive impairments by reducing mitochondrial-derived apoptosis through promotion of FUNDC1-related mitophagy via Rac1-ROS axis. ACS Chem Neurosci 12(13):2280–2307. https://doi.org/10.1021/acschemneuro.0c00682
Akhtar MW, Sanz-Blasco S, Dolatabadi N, Parker J, Chon K, Lee MS, Soussou W, McKercher SR, Ambasudhan R, Nakamura T, Lipton SA (2016) Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat Commun 7:10242. https://doi.org/10.1038/ncomms10242
Fritsch LE, Moore ME, Sarraf SA, Pickrell AM (2020) Ubiquitin and receptor-dependent mitophagy pathways and their implication in neurodegeneration. J Mol Biol 432(8):2510–2524. https://doi.org/10.1016/j.jmb.2019.10.015
Cai Q, Jeong YY (2020) Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 9(1):150. https://doi.org/10.3390/cells9010150
Anzell AR, Maizy R, Przyklenk K, Sanderson TH (2018) Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol Neurobiol 55(3):2547–2564. https://doi.org/10.1007/s12035-017-0503-9
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141. https://doi.org/10.1038/ncb2152
Mercer TJ, Tooze SA (2021) The ingenious ULKs: expanding the repertoire of the ULK complex with phosphoproteomics. Autophagy 14:1–3. https://doi.org/10.1080/15548627.2021.1968615
Fracchiolla D, Sawa-Makarska J, Martens S (2017) Beyond Atg8 binding: the role of AIM/LIR motifs in autophagy. Autophagy 13(5):978–979. https://doi.org/10.1080/15548627.2016.1277311
Wang R, Tan J, Chen T, Han H, Tian R, Tan Y, Wu Y, Cui J, Chen F, Li J, Lv L, Guan X, Shang S, Lu J, Zhang Z (2019) ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J Cell Biol 218(1):267–284. https://doi.org/10.1083/jcb.201804165
Lysyk L, Brassard R, Touret N, Lemieux MJ (2020) PARL protease: a glimpse at intramembrane proteolysis in the inner mitochondrial membrane. J Mol Biol 432(18):5052–5062. https://doi.org/10.1016/j.jmb.2020.04.006
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205(2):143–153. https://doi.org/10.1083/jcb.201402104
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565):309–314. https://doi.org/10.1038/nature14893
Chu CT (2019) Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis 122:23–34. https://doi.org/10.1016/j.nbd.2018.07.015
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, Ma HK, Jiang MD, Xu TT, Xu J, Shen Y, Song B, Gao PJ, Han WQ, Zhang W (2020) HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 36:101671. https://doi.org/10.1016/j.redox.2020.101671
Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K (2021) Molecular mechanisms and physiological functions of mitophagy. EMBO J 40(3):e104705. https://doi.org/10.15252/embj.2020104705
Marinković M, Šprung M, Novak I (2021) Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 17(5):1232–1243. https://doi.org/10.1080/15548627.2020.1755120
Wu X, Zheng Y, Liu M, Li Y, Ma S, Tang W, Yan W, Cao M, Zheng W, Jiang L, Wu J, Han F, Qin Z, Fang L, Hu W, Chen Z, Zhang X (2021) BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 17(8):1934–1946. https://doi.org/10.1080/15548627.2020.1802089
Li R, Toan S, Zhou H (2020) Role of mitochondrial quality control in the pathogenesis of nonalcoholic fatty liver disease. Aging 12(7):6467–6485. https://doi.org/10.18632/aging.102972
Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, Billiar TR, Tao WA, Zhang Z (2016) BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem 291(41):21616–21629. https://doi.org/10.1074/jbc.M116.733410
Dhingra A, Jayas R, Afshar P, Guberman M, Maddaford G, Gerstein J, Lieberman B, Nepon H, Margulets V, Dhingra R, Kirshenbaum LA (2017) Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med 112:411–422. https://doi.org/10.1016/j.freeradbiomed.2017.08.010
Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Q, Du L, Zheng M, Zhang C, Liu J, Chen Q (2016) Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5:e21407. https://doi.org/10.7554/eLife.21407
Xu Y, Shen J, Ran Z (2020) Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 16(1):3–17. https://doi.org/10.1080/15548627.2019.1603547
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, Duan L, Wang X, Liu L, Liu X, Shen Y, Zhu Y, Chen Q (2014) A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54(3):362–377. https://doi.org/10.1016/j.molcel.2014.02.034
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhao B, Feng D (2014) ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15(5):566–575. https://doi.org/10.1002/embr.201438501
Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L, Chen Q (2016) Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12(4):689–702. https://doi.org/10.1080/15548627.2016.1151580
Onphachanh X, Lee HJ, Lim JR, Jung YH, Kim JS, Chae CW, Lee SJ, Gabr AA, Han HJ (2017) Enhancement of high glucose-induced PINK1 expression by melatonin stimulates neuronal cell survival: Involvement of MT /Akt/NF-κB pathway. J Pineal Res 63(2):e12427. https://doi.org/10.1111/jpi.12427
Su CJ, Shen Z, Cui RX, Huang Y, Xu DL, Zhao FL, Pan J, Shi AM, Liu T, Yu YL (2020) Thioredoxin-Interacting Protein (TXNIP) regulates Parkin/PINK1-mediated mitophagy in dopaminergic neurons under high-glucose conditions: implications for molecular links between Parkinson’s disease and diabetes. Neurosci Bull 36(4):346–358. https://doi.org/10.1007/s12264-019-00459-5
Funding
This work was supported by grants from Capital’s Funds for Health Improvement and Research (2022-2-2232) to Ling-Ling Ding.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The first draft of the manuscript was written by J-SL, J-QN, L-LD. The analysis and sorting of references were performed by J-SL, Z-YC, W-JL, R-LZ. Figures were produced by J-SL, R-YY, M-JC. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, JS., Ning, JQ., Chen, ZY. et al. The Role of Mitochondrial Quality Control in Cognitive Dysfunction in Diabetes. Neurochem Res 47, 2158–2172 (2022). https://doi.org/10.1007/s11064-022-03631-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-022-03631-y