
Abstract
Ageing is the greatest risk factor for dementia, although physiological ageing by itself does not lead to cognitive decline. In addition to ageing, APOE ε4 is genetically the strongest risk factor for Alzheimer’s disease and is highly expressed in astrocytes. There are indications that human astrocytes change with age and upon expression of APOE4. As these glial cells maintain water and ion homeostasis in the brain and regulate neuronal transmission, it is likely that age- and APOE4-related changes in astrocytes have a major impact on brain functioning and play a role in age-related diseases. In this review, we will discuss the molecular and morphological changes of human astrocytes in ageing and the contribution of APOE4. We conclude this review with a discussion on technical issues, innovations, and future perspectives on how to gain more knowledge on astrocytes in the human ageing brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ageing affects every part of the human body differently, with some showing evident signs of this process like the skin undergoing atrophy [1]. On the contrary, the brain is fairly resilient to this process of decay, as physiological ageing does not lead to major neuronal and glial cell death or severe cognitive impairment [2,3,4]. Nonetheless, ageing is the most important risk factor for neurodegenerative diseases such as Alzheimer’s Disease (AD). This suggests that more subtle changes occur with advancing age, which makes the brain more vulnerable to age-related diseases, i.e. pathological ageing. In this review, we focus on age-related changes in human astrocytes and implications for astrocyte function.
Astrocytes are a major glial cell type in the human brain, harbouring homeostatic and neuromodulating functions (Fig. 1a) [5]. These cells maintain ion homeostasis by regulating water transport and potassium, calcium, and chloride levels. In addition they maintain homeostasis of reactive oxygen species (ROS) by releasing antioxidants like glutathione, which is important in the context of ageing [6]. Depending on the demand of neurons, astrocytes provide metabolic substrates, such as glycogen and lactate [7]. Moreover, they maintain not only neurotransmitter homeostasis via transporters like glutamate and GABA transporters, but also actively shape neuronal networks through regulation of synapse formation and pruning, together with microglia [8,9,10,11]. With their endfeet, they form part of the blood–brain barrier (BBB) and the brain-cerebrospinal fluid (CSF) barrier in the pial layer [6, 12]. Finally, astrocytes are, together with microglia, responsible for the immune response of the brain upon an infection, a trauma, or brain disease [6]. Given this extensive set of functions performed by astrocytes, we expect that age-related changes in astrocytes have a major impact on brain functioning.

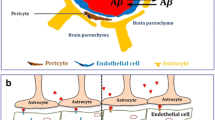
Ageing of astrocytes in the human brain. a In the adult human brain, astrocytes are part of the blood–brain barrier (BBB), maintain ion homeostasis, support and regulate neuronal transmission, and are involved in immune response of the brain. b Human APOE ε4/ε4 astrocytes in vitro display an aged molecular profile. BBB permeability is increased and cholesterol metabolism is altered. There is less secretion of neurotrophic BDNF and neurotransmitter homeostasis is affected by accumulation of EAAT1 and GLUL. Complement and cytokine secretion indicate an inflammatory profile. These changes are overlapping with aged astrocytes. In addition, APOE ε4/ε4 astrocytes increase the expression of matrisome-related molecules and are less capable of clearing Aβ. This early ageing phenotype possibly underlies the increased risk of developing AD in ε4 carriers. c Ageing of the human brain induces molecular changes in astrocytes. Molecules involved in the BBB are upregulated (ITGB4, AQP4, GJA1) and BBB permeability increases. The release of the antioxidant glutathione is reduced in aged astrocytes, leading to a decreased protection against ROS. Metabolic support does not seem to change with ageing. Regarding neuronal transmission, aged astrocytes secrete less neurotrophic BDNF and are less efficient in maintaining neurotransmitter homeostasis due to reduction of mGluR3 expression and decreased conversion of glutamate into glutamine. Molecules involved in synaptogenesis and pruning are all upregulated (THBS, GPC, MEGF10, C1q, C3). Also, aged astrocytes display an inflammatory profile indicated by increased GFAP and complement levels. ITGB4 integrin beta 4; AQP4 aquaporin-4; GJA1 connexin 43; ROS reactive oxygen species; BDNF brain-derived neurotrophic factor; THBS thrombospondins; GPC glypicans; MEGF10 multiple epidermal growth factor-like domains protein 10; GFAP glial fibrillary acidic protein; EAAT1 excitatory amino acid transporter 1; GLUL glutamine synthetase; Aβ amyloid beta; AD Alzheimer’s disease; ? means contradictory results; – means not changed
Besides this array of functions, human astrocytes are heterogenous in morphology and transcriptional profile. In the adult human cortex, four types of astrocytes have been described based on morphology: interlaminar astrocytes, protoplasmic astrocytes, varicose projection astrocytes, and fibrous astrocytes [5]. These subtypes are distinguished based on the glial fibrillary acidic protein (GFAP), which is the main intermediate filament (IF) protein in astrocytes [13]. It should be kept in mind that immunolabeling of GFAP only reveals the filamentous skeleton of an astrocyte and therefore does not reflect the full morphology. The fine processes that make up most of the astrocytes cannot be visualized with GFAP immunostaining. Furthermore, GFAP shows transcriptional variation as some astrocytes express low levels of GFAP, while others have a higher baseline GFAP expression [14,15,16]. Likewise, other astrocyte markers present regional variation. For example, cortical astrocytes express relative high levels of ALDH1L1 and low levels of GFAP [17, 18]. Still, GFAP is often used as the golden standard to identify astrocytes, but region-specific expression patterns indicate that a proportion of astrocytes is missed if only GFAP as a marker is used. This morphological and transcriptional heterogeneity should be considered when studying astrocytes in the human brain [14, 19]. Thus, identifying astrocytes solely based on GFAP expression will bias studies towards a specific subpopulation and this is a challenge that needs to be faced to enhance our understanding of astrocytes.
During physiological ageing, the brain changes including its astrocytes. As cell numbers do not change, subtle alterations must underlie the increased vulnerability for pathological ageing processes such as AD. On the contrary, in AD there are unquestionable changes in cell numbers due to degeneration which leads to severe cognitive decline and these pathological processes cannot be ascribed to physiological ageing processes [2]. Thus, another stimulus will likely tip the balance from physiological to pathological ageing. A possible candidate is apolipoprotein E4 (APOE4), the largest genetic risk factor for AD [20, 21]. This protein is highly expressed by astrocytes and therefore it is plausible that astrocytic changes underly the increased risk of AD [22]. In this review, we will describe transcriptional and IF-related morphological changes of human astrocytes during physiological ageing and induced by APOE4. We will compare these changes to pathological ageing in AD and hypothesize how these are interconnected.
Molecular Profile of Human Astrocytes in Physiological Ageing
In the last decade, gene expression profiling studies (microarray and RNA-sequencing) provided detailed insight in cell-type-specific transcriptomic changes across different brain regions in the ageing human brain [14, 23,24,25,26,27]. When analysing gene expression in various neural cell populations from different brain regions and different ages, changes in neuron-specific gene expression cluster per brain region [27,28,29]. Instead, changes in glia-specific gene expression, among which astrocyte genes, show a stronger correlation with age [25,26,27, 30].
To date, only a few transcriptomic studies have described astrocyte-specific gene expression in the human brain (Table 1). Two studies analysed the transcriptome of individual cell types, one in laser-captured GFAP positive astrocytes [30], the other in several astrocyte subpopulations identified by single nuclear RNAseq [26]. Both studies are focussed on AD pathology and only have data from elderly (> 71 years old) [26, 30], thus lacking a comparison to younger individuals. Therefore, it is hard to say anything about age-related changes in these studies. Habib et al. (2020) used the human dataset from Mathys et al. (2019) to compare with their mouse RNAseq data and identified a continuous range of astrocyte subpopulations categorized by GFAP expression [14]. The other four transcriptomic studies, Kumar et al. (2013) [23], Simon et al. (2018) [22, 31, 32], Soreq et al. (2019) [33,34,35], and Wruck et al. (2020) [33, 36,37,38,39,40,41], made use of bulk brain tissue transcriptomics. Although an astrocyte-specific human ageing study has not yet been performed, for this review we extracted astrocyte-specific changes from these eight studies.
Interestingly, of the 10 brain regions studied by Soreq et al. (2017) this age-related loss of region-specificity is most pronounced in the hippocampus and substantia nigra, two key areas in age-related neurodegenerative diseases such as AD and Parkinson’s disease (PD) [27]. Gene ontology (GO) and pathway analyses of differentially expressed genes (DEGs) can give insight into broader functional changes that are related to the transcriptional changes. Astrocyte-specific DEGs upregulated with age were enriched in pathways such as “inflammation”, “cell–cell adhesion”, “cell morphogenesis”, and “extracellular matrix organization” [25, 27]. A decreased expression with age is only shown by a few astrocyte-specific genes, which are related to the glutamate system in pathways such as “glutamate receptor signalling” and “synaptic transmission, glutamatergic” [25]. These pathways hint towards altered homeostatic functions in human aged astrocytes. Next, we will discuss these groups of astrocyte functions that changed based on human transcriptional studies. Besides, we will discuss whether astrocyte functions that are changed in rodent ageing studies, are also represented in human (transcriptional) studies.
Altered Homeostatic Functions of Aged Human Astrocytes
Astrocytes are involved in molecular, metabolic, and cellular brain homeostasis [6]. We will describe age-related changes divided into three categories of homeostatic functions, namely neuronal support functions, regulation of synapse formation and function, and maintaining the BBB (Fig. 1c).
Neuronal Support Functions
One of the homeostatic functions that astrocytes exhibit is the protection of neurons against free radicals and oxidative stress. The oxidative stress theory of ageing states that accumulation of ROS-induced damage underlie age-related changes [47,48,49]. Astrocytes release glutathione, which is an endogenous antioxidant that protects neurons from free radicals such as ROS [50]. In the aged human brain, glutathione levels are reduced in astrocytes thereby hampering this homeostatic function [51, 52]. Another homeostatic function of astrocytes is providing metabolites to neurons. Lactate is produced by astrocytes via glycolysis and when secreted it can be used as an energy source by neurons [7]. Studies on lactate levels in the aged human brain have been inconsistent, reporting either an increase or a decrease with advancing age [7, 51, 53]. Astrocytes also provide neurons with cholesterol to maintain membrane integrity [54]. Whereas rodent studies show a reduction in cholesterol synthesis [25, 27, 55], no human studies are known to indicate that this process is affected by ageing in astrocytes. To conclude, homeostatic functions regarding neuronal support seem not to be majorly changed in aged human astrocytes.
Regulation of Synapse Formation and Function
Astrocytes have thin protrusions that intimately interact with synapses, forming the tripartite synapse [56]. These perisynaptic processes express proteins that are involved in synaptic transmission, among which metabotropic glutamate receptors (mGluRs), glutamate transporters, and glutamine synthetase (GLUL), which bind, transport, and convert glutamate, respectively. MGluR3 and mGluR5 are the most abundant mGluRs on astrocytes in the human brain [57, 58]. They are activated by glutamate overspill from the synapse, resulting in local calcium concentration increase and subsequent release of gliotransmitters [57]. The mGluR3 shows a decrease with advancing age [25, 59] and the enzyme GLUL seems less efficient in the aged human brain as demonstrated by an increased glutamine/glutamate ratio [60]. Astrocytes also secrete neurotrophic molecules to support neurogenesis and synaptogenesis. For example, brain-derived neurotrophic factor (BDNF), involved in synaptic plasticity, is decreased in the ageing human brain [61]. Such changes in neurotropic function and glutamate homeostasis might hamper the capacity of astrocytes to regulate synaptic processes in the aged human brain.
In the developing and healthy adult brain, astrocytes play a role in synapse formation by expressing molecules such as thrombospondins, SPARC, Hevin, and glypicans to regulate cell–cell interaction [10, 11, 62]. Subsequently, fine-tuning of synaptic transmission is done by synaptic pruning, a task mainly performed by microglia through complement-mediated synapse removal involving C1q and C3 [9], but astrocytes can also engulf synapses through phagocytosis via MERTK and MEGF10 [8]. In human astrocytes, glypicans, thrombospondins, and MEGF10 are upregulated with age in many brain regions, except in the frontal cortex and hippocampus where MEGF10 is downregulated [27, 63]. Furthermore, both C1q and C3 are upregulated in the aged human brain, which overall hints towards increased synaptic pruning in the aged human brain [64, 65].
Blood–Brain-Barrier Regulation
Together with endothelial cells and pericytes, astrocytes are part of the BBB, which limits the influx of blood components, pumps out cerebral waste materials, and regulates the movement of amino acids and glucose into the cerebral parenchyma to support the function and survival of brain cells. One of the molecules in astrocytes that is essential for the formation of the BBB is integrin β4 (ITGB4). This gene, which is involved in cell–cell adhesion and extracellular matrix (ECM) interaction [22, 66], is among the 10 genes most significantly correlated with age in the human prefrontal cortex [25]. Since in rodent studies an upregulation of Itgb4 is linked to reactive astrocytes [67, 68], this could be indicative of an inflammatory profile of these aged human astrocytes.
Because astrocytes have terminal processes on both synapses and the brain vasculature, they can modulate neuronal activity and cerebral blood flow, so-called ‘neurovascular coupling’ [69]. Moreover, the vascular endfeet, which are interconnected by gap junctions through connexin 43 and 30 (GJA1; GJB6), express the potassium channel Kir4.1 and water channel aquaporin-4 (AQP4) to regulate ion concentrations and water balance [5]. Transcriptomic data shows that both AQP4 and GJA1 are upregulated with ageing in the frontal, occipital, and temporal cortices, hippocampus, putamen, medulla, white matter, and cerebellum [14, 27]. This upregulation of AQP4 is confirmed on protein level in the frontal and occipital cortex [70, 71]. In a broader perspective, BBB integrity, measured by magnetic resonance imaging (MRI), shows no significant changes with increasing age in the frontal and temporal cortex, however, in the hippocampus there is an age-dependent progressive loss of BBB integrity [72]. This is consistent with the meta-analysis of Farrall and Wardlaw (2009) that shows an increased permeability with ageing [73]. If and how human astrocytes are implicated in this loss of BBB integrity is to date not known.
In conclusion, there are multiple indications that astrocyte homeostatic functions are altered in the aged human brain, which is in corroboration with rodent studies [74,75,76]. These homeostatic changes could go hand in hand with an increase in the inflammatory function of astrocytes, which is discussed below.
Inflammatory Profile of Aged Human Astrocytes
Neuroinflammation is associated with the ageing brain and both astrocytes and microglia are involved [77,78,79]. Reactive astrogliosis is a process in which astrocytes activate molecularly defined programs resulting in biochemical, morphological, metabolic, and transcriptional changes, which leads to a gain of new functions or alterations in homeostatic functions [15, 80, 81]. One gene that is significantly upregulated in reactive astrocytes is GFAP [15]. Immunostainings with antibodies against this IF protein can visualize the hypertrophic morphology typical for reactive astrocytes, and therefore GFAP is often used as a marker for astrocyte reactivity [15, 80]. Expression of GFAP in the brain has been studied across human lifespan in various brain regions (Table 2). It is consistently seen that the expression of GFAP, both on mRNA as well as protein level, increases with age, especially around the age of 70 [82, 83]. This increase is most prominent in the hippocampus, but also present in frontal, temporal, and entorhinal cortices, and substantia nigra [82,83,84,85]. Based on the GFAP immunoreactivity pattern, the astrocyte IF network changes morphologically from long, thin processes in younger individuals, to short, stubby processes in older individuals (Table 2) [85, 86]. While this resembles the hypertrophic morphology of reactive astrocytes [80, 87], systematic human studies into this morphological change with various methods across different brain regions are limited. Both the upregulation of GFAP and the morphological changes would endorse the “inflammageing” theory, which states that ageing induces a chronic, low-grade inflammatory status [88]. However studies should not only rely on an increase in GFAP in the human ageing brain to prove a reactive astrocyte phenotype [80, 89].
Indications from Mouse Ageing Studies
In search for age-related reactive astrocyte markers, we reviewed several hallmark papers on transcriptomic changes in mouse astrocytes in response to CNS injuries and ageing (Table 3).
We were the first to analyse DEGs between young adult and aged mouse astrocytes and showed that genes with increased expression in cortical aged astrocytes were implicated in biological processes such as ‘defence response’ and ‘antigen presentation’ [90]. These processes were also upregulated in astrocytes after induction of neuroinflammation by systemic lipopolysaccharide (LPS) injection [85], a subtype identified by Liddelow et al. (2017) as neurotoxic A1 astrocytes [86]. Expression profiling of astrocytes induced by ischaemia revealed a different subtype of reactive astrocyte, which was termed the neuroprotective A2 astrocyte [86]. The minority of genes identified in both A1 and A2 astrocytes are termed pan-reactive genes, which include Gfap, Vimentin (Vim), Serpina3n, and Cxcl10.
A more recent study comparing the transcriptional changes with age in three brain areas showed that the most highly up-regulated genes in all regions were reactive astrocyte-associated genes, mostly characteristic of the ‘A1 reactive astrocyte genes’ [79]. These genes included those involved in the complement (C3 and C4b), antigen presentation (H2-D1 and H2-K1), peptidase inhibitor (Serpina3n) and cytokine (Cxcl10) pathways. Nevertheless, a number of aged astrocytes also expressed a combination of A1 and A2 genes. A third study, although looking at four different brain regions and taking a different astrocyte isolation approach, also found similar genes upregulated with age, including pan-reactive gene Serpina3n and A1 genes C3 and C4b [55], which are involved in synapse elimination [9, 91]. These last two studies both screened their DEGs for gene families involved with synapses and both found Sparc to be upregulated in aged astrocytes, which suggests an active role in decreasing synapse function [92]. Although a fourth study analysed astrocytes isolated from whole brain and of five ages ranging from young adult to middle-age, also Serpina3n, C4b and Cxcl10 were significantly upregulated in astrocytes from the oldest compared to the youngest group [93]. Overall, aged murine astrocytes seem to become more reactive resembling the A1 phenotype. This reactive subtype is also present in different neurodegenerative diseases [94], however how expression of these reactivity-genes change with human ageing is largely unclear. One recent meta-analysis of human transcriptome data did analyse A1 and A2 signature genes, and though they found a reactive phenotype in aged astrocytes of the prefrontal cortex, A1 and A2 genes were equally over-represented [43]. Overall, it remains to be investigated how these genes behave throughout the ageing human brain and whether all astrocyte subpopulations respond similarly to neurotoxic stimuli. Moreover, the gene signature of ‘neurotoxic A1 astrocytes’ in humans may differ from those in mice, as it was recently shown that the transcriptional signatures of the human AD response in astrocytes was remarkably different from those observed in mice [95].
Ageing-Induced Shift in Astrocyte Subpopulations
Astrocytes are a heterogeneous cell population and the composition shifts with ageing. The GFAP-high subpopulation as described above increases with age, at the expense of the homeostatic GFAP-low subpopulation [14]. Another subpopulation consists of the disease-associated astrocytes (DAAs). Albeit that these cells not only are present in disease, but also in the human brain during physiological ageing [14, 79]. DAAs show an upregulation of a subset of genes, including GFAP, SERPINA3n, and VIM [14]. This subset of upregulated genes comes together in processes such as endocytosis, complement cascade, development and differentiation, metabolic pathways, and inflammatory signalling. This DAA-gene-expression-pattern shows similarity to the previously in mice described A1 astrocytes [79]. The number of DAAs significantly increases in an early stage of AD and this shift in the astrocyte population composition is possibly a driving force of the switch between physiological to pathological ageing [14, 26]. This raises the question: What causes the shift in composition of astrocyte subpopulations? A possible candidate is the APOE gene, more precisely the E4 isoform which is the largest genetic risk factor for late-onset AD and mainly expressed in astrocytes [20, 22]. The effect of different APOE variants on astrocyte transcriptomics and its contribution to pathological ageing will be discussed in the next section.
APOE4 in Human Astrocytes: Accelerating the Shift from Physiological to Pathological Ageing?
There are three different APOE alleles due to single nucleotide polymorphisms (SNPs) in the APOE gene: ε2, ε3 and ε4. This results in three APOE protein isoforms with one or two differences in amino acids [96]. The ε3 allele is the most common variant and 77% of the population has this allele. The ε4 allele has a high prevalence in AD patients (40%) compared to the general population (15%) and is the main genetic risk factor for late-onset AD. These changes in the APOE4 protein lead to functional changes that are involved in the pathogenesis of AD [20, 97]. People with two ε4 alleles have a 14.9 times higher chance of developing AD, show a decrease in AD age of onset, and present with a steeper progress in cognitive decline compared to people with two ε3 alleles [20, 21].
APOE is a glycoprotein involved in the lipid metabolism of the brain. In homeostatic conditions, it is secreted predominantly by astrocytes and binds to lipids forming a lipoprotein particle. These particles bind on cell-surface APOE receptors (APOERs), primarily of the LDLR-family. For example, astrocyte-secreted APOE forms a lipoprotein with cholesterol, which binds to neuronal APOERs thereby supporting neuronal functioning [54]. The amount of APOE protein is SNP-dependent: ε2/ε2 results in the highest and ε4/ε4 in the lowest levels of APOE in serum, CSF, and interstitial fluid (ISF) [24]. In human astrocytes specifically, it has been shown that APOE mRNA as well as intracellular and secreted levels of APOE protein are lower in ε4/ε4 compared to ε3/ε3 astrocytes [98, 99]. Also structurally, APOE3 and APOE4 differ significantly in isoform-dependent lipid-binding capacity, leading to altered lipoprotein sizes [100]. Besides the homeostatic functions, APOE is also involved in amyloid β (Aβ) clearance and the efficiency of clearance is isoform dependent [98, 101]. APOE ε4 carriers show more accumulation of APOE and Aβ in plaques and clearance of Aβ is impaired [102, 103]. These differences are thought to underlie the effect of APOE4 on the increased risk to develop AD. However, it is not a direct effect of APOE4 on neurons, as astrocyte-conditioned medium containing APOE4 does not cause neuronal degeneration [104]. This suggests that, besides its role in Aβ clearance, other transcriptional and functional changes in astrocytes inflicted by APOE genotype may play a role in the increased risk to develop AD.
APOE ε4 carriers without AD pathology already show differences in the brain compared to non-carriers. There are altered levels of complement proteins and other inflammatory markers in the CSF, such as reactive astrocyte markers (CCL4, S100β, YKL40, GFAP), compared to non-carriers of the same age [105]. Brain proteome analysis also shows that the GO term “regulation of inflammatory response” is positively correlated with APOE ε4/ε4 genotype, while “synaptic transmission” and “post-synaptic membrane” are negatively correlated [20]. This is reflected in the inverse correlation of APOE ε4 alleles with spine density in the dentate gyrus and hippocampal volume [106, 107]. APOE ε4 carriers have an increased BBB breakdown in the hippocampus, which is also the brain region presenting age-related BBB permeability [108]. This is most likely mainly due to the effect of astrocyte-secreted APOE4 on pericytes and endothelial cells [109, 110].
Focussing on astrocytes specifically, in vitro studies show several differences between APOE ε4/ε4 and ε3/ε3 astrocytes (Fig. 1b). Regarding homeostatic functions, neuronal support and regulation of synaptic transmission are affected by APOE genotype. Cholesterol synthesis is the most significant positively enriched GO pathway in APOE ε4/ε4 astrocytes and this is reflected in increased levels of intracellular and secreted cholesterol in these cells [98, 99]. This pathway is also enriched in the GFAP-high and DAA astrocyte subpopulations, which both increase in number with ageing [14]. Another characteristic of APOE ε4/ε4 astrocytes is the upregulation of matrisome-related signals [99]. Matrisome is defined as a combination of ECM proteins (such as glycoproteins and collagens) and ECM associated factors (such as integrins, proteases, glypicans, plexins) [111]. The upregulation of molecules such as integrins also occurs in astrocytes in the aged human brain [25]. Glutamate receptor signalling is negatively correlated with age and the fact that the APOE genotype affects glutamate transporters and enzymes, corroborates the role of APOE4 in pathological ageing [25]. APOE ε4/ε4 astrocytes show nuclear accumulation of excitatory amino acid transporter 1 (EAAT1) and cytoplasmic accumulation of GLUL [112]. Another homeostatic function related to the BDNF metabolism is regulated by APOE in a genotype-dependent way, as APOE ε4/ε4 astrocytes have lower levels of intracellular BDNF than ε3/ε3 astrocytes [113]. This suggests that an APOE ε4/ε4 genotype amplifies the disruption of neuronal support and synaptic transmission functioning caused by ageing.
One study shows that astrocytes from an APOE ε4/ε4 AD patient are more fibroblast-like compared to the arborized morphology of astrocytes of a control subject that is an APOE ε3 carrier [112]. Besides, APOE ε4/ε4 astrocytes show a general inflammatory profile, with high levels of secreted proteins such as SDF-1a (CXCL12), Gro-alpha/KC (CXCL1), MIP-1b (CCL4), Eotaxin (CCL11), IP-10 (CXCL10) and RANTES (CCL5), cytokines (IL-8, LIF and IL-6), and growth factors (VEGF-A, HGF and VEGF-D [99]. This corresponds to the higher levels of inflammatory markers in the CSF of APOE ε4 carriers [105].
Next to these baseline differences between APOE ε4/ε4 and ε3/ε3 astrocytes, molecular differences are apparent in AD pathology. In Braak stage I-II subjects, the following groups of genes are downregulated in APOE ε4 carriers: “cell signalling and communication”, “cytoskeleton”, “metabolism”, “DNA damage response”, and “transcription”. In contrast, “immune response” shows an upregulation in APOE ε4-carrying AD patients, on top of the consensus of reactive astrocytes in AD [30]. In vitro, APOE ε4/ε4 astrocytes are less resistant to high levels of Tau leading to cell death, in comparison to APOE ε3/ε3 astrocytes [114]. In addition to the age- and APOE-related changes that have been described so far, astrocytes dramatically change in AD. In the final section, we will review the age- and APOE4-related astrocyte changes in the context of AD for which both conditions are the largest risk factors.
Linking Ageing, APOE4, and Alzheimer’s Disease in Human Astrocytes
Neuroinflammation is one of the major hallmarks of AD, in addition to Aβ plaques and neurofibrillary tangles [115]. Reactive astrocytes are already present in the prodromal phase of AD, indicated by high CSF levels of chitinase-3-like protein 1 (YKL-40), IL-15, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion protein 1 (VCAM-1) [116]. Also, hypertrophic astrocytes and increased expression of GFAP, S100b, MAOB, and VIM have been shown in AD patients [117,118,119,120,121]. Both aged astrocytes and APOE4 astrocytes show an inflammatory phenotype, which indicates that this is underlying the increased risk of developing AD with age and with APOE ε4 genotype.
Glutamatergic neurotransmission is further affected when AD pathology becomes apparent. Expression of EAAT1 and EAAT2 is inversely related to AD pathology [30, 122, 123]. And in contrast to mGluR3, which is downregulated in ageing and AD, mGluR5 is upregulated in AD possibly due to effects of Aβ plaques via activation of NF-κB pathway [26, 124]. This change in mGluR expression increases calcium signalling in astrocytes, which is a generally observed phenotype in AD [30, 125, 126]. Regarding calcium signalling, rodent studies have shown that APOE ε4/ε4 astrocytes have an increased calcium signalling which could mean that these astrocytes are less capable of compensating the ageing-induced changes thereby switching to pathological ageing/AD [127].
BBB integrity loss is something seen in both aged as well as APOE ε4 carrying individuals. In AD patients, this phenotype is also present and the BBB integrity loss spreads to other brain regions rather than only hippocampus which is the case in ageing [72, 128, 129]. Importantly, BBB leakage is significantly correlated with cognitive decline [130]. This is (partly) caused by the downregulation of tight junction proteins in astrocytes, which are necessary for the integrity of the BBB [30]. AQP4, another protein involved in the BBB via perivascular endfeet, is increased in the hippocampus and temporal cortex of AD patients, but not the frontal cortex [42]. Reduction of AQP4 perivascular localization is associated with Aβ plaque density and cognitive decline [70]. In ageing, AQP4 is also upregulated, but not changed in localization indicating that mislocalization rather than upregulation is causative of pathological ageing.
GJB6 is specifically upregulated in early AD pathology compared to controls without pathology, while GJA1 is upregulated both in ageing and AD [26, 27]. However, Simpson et al. show significant transcriptional changes in astrocytes in the temporal cortex progressing from early to advanced stages of AD, amongst which a downregulation of the pathway “gap junctions” [30]. These contradicting studies have been performed in different stages of AD pathology indicating that the upregulation seen in ageing and early stages proceeds towards a downregulation in later stages of the disease [131].
Discussion
In this review, we have described the molecular changes in astrocytes that occur with ageing in the human brain. However, there is only a limited number of studies investigating astrocyte-specific changes in the ageing human brain. Therefore, we not only described the enriched pathways, but also described specific proteins indicated to be changed in ageing by rodent studies and examined them in human studies. Although, rodent studies cannot be directly extrapolated to humans as the genetic variation is larger is human population. There are several limitations regarding the study of astrocytes in human brain development and ageing. There are roughly four sources of human astrocytes: fixed and fresh post-mortem brain tissue, surgical brain tissue, and in vitro human models. Fixed post-mortem brain tissue has allowed looking at cytoarchitecture of the human brain. However, this method only allows for static observations of astrocytes in a certain condition and mainly represents end-stage conditions of for example AD. Importantly, there is a limited availability of human post-mortem tissue of healthy young individuals making it difficult to make comparisons over time. Acute isolation of astrocytes from fresh post-mortem or surgical human brain tissue would overcome the first limitation and would allow for functional assays. However, it has been proven challenging to isolate and culture human post-mortem astrocytes [Hol & Middeldorp, unpublished data]. First of all, culture condition should be serum-free, as astrocytes are only exposed to serum in vivo in the case of BBB breakdown or injury and will therefore obtain a reactive phenotype when encountering serum [22]. Second, until the previous decade, a human astrocyte-specific surface marker was lacking to obtain a pure astrocyte culture [132]. In 2016, the Barres lab published a protocol to isolate human astrocytes with HepaCAM, a cell-adhesion glycoprotein, and culture them in a serum-free environment [22]. This method overcomes two of the limitations of fixed human post-mortem brain tissue, but the limited availability of human brain specimens makes this method not appropriate for extensive functional experiments.
With the development of a method to dedifferentiate human somatic cells into induced pluripotent stem cells (iPSCs), a whole new range of models has been established [133]. These human iPSCs can be differentiated into any cell type, among which astrocytes and neurons [134,135,136,137]. Human iPSC-derived astrocytes display key molecular and functional features of adult astrocytes, such as expression of GFAP, S100B, GLUL, CD44, and GJA1, calcium signalling, glutamate uptake, and support of synaptogenesis [134, 137]. In addition to differentiation into single cell types, iPSCs can also be differentiated into complex 3D neural tissues such as cerebral organoids [138, 139]. Cerebral organoids rely on intrinsic self-organizational capacity of cells to develop into several brain regions such as cerebral cortex and hippocampus [140]. These regions comprise numerous cell types, ranging from neurons, astrocytes, and oligodendrocytes, to microglia [139, 141, 142]. The 3D organization of these cell types together makes this model unique to study cell–cell interactions. Astrocytes can also be isolated from cerebral organoids based on CD49f expression to study/manipulate them in isolation [143]. CD49f is a laminin receptor expressed on iPSC-derived astrocytes and human foetal/mature astrocytes as well as human endothelial cells [22, 143]. Therefore, this marker might be suitable to isolate astrocytes from human brain tissue, in addition to HepaCAM, but this still needs confirmation.
These new models open up the possibility to study ageing and age-related neurodegenerative disease. This is approach is especially suitable for experiments into genetic components of ageing and age-related neurodegenerative diseases, such as APOE. In particular to study the effect of physiological ageing on APOE ε4/ε4 astrocytes as there is essentially no healthy aged population with an APOE ε4/ε4 genotype. For example, cerebral organoids from APOE ε4 carrying AD patients show increased tau expression and phosphorylation [33]. However, it is unclear whether iPSC and iPSC-derived cells of aged individuals preserve ageing-associated characteristics in the epigenome [144]. This makes it more complicated to study physiological ageing in iPSC-derived models. To overcome this, ageing can be artificially induced in these models, by e.g. overexpression of progerin, a protein associated with premature ageing in progeria patients or by inducing oxidative stress which mimics ageing conditions [145, 146]. With these advancing techniques, more possibilities arise to study physiology and pathology of human astrocytes.
Concluding Remarks and Future Perspective
Human astrocytes are an heterogenous population which are not solely defined by GFAP expression. Across subpopulations, aged human astrocytes are morphologically and transcriptionally different from young astrocytes implying age-related functional changes. Changes induced by ageing are similar to APOE4-induced changes in human astrocytes (Fig. 1), which could make the latter more vulnerable to age-related diseases such as AD. However, there are few studies into ageing of human astrocytes, mostly due to technical challenges and limited availability of tissue from young and old healthy individuals. State-of-the-art iPSC technology may help to fill the knowledge gap on transcriptional, morphological, and particularly functional changes on human ageing astrocytes.
References
Chambers ES, Vukmanovic-Stejic M (2020) Skin barrier immunity and ageing. Immunology 160:116–125. https://doi.org/10.1111/imm.13152
Wilson RS, Wang T, Yu L et al (2020) Normative cognitive decline in old age. Ann Neurol 87:816–829. https://doi.org/10.1002/ana.25711
Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B (2008) Neocortical glial cell numbers in human brains. Neurobiol Aging 29:1754–1762. https://doi.org/10.1016/j.neurobiolaging.2007.04.013
Fabricius K, Jacobsen JS, Pakkenberg B (2013) Effect of age on neocortical brain cells in 90+ year old human females-a cell counting study. Neurobiol Aging 34:91–99. https://doi.org/10.1016/j.neurobiolaging.2012.06.009
Oberheim NA, Takano T, Han X et al (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29:3276–3287. https://doi.org/10.1523/JNEUROSCI.4707-08.2009
Verkhratsky A, Nedergaard M (2018) Physiology of Astroglia. Physiol Rev 98:239–389. https://doi.org/10.1152/physrev.00042.2016
Harris JL, Choi IY, Brooks WM (2015) Probing astrocyte metabolism in vivo: proton magnetic resonance spectroscopy in the injured and aging brain. Front Aging Neurosci 7:1–8. https://doi.org/10.3389/fnagi.2015.00202
Chung WS, Clarke LE, Wang GX et al (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. https://doi.org/10.1038/nature12776
Stevens B, Allen NJ, Vazquez LE et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Christopherson KS, Ullian EM, Stokes CCA et al (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. https://doi.org/10.1016/j.cell.2004.12.020
Kucukdereli H, Allen NJ, Lee AT et al (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1104977108
Cabezas R, Avila M, Gonzalez J et al (2014) Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci 8:211. https://doi.org/10.3389/fncel.2014.00211
Middeldorp J, Boer K, Sluijs JA et al (2010) GFAPdelta in radial glia and subventricular zone progenitors in the developing human cortex. Development 137:313–321. https://doi.org/10.1242/dev.041632
Habib N, McCabe C, Medina S et al (2020) Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23:701–706. https://doi.org/10.1038/s41593-020-0624-8
Hol EM, Pekny M (2015) Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol 32:121–130. https://doi.org/10.1016/j.ceb.2015.02.004
Van Den Berge SA, Middeldorp J, Zhang CE et al (2010) Longterm quiescent cells in the aged human subventricular neurogenic system specifically express GFAP-δ. Aging Cell 9:313–326. https://doi.org/10.1111/j.1474-9726.2010.00556.x
Waller R, Woodroofe MN, Wharton SB et al (2016) Gene expression profiling of the astrocyte transcriptome in multiple sclerosis normal appearing white matter reveals a neuroprotective role. J Neuroimmunol 299:139–146. https://doi.org/10.1016/j.jneuroim.2016.09.010
Macikova I, Perzelova A, Mraz P et al (2009) GFAP-positive astrocytes are rare or absent in primary adult human brain tissue cultures. Biologia (Bratisl) 64:833–839. https://doi.org/10.2478/s11756-009-0136-1
Matias I, Morgado J, Gomes FCA (2019) Astrocyte heterogeneity: impact to brain aging and disease. Front Aging Neurosci 11:1–18. https://doi.org/10.3389/fnagi.2019.00059
Dai J, Johnson ECB, Dammer EB et al (2018) Effects of APOE genotype on brain proteomic network and cell type changes in Alzheimer’s disease. Front Mol Neurosci 11:1–14. https://doi.org/10.3389/fnmol.2018.00454
Ferri E, Gussago C, Casati M et al (2019) Apolipoprotein E gene in physiological and pathological aging. Mech Ageing Dev 178:41–45. https://doi.org/10.1016/j.mad.2019.01.005
Zhang Y, Sloan SA, Clarke LE et al (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. https://doi.org/10.1016/j.neuron.2015.11.013
Kumar A, Gibbs JR, Beilina A et al (2013) Age-associated changes in gene expression in human brain and isolated neurons. Neurobiol Aging 34:1199–1209. https://doi.org/10.1016/j.neurobiolaging.2012.10.021
Trabzuni D, Ryten M, Walker R et al (2011) Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J Neurochem 119:275–282. https://doi.org/10.1111/j.1471-4159.2011.07432.x
Wruck W, Adjaye J (2020) Meta-analysis of human prefrontal cortex reveals activation of GFAP and decline of synaptic transmission in the aging brain. Acta Neuropathol Commun 8:1–18. https://doi.org/10.1186/s40478-020-00907-8
Mathys H, Davila-Velderrain J, Peng Z et al (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570:332–337. https://doi.org/10.1038/s41586-019-1195-2
Soreq L, Rose J, Soreq E et al (2017) Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep 18:557–570. https://doi.org/10.1016/j.celrep.2016.12.011
Verkhratsky A, Zorec R, Rodriguez-Arellano JJ, Parpura V (2019) Neuroglia in ageing. Adv Exp Med Biol 1175:181–197. https://doi.org/10.1007/978-981-13-9913-8_8
Lupo G, Gaetani S, Cacci E et al (2019) Molecular signatures of the aging brain: finding the links between genes and phenotypes. Neurotherapeutics 16:543–553. https://doi.org/10.1007/s13311-019-00743-2
Simpson JE, Ince PG, Shaw PJ et al (2011) Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimer’s pathology and APOE genotype. Neurobiol Aging 32:1795–1807. https://doi.org/10.1016/j.neurobiolaging.2011.04.013
Miller JA, Guillozet-Bongaarts A, Gibbons LE et al (2017) Neuropathological and transcriptomic characteristics of the aged brain. Elife 6:e31126. https://doi.org/10.7554/eLife.31126
Hokama M, Oka S, Leon J et al (2014) Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex 24:2476–2488. https://doi.org/10.1093/cercor/bht101
Lu T, Aron L, Zullo J et al (2014) REST and stress resistance in ageing and Alzheimer’s disease. Nature 507:448–454. https://doi.org/10.1038/nature13163
NABEC: North American Brain Expression Consortium. https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001300.v1.p1
UK Brain Expression Consortium (UKBEC). https://ukbec.wordpress.com/
Narayan S, Tang B, Head SR et al (2008) Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res 1239:235–248. https://doi.org/10.1016/j.brainres.2008.08.023
Barnes MR, Huxley-Jones J, Maycox PR et al (2011) Transcription and pathway analysis of the superior temporal cortex and anterior prefrontal cortex in schizophrenia. J Neurosci Res 89:1218–1227. https://doi.org/10.1002/jnr.22647
Lanz TA, Joshi JJ, Reinhart V et al (2015) STEP levels are unchanged in pre-frontal cortex and associative striatum in post-mortem human brain samples from subjects with schizophrenia, bipolar disorder and major depressive disorder. PLoS ONE 10:e0121744. https://doi.org/10.1371/journal.pone.0121744
Chen M, Puschmann TB, Wilhelmsson U et al (2017) Neural progenitor cells in cerebral cortex of epilepsy patients do not originate from astrocytes expressing GLAST. Cereb Cortex 27:5672–5682. https://doi.org/10.1093/cercor/bhw338
Hagenauer MH, Schulmann A, Li JZ et al (2018) Inference of cell type content from human brain transcriptomic datasets illuminates the effects of age, manner of death, dissection, and psychiatric diagnosis. PLoS ONE 13:e0200003. https://doi.org/10.1371/journal.pone.0200003
Cheng H, Xuan H, Green CD et al (2018) Repression of human and mouse brain inflammaging transcriptome by broad gene-body histone hyperacetylation. Proc Natl Acad Sci USA 115:7611–7616. https://doi.org/10.1073/pnas.1800656115
Simon MJ, Wang MX, Murchison CF et al (2018) Transcriptional network analysis of human astrocytic endfoot genes reveals region-specific associations with dementia status and tau pathology. Sci Rep 8:1–16. https://doi.org/10.1038/s41598-018-30779-x
Payán-Gómez C, Rodríguez D, Amador-Muñoz D, Ramírez-Clavijo S (2018) Integrative analysis of global gene expression identifies opposite patterns of reactive astrogliosis in aged human prefrontal cortex. Brain Sci. https://doi.org/10.3390/brainsci8120227
Somel M, Franz H, Yan Z et al (2009) Transcriptional neoteny in the human brain. Proc Natl Acad Sci USA 106:5743–5748. https://doi.org/10.1073/pnas.0900544106
Maycox PR, Kelly F, Taylor A et al (2009) Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol Psychiatry 14:1083–1094. https://doi.org/10.1038/mp.2009.18
Somel M, Guo S, Fu N et al (2010) MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res 20:1207–1218. https://doi.org/10.1101/gr.106849.110
Mattson MP, Magnus T (2006) Ageing and neuronal vulnerability. Nat Rev Neurosci 7:278–294. https://doi.org/10.1038/nrn1886
Mariani E, Polidori MC, Cherubini A, Mecocci P (2005) Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B 827:65–75. https://doi.org/10.1016/j.jchromb.2005.04.023
Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71:35–48. https://doi.org/10.1016/j.neuron.2011.06.031
Dringen R, Brandmann M, Hohnholt MC, Blumrich E-M (2015) Glutathione-dependent detoxification processes in astrocytes. Neurochem Res 40:2570–2582. https://doi.org/10.1007/s11064-014-1481-1
Emir UE, Raatz S, McPherson S et al (2011) Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed 24:888–894. https://doi.org/10.1002/nbm.1646
Venkateshappa C, Harish G, Mythri RB et al (2012) Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson’s disease. Neurochem Res 37:358–369. https://doi.org/10.1007/s11064-011-0619-7
Urrila AS, Hakkarainen A, Heikkinen S et al (2004) Stimulus-induced brain lactate: effects of aging and prolonged wakefulness. J Sleep Res 13:111–119. https://doi.org/10.1111/j.1365-2869.2004.00401.x
Zhao N, Liu CC, Qiao W, Bu G (2018) Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol Psychiatry 83:347–357. https://doi.org/10.1016/j.biopsych.2017.03.003
Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ (2018) The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep 22:269–285. https://doi.org/10.1016/j.celrep.2017.12.039
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215. https://doi.org/10.1016/s0166-2236(98)01349-6
Sun W, McConnell E, Pare J-F et al (2013) Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. https://doi.org/10.1126/science.1226740
Aronica E, Gorter JA, Ijlst-Keizers H et al (2003) Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci 17:2106–2118. https://doi.org/10.1046/j.1460-9568.2003.02657.x
García-Bea A, Walker MA, Hyde TM et al (2016) Metabotropic glutamate receptor 3 (mGlu3; mGluR3; GRM3) in schizophrenia: antibody characterisation and a semi-quantitative western blot study. Schizophr Res 177:18–27. https://doi.org/10.1016/j.schres.2016.04.015
Kaiser LG, Schuff N, Cashdollar N, Weiner MW (2005) Age-related glutamate and glutamine concentration changes in normal human brain: 1H MR spectroscopy study at 4 T. Neurobiol Aging 26:665–672. https://doi.org/10.1016/j.neurobiolaging.2004.07.001
Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S (2008) New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev 59:201–220. https://doi.org/10.1016/j.brainresrev.2008.07.007
Allen NJ, Bennett ML, Foo LC et al (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486:410–414. https://doi.org/10.1038/nature11059
Cáceres M, Suwyn C, Maddox M et al (2007) Increased cortical expression of two synaptogenic thrombospondins in human brain evolution. Cereb Cortex 17:2312–2321. https://doi.org/10.1093/cercor/bhl140
Cribbs DH, Berchtold NC, Perreau V et al (2012) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation 9:179. https://doi.org/10.1186/1742-2094-9-179
Stephan AH, Madison DV, Mateos JM et al (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33:13460–13474. https://doi.org/10.1523/JNEUROSCI.1333-13.2013
Su L, Lv X, Miao J (2008) Integrin beta 4 in neural cells. Neuromol Med 10:316–321. https://doi.org/10.1007/s12017-008-8042-1
Fasen K, Elger CE, Lie AA (2003) Distribution of alpha and beta integrin subunits in the adult rat hippocampus after pilocarpine-induced neuronal cell loss, axonal reorganization and reactive astrogliosis. Acta Neuropathol 106:319–322. https://doi.org/10.1007/s00401-003-0733-y
Milner R, Campbell IL (2006) Increased expression of the beta4 and alpha5 integrin subunits in cerebral blood vessels of transgenic mice chronically producing the pro-inflammatory cytokines IL-6 or IFN-alpha in the central nervous system. Mol Cell Neurosci 33:429–440. https://doi.org/10.1016/j.mcn.2006.09.004
Maragakis NJ, Rothstein JD (2006) Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2:679–689. https://doi.org/10.1038/ncpneuro0355
Zeppenfeld DM, Simon M, Haswell JD et al (2017) Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol 74:91–99. https://doi.org/10.1001/jamaneurol.2016.4370
Owasil R, O’Neill R, Keable A et al (2020) The pattern of AQP4 expression in the ageing human brain and in cerebral amyloid angiopathy. Int J Mol Sci. https://doi.org/10.3390/ijms21041225
Montagne A, Barnes SR, Sweeney MD et al (2015) Blood–brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302. https://doi.org/10.1016/j.neuron.2014.12.032
Farrall AJ, Wardlaw JM (2009) Blood–brain barrier: ageing and microvascular disease—systematic review and meta-analysis. Neurobiol Aging 30:337–352. https://doi.org/10.1016/j.neurobiolaging.2007.07.015
Lalo U, Palygin O, North RA et al (2011) Age-dependent remodelling of ionotropic signalling in cortical astroglia. Aging Cell 10:392–402. https://doi.org/10.1111/j.1474-9726.2011.00682.x
Popov A, Brazhe A, Denisov P et al (2020) Astrocytes dystrophy in ageing brain parallels impaired synaptic plasticity. bioRxiv. https://doi.org/10.1101/2020.08.05.237420
Gómez-Gonzalo M, Martin-Fernandez M, Martínez-Murillo R et al (2017) Neuron-astrocyte signaling is preserved in the aging brain. Glia 65:569–580. https://doi.org/10.1002/glia.23112
Loeffler DA, Brickman CM, Juneau PL et al (1997) Cerebrospinal fluid C3a increases with age, but does not increase further in Alzheimer’s disease. Neurobiol Aging 18:555–557. https://doi.org/10.1016/s0197-4580(97)00110-3
Gaya da Costa M, Poppelaars F, van Kooten C et al (2018) Age and sex-associated changes of complement activity and complement levels in a healthy Caucasian population. Front Immunol 9:2664. https://doi.org/10.3389/fimmu.2018.02664
Clarke LE, Liddelow SA, Chakraborty C et al (2018) Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA 115:E1896–E1905. https://doi.org/10.1073/pnas.1800165115
Escartin C, Galea E, Lakatos A et al (2021) Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. https://doi.org/10.1038/s41593-020-00783-4
Pekny M, Pekna M, Messing A et al (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345. https://doi.org/10.1007/s00401-015-1513-1
Nichols NR, Day JR, Laping NJ et al (1993) GFAP mRNA increases with age in rat and human brain. Neurobiol Aging 14:421–429. https://doi.org/10.1016/0197-4580(93)90100-p
David JP, Ghozali F, Fallet-Bianco C et al (1997) Glial reaction in the hippocampal formation is highly correlated with aging in human brain. Neurosci Lett 235:53–56. https://doi.org/10.1016/s0304-3940(97)00708-8
del Valle E, Navarro A, Astudillo A, Tolivia J (2003) Apolipoprotein D expression in human brain reactive astrocytes. J Histochem Cytochem 51:1285–1290. https://doi.org/10.1177/002215540305101005
Jyothi HJ, Vidyadhara DJ, Mahadevan A et al (2015) Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol Aging 36:3321–3333. https://doi.org/10.1016/j.neurobiolaging.2015.08.024
Cruz-Sánchez FF, Moral A, Tolosa E et al (1998) Evaluation of neuronal loss, astrocytosis and abnormalities of cytoskeletal components of large motor neurons in the human anterior horn in aging. J Neural Transm 105:689–701. https://doi.org/10.1007/s007020050088
Escartin C, Guillemaud O, Carrillo-de Sauvage MA (2019) Questions and (some) answers on reactive astrocytes. Glia 67:2221–2247. https://doi.org/10.1002/glia.23687
Franceschi C, Bonafè M, Valensin S et al (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908:244–254. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x
Verkhratsky A, Augusto-Oliveira M, Pivoriūnas A et al (2020) Astroglial asthenia and loss of function, rather than reactivity, contribute to the ageing of the brain. Pflugers Arch. https://doi.org/10.1007/s00424-020-02465-3
Orre M, Kamphuis W, Osborn LM et al (2014) Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging 35:1–14. https://doi.org/10.1016/j.neurobiolaging.2013.07.008
Sekar A, Bialas AR, de Rivera H et al (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530:177–183. https://doi.org/10.1038/nature16549
Allen NJ, Eroglu C (2017) Cell biology of astrocyte-synapse interactions. Neuron 96:697–708. https://doi.org/10.1016/j.neuron.2017.09.056
Pan J, Ma N, Yu B et al (2020) Transcriptomic profiling of microglia and astrocytes throughout aging. J Neuroinflammation 17:1–19. https://doi.org/10.1186/s12974-020-01774-9
Liddelow SA, Guttenplan KA, Clarke LE et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Zhou Y, Song WM, Andhey PS et al (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26:131–142. https://doi.org/10.1038/s41591-019-0695-9
Bu G (2009) Apolipoprotein e and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344. https://doi.org/10.1038/nrn2620
Corder EH, Saunders AM, Risch NJ et al (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7:180–184. https://doi.org/10.1038/ng0694-180
Lin YT, Seo J, Gao F et al (2018) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98:1141-1154.e7. https://doi.org/10.1016/j.neuron.2018.05.008
Tcw J, Liang SA, Qian L et al (2019) Cholesterol and matrisome pathways dysregulated in human APOE ∊4 Glia. SSRN Electron J. https://doi.org/10.2139/ssrn.3435267
Nelson TJ, Sen A (2018) Apolipoprotein E particle size is increased in Alzheimer’s disease. Alzheimer’s Dements 11:10–18. https://doi.org/10.1016/j.dadm.2018.10.005
Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein e and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. https://doi.org/10.1038/nrneurol.2012.263
Namba Y, Tomonaga M, Kawasaki H et al (1991) Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 541:163–166. https://doi.org/10.1016/0006-8993(91)91092-f
Castellano JM, Kim J, Stewart FR et al (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med 3:89ra57. https://doi.org/10.1126/scitranslmed.3002156
Wang C, Najm R, Xu Q et al (2018) Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector article. Nat Med 24:647–657. https://doi.org/10.1038/s41591-018-0004-z
Konijnenberg E, Tijms BM, Gobom J et al (2020) APOE ϵ4 genotype-dependent cerebrospinal fluid proteomic signatures in Alzheimer’s disease. Alzheimer’s Res Ther 12:1–11. https://doi.org/10.1186/s13195-020-00628-z
Ji Y, Gong Y, Gan W et al (2003) Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience 122:305–315. https://doi.org/10.1016/j.neuroscience.2003.08.007
Bussy A, Snider BJ, Coble D et al (2018) Effect of Apolipoprotein E4 on clinical, neuroimaging and biomarker measures in non-carrier participants in the Dominantly Inherited Alzheimer Network. Neurobiol Aging 75:42–50. https://doi.org/10.1016/j.neurobiolaging.2018.10.011
Montagne A, Nation DA, Sagare AP et al (2020) APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581:71–76. https://doi.org/10.1038/s41586-020-2247-3
Blanchard JW, Bula M, Davila-Velderrain J et al (2020) Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med 26:952–963. https://doi.org/10.1038/s41591-020-0886-4
Halliday MR, Rege SV, Ma Q et al (2016) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36:216–227. https://doi.org/10.1038/jcbfm.2015.44
Naba A, Clauser KR, Ding H et al (2016) The extracellular matrix: tools and insights for the “omics” era. Matrix Biol 49:10–24. https://doi.org/10.1016/j.matbio.2015.06.003
Jones VC, Atkinson-Dell R, Verkhratsky A, Mohamet L (2017) Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis 8:1–11. https://doi.org/10.1038/cddis.2017.89
Sen A, Nelson TJ, Alkon DL (2017) ApoE isoforms differentially regulates cleavage and secretion of BDNF. Mol Brain 10:1–7. https://doi.org/10.1186/s13041-017-0301-3
Koch G, Di Lorenzo F, Loizzo S et al (2017) CSF tau is associated with impaired cortical plasticity, cognitive decline and astrocyte survival only in APOE4-positive Alzheimer’s disease. Sci Rep 7:1–12. https://doi.org/10.1038/s41598-017-14204-3
Carter SF, Herholz K, Rosa-Neto P et al (2019) Astrocyte biomarkers in Alzheimer’s disease. Trends Mol Med 25:77–95. https://doi.org/10.1016/j.molmed.2018.11.006
Janelidze S, Mattsson N, Stomrud E et al (2018) CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 91:e867–e877. https://doi.org/10.1212/WNL.0000000000006082
Rodriguez-Vieitez E, Nordberg A (2018) Imaging neuroinflammation: quantification of astrocytosis in a multitracer PET approach. Methods Mol Biol 1750:231–251. https://doi.org/10.1007/978-1-4939-7704-8_16
Carter SF, Schöll M, Almkvist O et al (2012) Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-l-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med 53:37–46. https://doi.org/10.2967/jnumed.110.087031
Griffin WS, Stanley LC, Ling C et al (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86:7611–7615. https://doi.org/10.1073/pnas.86.19.7611
Vijayan VK, Geddes JW, Anderson KJ et al (1991) Astrocyte hypertrophy in the Alzheimer’s disease hippocampal formation. Exp Neurol 112:72–78. https://doi.org/10.1016/0014-4886(91)90115-s
Vanzani MC, Iacono RF, Caccuri RL, Berria MI (2005) Immunochemical and morphometric features of astrocyte reactivity vs. plaque location in Alzheimer’s disease. Medicina (B Aires) 65:213–218
Simpson JE, Ince PG, Lace G et al (2010) Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 31:578–590. https://doi.org/10.1016/j.neurobiolaging.2008.05.015
Sharma A, Kazim SF, Larson CS et al (2019) Divergent roles of astrocytic versus neuronal EAAT2 deficiency on cognition and overlap with aging and Alzheimer’s molecular signatures. Proc Natl Acad Sci USA 116:21800–21811. https://doi.org/10.1073/pnas.1903566116
Lim D, Iyer A, Ronco V et al (2013) Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 61:1134–1145. https://doi.org/10.1002/glia.22502
Planas-Fontánez TM, Dreyfus CF, Saitta KS (2020) Reactive astrocytes as therapeutic targets for brain degenerative diseases: roles played by metabotropic glutamate receptors. Neurochem Res 45:541–550. https://doi.org/10.1007/s11064-020-02968-6
Lim D, Ronco V, Grolla AA et al (2014) Glial calcium signalling in Alzheimer’s disease. Rev Physiol Biochem Pharmacol 167:45–65. https://doi.org/10.1007/112_2014_19
Larramona-Arcas R, González-Arias C, Perea G et al (2020) Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol Neurodegener 15:35. https://doi.org/10.1186/s13024-020-00382-8
Montagne A, Nation DA, Pa J et al (2016) Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol 131:687–707. https://doi.org/10.1007/s00401-016-1570-0
van de Haar HJ, Jansen JFA, van Osch MJP et al (2016) Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging 45:190–196. https://doi.org/10.1016/j.neurobiolaging.2016.06.006
van de Haar HJ, Burgmans S, Jansen JFA et al (2016) Blood–brain barrier leakage in patients with early Alzheimer disease. Radiology 281:527–535. https://doi.org/10.1148/radiol.2016152244
Mei X, Ezan P, Giaume C, Koulakoff A (2010) Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 171:92–105. https://doi.org/10.1016/j.neuroscience.2010.08.001
Foo LC, Allen NJ, Bushong EA et al (2011) Development of a method for the purification and culture of rodent astrocytes. Neuron 71:799–811. https://doi.org/10.1016/j.neuron.2011.07.022
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. https://doi.org/10.1016/j.cell.2006.07.024
Canals I, Hidalgo I, Ginisty A et al (2018) Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat Methods 16:134–134. https://doi.org/10.1038/s41592-018-0264-z
Zhang Y, Pak CH, Han Y et al (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78:785–798. https://doi.org/10.1016/j.neuron.2013.05.029
Ho SM, Hartley BJ, TCW Julia et al (2016) Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods 101:113–124. https://doi.org/10.1016/j.ymeth.2015.11.019
Tcw J, Wang M, Pimenova AA et al (2017) An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Reports 9:600–614. https://doi.org/10.1016/j.stemcr.2017.06.018
Lancaster MA, Knoblich JA (2014) Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc 9:2329–2340. https://doi.org/10.1038/nprot.2016.098
Ormel PR, Vieira de Sá R, van Bodegraven EJ et al (2018) Microglia innately develop within cerebral organoids. Nat Commun. https://doi.org/10.1038/s41467-018-06684-2
Lancaster MA, Renner M, Martin CA et al (2013) Cerebral organoids model human brain development and microcephaly. Nature 501:373–379. https://doi.org/10.1038/nature12517
Matsui TK, Matsubayashi M, Sakaguchi YM et al (2018) Six-month cultured cerebral organoids from human ES cells contain matured neural cells. Neurosci Lett 670:75–82. https://doi.org/10.1016/j.neulet.2018.01.040
Renner M, Lancaster MA, Bian S et al (2017) Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J 36:1316–1329. https://doi.org/10.15252/embj.201694700
Barbar L, Jain T, Zimmer M et al (2020) CD49f is a novel marker of functional and reactive human iPSC-derived astrocytes. Neuron. https://doi.org/10.1016/j.neuron.2020.05.014
Ravaioli F, Bacalini MG, Franceschi C, Garagnani P (2018) Age-related epigenetic derangement upon reprogramming and differentiation of cells from the elderly. Genes (Basel). https://doi.org/10.3390/genes9010039
Miller JD, Ganat YM, Kishinevsky S et al (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13:691–705. https://doi.org/10.1016/j.stem.2013.11.006
Hou J, Yun Y, Xue J et al (2019) D-galactose induces astrocytic aging and contributes to astrocytoma progression and chemoresistance via cellular senescence. Mol Med Rep 20:4111–4118. https://doi.org/10.3892/mmr.2019.10677
Funding
JM was supported by ZonMw (733050504) and EMH and JM by ZonMw (733050816)—The Netherlands Organisation for Health Research and Development, Dementia Research and Innovation Program “Memorabel” with additional support from Alzheimer Nederland. EMH and JM were supported by Alzheimer Nederland (WE.03-2017-04). ZonMw and Alzheimer Nederland projects are part of the cooperative “Deltaplan for Dementia”, the Dutch national platform to address and manage the growing problem of dementia. JM received support from the Brain Center UMC Utrecht, Rudolf Magnus Talent fellowship. MV is supported by the Dutch Research Council (NWO) Graduate Program (022.006.001).
Author information
Authors and Affiliations
Contributions
MV and JM conceived the outline of the review, MV drafted the article with support of JM. MV, JM and EH discussed content, MV designed the figure, JM and EH critically revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special issue: In Honor of Prof. Vladimir Parpura.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verkerke, M., Hol, E.M. & Middeldorp, J. Physiological and Pathological Ageing of Astrocytes in the Human Brain. Neurochem Res 46, 2662–2675 (2021). https://doi.org/10.1007/s11064-021-03256-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03256-7