
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, characterized by progressive loss of dopaminergic neurons that results in characteristic motor and non-motor symptoms. l-3,4 dihydroxyphenylalanine (l-DOPA) is the gold standard therapy for the treatment of PD. However, long-term use of l-DOPA leads to side effects such as dyskinesias and motor fluctuation. Since purines have neurotransmitter and co-transmitter properties, the function of the purinergic system has been thoroughly studied in the nervous system. Adenosine and adenosine 5′-triphosphate (ATP) are modulators of dopaminergic neurotransmission, neuroinflammatory processes, oxidative stress, excitotoxicity and cell death via purinergic receptor subtypes. Aberrant purinergic receptor signalling can be either the cause or the result of numerous pathological conditions, including neurodegenerative disorders. Many data confirm the involvement of purinergic signalling pathways in PD. Modulation of purinergic receptor subtypes, the activity of ectonucleotidases and ATP transporters could be beneficial in the treatment of PD. We give a brief summary of the background of purinergic signalling focusing on its roles in PD. Possible targets for pharmacological treatment are highlighted.
Similar content being viewed by others


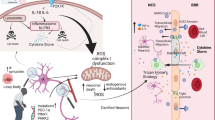
Introduction
Parkinsons’s Disease: Pathophysiological Background
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta that results in dopamine (DA) deficiency in the striatum. The ongoing degeneration of this peculiar pathway causes the characteristic motor symptoms such as resting tremor, rigidity, bradykinesia and postural instability [1, 2]. Besides dopaminergic neural degeneration, the presence of Lewy bodies (protein aggregates) due to misfolding of α-synuclein occurs in various regions of the affected brain [3]. In spite of many studies on the pathogenesis of PD, the precise mechanism underlying these events has not been unraveled yet. However, a genetic predisposition associated with disturbed proteostasis due to impaired ubiquitin–proteasome system, mitochondrial dysfunction, oxidative stress and neuroinflammation seems to play cardinal roles for the α-synuclein aggregation and the progression of pathology in PD [4,5,6,7]. Among these factors, the pathological, self-amplifying interaction between mitochondrial dysfunction and oxidative stress has been early recognized, which might be a key factor responsible for the selective vulnerability of dopaminergic neurons in PD, and one potential reason behind the clinical failures of neuroprotective therapies so far [8]. Dysfunction of the mitochondrial complex I results in an enhanced production of reactive oxygen species (ROS), which, in turn will inhibit complex I and other vital metabolic enzymes such as alpha-ketoglutarate dehydrogenase, whilst the latter also serves as a source of ROS generation in mitochondria [9, 10]. Simultaneous or preceding mitochondrial dysfunction exacerbates the effect of oxidative stress on pathological monoamine release from nerve terminals [11, 12]. This process leads to the formation of toxic, oxidative DA metabolites, such as dopamine quinone, which might further amplify the ongoing degeneration process [13]. Therefore, disease-modifying potential could be primarily expected from those novel multi-target therapies, which simultaneously target the above mentioned pivotal pathological pathways and prevent their pathological interaction [14, 15].
The Current Treatment of PD
As for the symptomatic treatment of PD, the clinical break-through came with the first clinical trials of DA replacement therapy using the high dosage of the DA precursor l-3,4 dihydroxyphenylalanine (l-DOPA) [16,17,18,19]. l-DOPA is able to cross the blood–brain barrier and converts into DA that engages specific DA receptor subtypes (D1 to D5) [20]. However, long-term use of l-DOPA leads to a dysbalance of striatal circuits of the motor system and leads to side effects such as l-DOPA induced dyskinesias and motor fluctuation in 50% of patients after 5 years of continuous treatment [21, 22]. The therapeutic management of these complications is difficult and there is a need for developing effective and new pharmacological therapies against motor fluctuation and dyskinesias [23].
Purinergic Signalling: Concept and Purinergic Receptors
The concept of purinergic signalling, being adenosine 5′-triphosphate (ATP) as an extracellular signalling molecule with neurotransmitter properties was proposed in the early 1970s [24, 25]. A couple of years later, purines were also described as co-transmitters and neuromodulators in the peripheral and central nervous system (CNS), as they are able to modulate other signalling pathways and neurotransmitter systems [26,27,28]. ATP is co-released with acetylcholine, catecholamines, γ-amino butyric acid (GABA), glutamate and DA in the CNS [29,30,31,32,33,34]. Extracellular ATP is released from cells under physiological conditions. The levels of extracellular ATP are controlled by ectonucleotidases that catalyze its degradation [35, 36].
There are two families of purinergic receptors, which are distinguished by their main agonists [37]. P1 receptors are G protein-coupled metabotropic receptors activated by adenosine and can be subdivided into four subtypes (A1, A2A, A2B, A3). P2 receptors are subdivided into two classes: P2X(1-7) ionotropic receptors, activated by ATP and G protein-coupled metabotropic P2Y(1-2,4,6,11-14) receptors, activated by ATP, adenosine diphosphate (ADP), uridine di- and triphosphate (UDP and UTP), or UDP-glucose depending on the receptor subtype [38,39,40]. ATP is able to bind to the extracellular ligand-binding site of P2X receptors and leading to conformational change that opens a permeable channel to Na+, K+ and Ca2+. The activation of these ionotropic receptors is important for Ca2+-induced intracellular signalling pathways [41,42,43]. Depending on the activated adenosine and P2 receptor subtype, the induced signalling pathway may vary. These activated receptors are able to make alterations in Ca2+ levels, which modulate the activity of several secondary messengers involved in physiological processes [44,45,46]. The final effects of purinergic receptor-mediated signalling depend on the cell type and other physiological (neurogenesis, proliferation, cell death, stem cell differentiation) or pathological cellular conditions (inflammatory, neurological, psychiatric, oncological, cognitive, neuromuscular and neuromotor diseases) [47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66]. Purinergic receptor activation may have para- or autocrine nature, which is characteristic for astrocytes in the regulation of neuronal activity [67]. Not only purinergic receptors but membrane nucleotide/nucleoside transporters, channels and ectonucleotidases also play important role in purinergic signalling [36, 68,69,70].
Adenosine is the predominant, presynaptic modulator of neurotransmitter release in the CNS, although ATP has presynaptic modulator effect as well [71,72,73]. Adenosine is produced by enzymatic breakdown of released ATP, but some CNS cells are able to release adenosine directly [74]. A1 and A2A receptors have higher affinity (activated by physiological extracellular levels of adenosine) and A2B and A3 receptors have lower affinity (activated by higher extracellular levels of adenosine) for the ribonucleoside [75,76,77]. The adenosine A1 and A2A receptors are highly expressed in the brain and CNS, where they have profound influence on neuronal activity. Adenosine A1 receptor is the dominant adenosine receptor subtype in the CNS. Adenosine A1 receptors can be found in various cortical and subcortical regions of the brain, while A2A receptors are mainly expressed in the striatum [78,79,80,81] (Table 1). In contrast, adenosine A2B and A3 receptors are mainly found in peripheral tissues, even though low levels of these receptors are also expressed in some regions of the brain [82,83,84].
There is a heterogeneous distribution of P2 purinergic receptors in the CNS as well. For instance P2X1 receptors are predominantly expressed in the cerebellum, while P2X3 receptors are expressed in the brainstem [85, 86], and they can be found in the basal ganglia with variable expression level [87] (Table 2). Various P1 and P2 receptor subtypes are also expressed by microglia, astrocytes and oligodendrocytes [88,89,90,91,92,93]. Extracellular nucleotides act as messengers between neuronal and non-neuronal cells, thereby integrating functional activity between neurons, glial and vascular cells in the CNS [94,95,96,97,98]. Adenosine and ATP—as key players in neuron–glia interaction and microglial activation—are modulators of neuroinflammatory processes, oxidative stress, excitotoxicity and cell death [99,100,101,102]. Aberrant purinergic receptor signalling can be the cause or result of numerous pathological conditions, including neurodegenerative disorders [103]. Here, we explore the importance of purinergic signalling in PD to suggest potential targets for novel therapies.
Purinergic Signalling Involvement in PD
Purinergic Gene Polymorphisms in PD
Two ADORA2A (A2A receptor) polymorphisms (rs71651683, a 5′ variant or rs5996696, a promoter region variant) were inversely associated with genetic PD risk, moreover, there was evidence of interaction with coffee consumption [104]. CYP1A2a is an enzyme, which is responsible for caffeine metabolism, two polymorphisms (rs762551 or rs5996696) of the enzyme in homozygous coffee drinkers reduced PD risk [104]. Humans with R1628P variant (LRRK2 risk variant) who did not take caffeine had a 15 times increased risk of PD [105]. GRIN2A encodes an N-methyl-d-aspartate-2A (NMDA) glutamate receptor subunit involved in central excitatory neurotransmission, which is associated with A2A receptor activation. Carriers of GRIN2A rs4998386-T allele had a lower risk of PD, than carriers of rs4998386-CC genotype among heavy coffee drinkers [106]. There is evidence that creatine is able to hasten PD progression in GRIN2A coffee drinkers, which demonstrates an example of a genetic factor interacting with environmental factors exemplifying the complexity of environment–gene interactions in the progression of PD [107]. In addition, P2X7 receptor 1513A>C polymorphism is a risk factor for sporadic PD, late-onset PD and male PD in Han Chinese population [108].
Adenosine Receptor-Mediated Signalling in PD
A2A receptors are enriched in dopaminergic brain areas (the highest expression of these receptors are in the striatum), thus pointing to a significant role of purines in motor control [109]. A2A and DA D2 receptors are mainly expressed in the neurons of the indirect pathway of striatal circuits projecting to the globus pallidus, in contrast to A1 and DA D1 receptors, which are mainly found on the neurons of the direct pathway of motor control projecting to the internal globus pallidus and substantia nigra pars reticulata. The main adenosine signalling mechanism is via the cyclic adenosine monophosphate (cAMP)-dependent pathway. Activated A2A receptors stimulate the enzymatic function of adenyl cyclase that increases cAMP levels and depresses the signalling mediated by D2 receptors. Activation of protein Gi-coupled DA D2 receptors leads to reduction in the cAMP level. There is a reciprocal situation in the direct pathway of motor control with protein Gs-coupled D1 and protein Gi/o-coupled A1 receptors. Generally, adenosine acts as a negative modulator of D1- and D2-mediated actions in the direct and indirect pathways [110,111,112].
The antagonistic functional interaction between adenosine A2A and DA D2 receptors may depend on the formation of receptor heterodimers (A2A-D2 heteroreceptor complexes) in the striatum thereby balancing the inhibitory and excitatory impulses in the striatal circuits [112]. Not only dopaminergic mechanisms, but non-dopaminergic modes of action of A2A receptors may involve interactions with various non-dopaminergic receptors, possibly by forming heterodimeric and/or multimeric receptor complexes [23]. Thus, adenosine A2A receptors may adjust the actions of striatal adenosine A1 receptors (A1-A2A heteroreceptor complexes), metabotropic glutamate receptors (mGlu) 5 (A2A-mGlu5 heteroreceptor complexes), cannabinoid receptor type 1 (CB1) receptors (A2A-CB1 heteroreceptor complexes) and serotonin 1A (5-HT1A) receptors [113,114,115]. Moreover, studies also suggested the presence of multimeric A2A-D2-mGlu5 and A2A-CB1-D2 receptor complexes in the striatum [116, 117]. These functional interactions between receptors may modulate the activity of striatal efferent neurons and influence motor behavior [23]. In general, adenosine tone appears as a key for the fine tune control of DA dependent actions in the basal ganglia and affects non-dopaminergic mechanisms also [20].
Adenosine receptor antagonists (especially non-selective A2A receptor antagonists, such as methylxanthines, caffeine, or selective A2A antagonists) have been shown to enhance therapeutic effect of l-DOPA in a wide range of animal models of PD [118,119,120,121]. A2A homoreceptor complexes are in balance with DA D2 homoreceptor complexes in intact striatum [122,123,124,125,126]. Dysbalance of striatal circuits leads to motor inhibition and disruption of this balance in PD leads to increased signalling via A2A receptors and decreased signalling via DA D2 receptors. These changes explain the beneficial effect of A2A receptor antagonists on increasing motor functions without worsening l-DOPA-induced dyskinesias [20, 127].
A2A receptor antagonists have been used in clinical trials in patients with PD (Table 3). Istradefylline is a xanthine-based compound with increased selectivity for A2A receptors against A1 receptors, which is used concomitantly with l-DOPA [128]. The drug was not approved in the USA because there was no significant reduction in off time compared to l-DOPA treatment [129]. In contrast, istradefylline was approved in Japan in 2013 with the trade name Nouriast® to enhance the antiparkinsonian effect of l-DOPA with less long-term side effects [130, 131]. Preladenant is a second-generation A2A receptor antagonist, which failed in phase III clinical trials in the treatment of PD because the compound was not superior to placebo in reducing off state [132, 133]. Vipadenant is a triazolopyromidine-based drug, which has increased selectivity for A2A receptors versus A1 and A3 receptors [134]. Its development as an antiparkinsonian medication was stopped; however, A2A receptor antagonists have considerable potential in novel immune-oncology and cardiology therapies [113, 135,136,137]. Another adenosine A2A receptor antagonist, tozadenant was safe, well tolerated and effective in reducing off time in PD patients in phase II trial but phase III clinical trial was discontinued because of serious adverse events (agranulocytosis) [23, 133, 138]. There have been many drug trials for selective A2A receptor antagonists. Most of them were shown to be safe, well tolerated and beneficial; however, the majority did not reach the regulatory threshold for efficacy to be approved as PD drugs [139, 140]. Development of bivalent drugs (able to bind to two receptors simultaneously) to target A2A-D2 heteroreceptor complexes acting on A2A and DA D2 receptors may be a good therapeutic approach in the future. Heterobivalent drugs offers the opportunity to target the orthosteric sites of the receptors in the heterodimer with a higher affinity and a higher specificity versus corresponding homomers and reduce the dose required for therapy and, accordingly, the side effects [20].
Adenosine A2A receptor antagonists may also involve direct or indirect actions at microglia and inflammatory processes. Pre-treatment of slices from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-injected mice with preladenant facilitates the ability of activated microglia to respond to tissue damage [141]. The nonselective A1/A2A adenosine receptor antagonist caffeine and the selective A2A receptor antagonist (KW-6002) had anti-inflammatory potential in a rat model of lipopolysaccharide (LPS)-induced neuroinflammation [142].
The Role of A2A Receptors in Synucleopathy
Increased striatal A2A receptor expression was observed as an early pathological event in PD and increased A2A receptor expression was detected after hippocampal injection of α-synuclein in mice [143, 144]. A2A receptor-knock out mice showed resistance against α-synuclein induced insults [145]. A2A receptor antagonism restrained hyperactivation of NMDA-glutamate receptors and decreased the aggregation of α-synucleins [146]. Based upon these results, A2A receptors seem to have role in the pathological process of synucleinopathy [111].
P2 Receptor-Mediated Signalling in PD
P2 ionotropic and metabotropic receptors are widely expressed in basal ganglia and in various cell types, such as neurons and astrocytes [87, 147, 148]. 6-Hydroxidopamine (6-OHDA) induced lesions of nigral dopaminergic neurons generate a significant decrease in the expression of P2X and P2Y receptor proteins from striatal spiny neurons and GABAergic interneurons, thus confirming the involvement of P2 receptors and extracellular ATP in the striatal circuits [87]. P2Y1 and P2X1-4, 6 receptor protein subtypes are expressed in dopaminergic neurons with co-expression of P2X1 with DA D1 receptors, therefore stimulation of P2 receptors by ATP induces an increased release of DA in the striatum [149,150,151,152]. In a neuronal cell model, extracellular ATP induced a significant increase in intracellular α-synuclein levels, which was the result of lysosome dysfunction caused by P2X1 receptor activation [153].
Many data have implicated the role of P2X7 receptor in PD. P2X7 receptor antagonism with A-438059 or Brilliant Blue G (BBG) prevented DA deficit in the striatum and 6-OHDA-induced hemiparkinsonian behavior [154, 155]. However, P2X7 receptor deficiency or inhibition did not promote the survival of dopaminergic neurons in rotenone and MPTP induced animal models of PD [156]. It is presumed that there is a massive release of ATP during cell death in the lesioned striatum and substantia nigra, which activates cell death pathways via purinergic receptors and is able to activate further purinergic subtypes [20]. Permanent purinergic receptor activation and ATP release seem to play a key role in the neuronal death, which exacerbates α-synuclein aggregation in PD [87]. The accumulation of α-synuclein might overwhelm the capacity of intracellular protein-degradation mechanisms and induce neuroinflammation, which creates a positive feedback loop promoting the degeneration of dopaminergic cells [7]. α-Synuclein-induced intracellular free calcium mobilization in neuronal cells depends on the activation of purinergic P2X7 receptors. In the same study, activation of P2X7 receptors lead to ATP release with the recruitment of the pore forming protein pannexin1, whilst α-synuclein decreased the activity of extracellular ecto-ATPase which is responsible for ATP degradation [157]. Stimulation of the microglial P2X7 receptor by extracellular α-synuclein increased oxidative stress, which was prevented with the use of P2X7 receptor antagonist [158].
DA neurotransmission has been linked to calcium signalling. There is data that P2Y1 receptor is involved in the regulation of calcium signalling [159]. Neurodegeneration induced by 6-OHDA in nigrostriatal dopaminergic neurons was reduced by pretreatment with diadenosine tetraphosphate (AP4A, an endogenous diadenosine polyphosphate) possibly through an anti-apoptotic mechanism and the activation of P2Y1 and P2Y4 receptors [160]. Recently, expression levels of P2Y6 receptor in PD patients younger than 80 years were higher than healthy controls and multiple system atrophy (MSA) patients and P2Y6 receptor could thereby be a potential clinical biomarker of PD. P2Y6 receptor was also upregulated in LPS-treated microglial cells and involved in proinflammatory cytokine release through UDP secretion [161]. Another study showed that expression of P2Y6 receptor on neuronal SH-SY5Y cell is associated with the progression of oxidative stress and cell death induced by 1-methyl-4-phenylpyridinium (MPP+) [162]. In vivo, LPS induced microglial activation and delayed neuronal loss was prevented by selective inhibition of P2Y6 receptor with MRS2578 [163]. Based on these studies P2Y6 receptor subtype seems to be involved in the process of neuroinflammation in PD and blocking UDP/P2Y6 receptor signalling could reverse these pathological processes [161].
Conclusion
In general, many data confirm the involvement of purinergic signalling pathways in PD. Modulation of purinergic receptor subtypes, the activity of ectonucleotidases and ATP transporters could be beneficial in the treatment of PD. Antagonism of A2A, P2X1, P2X7 and P2Y6 receptor subtypes is a promising weapon against PD via various ways: reducing l-DOPA induced dyskinesia, influencing neuroinflammation, preventing α-synuclein aggregation, reducing microglia activation. Development of new bivalent compounds to target A2A-D2 heteroreceptor complexes, which are orally bioavailable and can cross the blood–brain barrier could be a potential therapeutic tool. In addition, multi-target compounds targeting self-amplifying circuits controlled by purinergic and non-purinergic receptors could be a viable strategy to obtain the desired disease-modifying effect [164]. Additional studies and better quality PD animal models are required for the deeper understanding of underlying unknown pathological processes in PD and the role of purinergic signalling in it.
Abbreviations
- ADORA2A:
-
Adenosine A2A receptor
- ADP:
-
Adenosine 5′-diphosphate
- AP4A:
-
Diadenosine tetraphosphate
- ATP:
-
Adenosine 5′-triphosphate
- cAMP:
-
Cyclic adenosine monophosphate
- CB1 :
-
Cannabinoid receptor type 1
- DA:
-
Dopamine
- GABA:
-
γ-Amino butyric acid
- GRIN2A:
-
Glutamate ionotropic receptor NMDA type subunit 2A
- 5-HT1A :
-
5-Hydroxytryptamine/serotonin receptor 1A
- l-DOPA:
-
l-3,4 dihydroxyphenylalanine
- LPS:
-
Lipopolysaccharide
- LRRK2:
-
Leucine-rich repeat kinase 2
- 6-OHDA:
-
6-hydroxydopamine
- mGlu:
-
Metabotropic glutamate receptor
- MPP+ :
-
1-Methyl-4-phenylpyridinium
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSA:
-
Multiple system atrophy
- NMDA:
-
N-methyl-d-aspartate
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- UDP:
-
Uridine 5′-diphosphate
- UTP:
-
Uridine 5′-triphosphate
References
Dehay B, Bourdenx M, Gorry P et al (2015) Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol 14:855–866
Olanow CW, Kieburtz K, Odin P et al (2014) Continuous intrajejunal infusion of levodopa–carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol 13:141–149
Braak H, Del Tredici K, Rüb U et al (2013) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Ghavami S, Shojaei S, Yeganeh B et al (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 112:24–49
Osellame LD, Duchen MR (2013) Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 9:1633–1635
Tansey MG, Goldberg MS (2010) Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis 37:510–518
Zhang G, Xia Y, Wan F et al (2018) New perspectives on roles of alpha-synuclein in Parkinson’s disease. Front Aging Neurosci 10:370
Tretter L, Sipos I, Adam-Vizi V (2004) Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochem Res 29:569–577
Tretter L, Adam-Vizi V (2004) Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci 24:7771–7778
Adam-Vizi V, Tretter L (2013) The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem Int 62:757–763
Milusheva E, Sperlagh B, Shikova L et al (2003) Non-synaptic release of [3H]noradrenaline in response to oxidative stress combined with mitochondrial dysfunction in rat hippocampal slices. Neuroscience 120:771–781
Milusheva E, Baranyi M, Kittel Á et al (2005) Increased sensitivity of striatal dopamine release to H2O2 upon chronic rotenone treatment. Free Radic Biol Med 39:133–142
Baranyi M, Milusheva E, Vizi ES et al (2006) Chromatographic analysis of dopamine metabolism in a Parkinsonian model. J Chromatogr 1120:13–20
Milusheva E, Baranyi M, Kormos E et al (2010) The effect of antiparkinsonian drugs on oxidative stress induced pathological [3H]dopamine efflux after in vitro rotenone exposure in rat striatal slices. Neuropharmacology 58:816–825
Baranyi M, Porceddu PF, Gölöncsér F et al (2016) Novel (hetero)arylalkenyl propargylamine compounds are protective in toxin-induced models of Parkinson’s disease. Mol Neurodegener 11:6
Birkmayer W, Hornykiewicz O (1962) The l-dihydroxyphenylalanine (l-DOPA) effect in Parkinson’s syndrome in man: on the pathogenesis and treatment of Parkinson akinesis. Arch Psychiatr Nervenkrankh Z Gesamte Neurol Psychiatr 203:560–574
Birkmayer W, Hornykiewicz O (1964) Additional experimental studies on l-DOPA in Parkinson’ syndrome and Reserpine Parkinsonism. Arch Psychiatr Nervenkrankh 206:367–381
Cotzias GC, Van Woert MH, Schiffer LM (1967) Aromatic amino acids and modification of Parkinsonism. N Engl J Med 276:374–379
Cotzias GC, Papavasiliou PS, Gellene R (1969) Modification of Parkinonism-chronic treatment with l-DOPA. N Engl J Med 280:337–345
Navarro G, Borroto-Escuela DO, Fuxe K et al (2016) Purinergic signaling in Parkinson’s disease. Relevance for treatment. Neuropharmacology 104:161–168
Lang AE (2009) When and how should treatment be started in Parkinson disease? Neurology 72:S39–S43
Olanow CW, Stern MB, Sethi K (2009) The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72:S1–S136
Pinna A, Serra M, Morelli M et al (2018) Role of adenosine A2A receptors in motor control: relevance to Parkinson’s disease and dyskinesia. J Neural Transm (Vienna) 125:1273–1286
Burnstock G, Campbell G, Satchell D et al (1970) Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br J Pharmacol 40:668–688
Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24:509–581
Burnstock G (1976) Do some nerve cells release more than one transmitter? Neuroscience 1:239–248
Burnstock G (1997) The past, present and future of purine nucleotides as signaling molecules. Neuropharmacology 36:1127–1139
Burnstock G (2009) Purinergic cotransmission. Exp Physiol 94:20–24
Potter P, White TD (1980) Release of adenosine 5′-triphosphate from synaptosomes from different regions of rat brain. Neuroscience 5:1351–1356
Poelchen W, Sieler D, Wirkner K et al (2001) Co-transmitter function of ATP in central catecholaminergic neurons of the rat. Neuroscience 102:593–602
Sperlágh B, Sershen H, Lajtha A et al (1998) Co-release of endogenous ATP and [3H]noradrenaline from rat hypothalamic slices: origin and modulation by α2-adrenoreceptors. Neuroscience 82:511–520
Jo YH, Role LW (2002) Coordinate release of ATP and GABA at in vitro synapses of lateral hypothalamic neurons. J Neurosci 22:4794–4804
Mori M, Heuss C, Gahwiler BH et al (2001) Fast synaptic transmission mediated by P2X receptors in CA3 pyramidal cells of rat hippocampal slice cultures. J Physiol 535:115–123
Krügel U, Kittner H, Franke H et al (2003) Purinergic modulation of neuronal activity in the mesolimbic dopaminergic system in vivo. Synapse 47:134–142
Zimmermann H (2001) Ectonucleotidases: some developments and a note on nomenclature. Drug Dev Res 52:44–56
Zimmermann H (2006) Ectonucleotidases in nervous system. Purinergic Signal Neuron–Glia Interact 276:113–130
Burnstock G (1978) A basis for distinguishing two types of purinergic receptor. In: Straub RW, Bolis L (eds) Cell membrane receptors for drugs and hormones: a multidisciplinary approach. Raven Press, New York, pp 107–118
Burnstock G (2007) Purine and pyrimidine receptors. Cell Mol Life Sci 64:1471–1483
Ciruela F, Albergaria C, Soriano C et al (2010) Adenosine receptors interacting proteins (ARIPs): behind the biology of adenosine signaling. Biochim Biophys Acta 1798:9–20
Burnstock G (2014) Purinergic signalling: from discovery to current developments. Exp Physiol 99:16–34
Khakh BS, Burnstock G, Kennedy C et al (2001) International Union Of Pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev 53:107–118
Surprenant A, North RA (2009) Signaling at purinergic P2X receptors. Annu Rev Physiol 71:333–359
Puchalowicz K, Baranowska-Bosiacka I, Dziedziejko V et al (2015) Purinergic signaling and the functioning of the nervous system cells. Cell Mol Biol Lett 20:867–918
Dubyak GR, el-Moatassim C (1993) Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol 265:C577–C606
Abbracchio MP, Burnstock G, Boeynaems JM et al (2006) International Union of Pharmacology LVIII: update on the P2Y G protein coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
Oliveira A, Illes P, Ulrich H (2016) Purinergic receptors in embryonic and adult neurogenesis. Neuropharmacology 104:272–281
Beamer E, Gölöncsér F, Horváth G et al (2016) Purinergic mechanisms in neuroinflammation: an update from molecules to behavior. Neuropharmacology 104:94–104
Madeira MH, Boia R, Ambrósio AF et al (2017) Having a coffee break: the impact of caffeine consumption on microglia-mediated inflammation in neurodegenerative diseases. Mediat Inflamm 2017:1–12
Przybyla T, Sakowicz-Burkiewicz M, Pawelczyk T (2018) Purinergic signaling in B cells. Acta Biochim Pol 65:1–7
Allard B, Beavis PA, Darcy PK et al (2016) Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol 29:7–16
Vijayan D, Young A, Teng M et al (2017) Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer 17:709–724
Whiteside TL (2017) Targeting adenosine in cancer immunotherapy: a review of recent progress. Expert Rev Anticancer Ther 17:527–535
Kazemi MH, Raoofi Mohseni S, Hojjat-Farsangi M et al (2018) Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J Cell Physiol 233:2032–2057
Burnstock G, Fredholm BB, Verkhratsky A (2011) Adenosine and ATP receptors in the brain. Curr Top Med Chem 11:973–1011
Stockwell J, Jakova E, Cayabyab FS (2017) Adenosine A1 and A2A receptors in the brain: current research and their role in neurodegeneration. Molecules 22:676
Lindberg D, Shan D, Ayers-Ringler J et al (2015) Purinergic signaling and energy homeostasis in psychiatric disorders. Curr Mol Med 15:275–295
Csóka B, Töro G, Vindeirinho J et al (2017) A2A adenosine receptors control pancreatic dysfunction in high-fat-diet-induced obesity. FASEB J 31:4985–4997
Parpura V, Fisher ES, Lechleiter JD et al (2017) Glutamate and ATP at the interface between signaling and metabolism in astroglia: examples from pathology. Neurochem Res 42:19–34
Tozzi M, Novak I (2017) Purinergic receptors in adipose tissue as potential targets in metabolic disorders. Front Pharmacol 8:878
Labazi H, Teng B, Mustafa SJ (2018) Functional changes in vascular reactivity to adenosine receptor activation in type I diabetic mice. Eur J Pharmacol 820:191–197
Ortiz R, Ulrich H, Zarate CA et al (2015) Purinergic system dysfunction in mood disorders: a key target for developing improved therapeutics. Prog Neuropsychopharmacol Biol Psychiatry 57:117–131
Krügel U (2016) Purinergic receptors is psychiatric disorders. Neuropharmacology 104:212–225
Cheffer A, Castillo AR, Corrêa-Velloso JC et al (2017) Purinergic system in psychiatric diseases. Mol Psychiatry 23:94–106
Illes P, Verkhratsky A (2016) Purinergic neurone–glia signalling in cognitive-related pathologies. Neuropharmacology 104:62–75
Burnstock G, Arnett TR, Orriss IR (2013) Purinergic signaling in the musculoskeletal system. Purinergic Signal 9:541–572
Safarzadeh E, Jadidi-Niaragh F, Motallebnezhad M et al (2016) The role of adenosine and adenosine receptors in the immunopathogenesis of multiple sclerosis. Inflamm Res 65:511–520
Pascual O, Casper KB, Kubera C et al (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116
Scemes E, Suadicani SO, Dahl G et al (2007) Connexin and pannexin mediated cell-cell communication. Neuron–Glia Biol 3:199–208
Abbrachio MP, Burnstock G, Verkhratsky A et al (2009) Purinergic signaling in the nervous system: an overview. Trends Neurosci 32:19–29
Lapato AS, Tiwari-Woodruff SK (2017) Connexins and pannexins: at the junction of neuro-glial homeostasis and disease. J Neurosci Res 96:31–44
Vizi ES, Knoll J (1976) The inhibitory effect of adenosine and related nucleotides on the release of acetylcholine. Neuroscience 1:391–398
Dunwiddie TV (1985) The physiological role of adenosine in the central nervous system. Int Rev Neurobiol 27:63–139
Cunha RA, Ribeiro JA (2000) ATP as a presynaptic modulator. Life Sci 68:119–137
Wall MJ, Dale N (2007) Auto-inhibition of rat parallel fibre-Purkinje cell synapses by activity-dependent adenosine release. J Physiol 581:553–565
Fredholm BB, IJzerman AP, Jacobson KA et al (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63:1–34
Chen JF, Pedata F (2008) Modulation of ischemic brain injury and neuroinflammation by adenosine A2A receptors. Curr Pharm Des 14:1490–1499
Pedata F, Dettori I, Coppi E et al (2016) Purinergic signalling in brain ischemia. Neuropharmacology 104:105–130
Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79:463–484
Augood SJ, Emson PC (1994) Adenosine A2A receptor mRNA is expressed by enkephalin cells but not somatostatin cells in rat striatum: a co-expression study. Mol Brain Res 22:204–210
Dixon AK, Gubitz AK, Sirinathsinghji DJ et al (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118:1461–1468
Sebastião AM, Ribeiro JA (2009) Adenosine receptors and the central nervous system. Handb Exp Pharmacol 193:471–534
Feoktistov I, Biaggioni I (1997) Adenosine A2B receptors. Pharmacol Rev 49:381–402
Hammarberg C, Schulte G, Fredholm BB (2003) Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem 86:1051–1054
Rivkees SA, Thevananther S, Hao H (2000) Are A3 adenosine receptors expressed in the brain? NeuroReport 11:1025–1030
Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240:31–304
Guo W, Xu X, Gao X et al (2008) Expression of P2X5 receptors in the mouse central nervous system. Neuroscience 128:697–712
Amadio S, Montilli C, Picconi B et al (2007) Mapping P2X and P2Y receptor proteins in striatum and substantia nigra: an immunohistological study. Purinergic Signal 3:389–398
Moore D, Chambers J, Waldvogel H et al (2000) Regional and cellular distribution of the P2Y1 purinergic receptor in the human brain: striking neuronal localization. J Comp Neurol 421:374–384
Miras-Portugal MT, Marìn-García P, Carrasquera LM et al (2007) Physiological role of extracellular nucleotides at the central nervous system: signaling through P2X and P2Y receptors. An R Acad Nac Farm 73:1127–1157
Verkhratsky A, Krishtal OA, Burnstock G (2009) Purinoreceptors in neuroglia. Mol Neurobiol 39:190–208
Fukumitsu N, Ishii K, Kimura Y et al (2005) Adenosine A1 receptor mapping of the human brain by PET with 8-dicyclopropylmethyl-1-11C-methyl-3-propylxanthine. J Nucl Med 46:32–37
Ishiwata K, Mishina M, Kimura Y et al (2005) First visualization of adenosine A(2A) receptors in the human brain by positron emission tomography with [11C]TMSX. Synapse 55:133–136
Sheth S, Brito R, Mukherjea D et al (2014) Adenosine receptors: expression, function and regulation. Int J Mol Sci 15:2024–2052
Abbracchio MP, Burnstock G (1998) Purinergic signaling: pathophysiological roles. Jpn J Pharmacol 78:113–145
Fields D, Burnstock G (2006) Purinergic signaling in neuron–glial interactions. Nat Neurosci Rev 7:423–436
Parpura V, Zorec R (2010) Gliotransmission: exocytotic release from astrocytes. Brain Res Rev 63:83–92
Matute C, Cavaliere F (2011) Neuroglial interactions mediated by purinergic signaling in the pathophysiology of CNS disorders. Semin Cell Dev Biol 22:252–259
Verderio C, Matteoli M (2011) ATP in neuron–glia bidirectional signaling. Brain Res Rev 66:106–114
Cunha RA (2016) How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem 139:1019–1055
Borea PA, Gessi S, Merighi S et al (2017) Pathological overproduction: the bad side of adenosine. Br J Pharmacol 174:1945–1960
Faas MM, Sáez T, de Vos P (2017) Extracellular ATP and adenosine: the Yin and Yang in immune responses? Mol Asp Med 55:9–19
Miras-Portugal MT, Sebastian-Serrano Á, de Diego GarcíaL et al (2017) Neuronal P2X7 receptor: involvement in neuronal physiology and pathology. J Neurosci 37:7063–7072
Burnstock G (2016) An introduction to the roles of purinergic signaling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104:4–17
Popat RA, Van Den Eeden SK, Tanner C et al (2011) Coffee, ADORA2A, and CYP1A2: the caffeine connection in Parkinson’s disease. Eur J Neurol 18:756–765
Kumar PM, Paing SS, Li H et al (2015) Differential effect of caffeine intake in subjects with genetic susceptibility to Parkinson’s disease. Sci Rep 5:15492
Yamada-Fowler N, Frekdrikson M, Söderkvist P (2014) Caffeine interaction with glutamate receptor gene GRIN2A: Parkinson’s disease in Swedish population. PLoS ONE 9:e99294
Simon DK, Wu C, Tilley BC et al (2017) Caffeine, creatine, GRIN2A and Parkinson’s disease progression. J Neurol Sci 375:355–359
Liu H, Han X, Li Y et al (2013) Association of P2X7 receptor gene polymorphisms with sporadic Parkinson’s disease in a Han Chinese population. Neurosci Lett 546:42–45
Schiffmann SN, Fisone G, Moresco R et al (2007) Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol 83:277–292
Fuxe K, Marcellino D, Genedani S et al (2007) Adenosine A(2A) receptors, dopamine D(2) receptors and their interactions in Parkinson’s disease. Mov Disord 22:1990–2017
Olivieira-Giacomelli Á, Naaldijk Y, Sardá-Arroyo L et al (2018) Purinergic receptors in neurological diseases with motor symptoms: targets for therapy. Front Pharmacol 9:325
Fuxe K, Borroto-Escuela DO, Marcellino D et al (2012) GPCR heteromers and their allosteric receptor–receptor interactions. Curr Med Chem 19:356–363
Armentero MT, Pinna A, Ferre S et al (2011) Past, present and future of A(2A) adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol Ther 132:280–299
Bogenpohl JW, Ritter SL, Hall RA et al (2012) Adenosine A2A receptor in the monkey basal ganglia: ultrastructural localization and colocalization with the metabotropic glutamate receptor 5 in the striatum. J Comp Neurol 520:570–589
Łukasiewicz S, Blasiak E, Faron-Gorecka A et al (2007) Fluorescence studies of homooligomerization of adenosine A2A and serotonin 5-HT1A receptors reveal the specificity of receptor interactions in the plasma membrane. Pharmacol Rep 59:379–392
Carriba P, Navarro G, Ciruela F et al (2008) Detection of heteromerization of more than two proteins by sequential BRET–FRET. Nat Methods 5:727–733
Navarro G, Carriba P, Gandía J et al (2008) Detection of heteromers formed by cannabinoid CB1, dopamine D2, and adenosine A2A G-protein-coupled receptors by combining bimolecular fluorescence complementation and bioluminescence energy transfer. Sci World J 8:1088–1097
Fuxe K, Ungerstedt U (1974) Action of caffeine and theophyllamine on supersensitive dopamine receptors: considerable enhancement of receptor response to treatment with DOPA and dopamine receptor agonists. Med Biol 52:48–54
Fredholm BB, Fuxe K, Agnati L (1976) Effect of some phosphodiesterase inhibitors on central dopamine mechanisms. Eur J Pharmacol 38:31–38
Kanda T, Jackson MJ, Smith LA et al (2000) Combined use of the adenosine A2A antagonist KW-6002 with L-DOPA or with selective D1 or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys. Exp Neurol 162:321–327
Fuzzati-Armentero MT, Cerri S, Levandis G et al (2015) Dual target strategy: combining distinct non-dopaminergic treatments reduces neuronal cell loss and synergistically modulates l-DOPA-induced rotational behavior in a rodent model of Parkinson’s disease. J Neurochem 134:740–747
Canals M, Burgueno J, Marcellino D et al (2004) Homodimerization of adenosine A2A receptors: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Neurochem 88:726–734
Lee SP, O’Dowd BF, George SR (2003) Homo- and hetero-oligomerization of G protein-coupled receptors. Life Sci 74:173–180
Guo W, Urizar E, Kralikova M et al (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J 27:2293–2304
Antonelli T, Fuxe K, Agnati L et al (2006) Experimental studies and theoretical aspects on A2A/D2 receptor interactions in a model of Parkinson’s disease. Relevance for l-DOPA induced dyskinesias. J Neurol Sci 248:16–22
Fuxe K, Marcellino D, Borroto-Escuela DO et al (2010) Adenosine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther 16:e18–e42
Fuxe K, Guidolin D, Agnati LF et al (2015) Dopamine heteroreceptor complexes as therapeutic targets in Parkinson’s disease. Expert Opin Ther Targets 19:377–398
Poewe W, Mahlknecht P, Jankovic J (2012) Emerging therapies for Parkinson’s disease. Curr Opin Neurol 25:448–459
Hauser RA (2011) Future treatments for Parkinson’s disease: surfing the PD pipeline. Int J Neurosci 121(Suppl 2):53–62
Zhu C, Wang G, Li J et al (2014) Adenosine A2A receptor antagonist istradefylline 20 versus 40 mg/day as augmentation for Parkinson’s disease: a meta-analysis. Neurol Res 36:1028–1034
Dungo R, Deeks ED (2013) Istradefylline: first global approval. Drugs 73:875–882
Hauser RA, Stocchi F, Rascol O et al (2015) Preladenant as an adjunctive therapy with levodopa in Parkinson disease: two randomized clinical trials and lessons learned. JAMA Neurol 72:1491–1500
Pinna A (2014) Adenosine A2A receptor antagonists in Parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 28:455–474
Gillespie RJ, Bamford SJ, Botting R et al (2009) Antagonists of the human A(2A) adenosine receptor. 4. Design, synthesis, and preclinical evaluation of 7-aryltriazolol[4,5-d]pyrimidines. J Med Chem 52:33–47
Sitkovsky MV, Hatfield S, Abbott R et al (2014) Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res 2:598–605
Hove-Madsen L, Prat-Vidal C, Llach A et al (2006) Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc Res 72:292–302
Llach A, Molina CE, Prat-Vidal C et al (2011) Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur Heart J 32:721–729
Hauser RA, Olanow CW, Kieburtz KD et al (2014) Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: a phase 2b, double-blind, randomised trial. Lancet Neurol 13:767–776
LeWitt PA, Guttman M, Tetrud JW et al (2008) Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol 63:295–302
Fernandez HH, Greeley DR, Zweig RM et al (2010) Istradefylline as monotherapy for Parkinson disease: results of the 6002-US-051 trial. Parkinsonism Relat Disord 16:16–20
Gyoneva S, Shapiro L, Lazo C et al (2014) Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson’s disease. Neurobiol Dis 67:191–202
Golembiowska K, Wardas J, Noworyta-Sokolowska K, Kaminska K et al (2013) Effects of adenosine receptor antagonists on the in vivo LPS-induced inflammation model of Parkinson’s disease. Neurotox Res 24:29–40
Villar-Menéndez I, Porta S, Buira SP et al (2014) Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol Dis 69:206–214
Hu Q, Ren X, Liu Y et al (2016) Aberrant adenosine A2A receptor signaling contributes to neurodegeneration and cognitive impairments in a mouse model of synucleinopathy. Exp Neurol 283:213–223
Kachroo A, Schwarzschild MA (2012) Adenosine A2A receptor gene disruption protects in an α-synuclein model of Parkinson’s disease. Ann Neurol 71:278–282
Ferreira DG, Batalha VL, Vicente Miranda H et al (2015) Adenosine A2A receptors modulate α-synuclein aggregation and toxicity. Cereb Cortex. https://doi.org/10.1093/cercor/bhv268
Pintor J, Diaz-Rey MA, Miras-Portugal MT (1993) Ap4A and ADP-beta-S binding to P2 purinoreceptors present on rat brain synaptic terminals. Br J Pharmacol 108:1094–1099
Rodriguez-Pascual F, Cortes R, Torres M et al (1997) Distribution of [3H]diadenosine tetraphosphate binding sites in rat brain. Neuroscience 77:247–255
Burnstock G (2008) Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov 7:575–590
Heine C, Wegner A, Grosche J et al (2007) P2 receptor expression in the dopaminergic system of the rat brain during development. Neuroscience 149:165–181
Krügel U, Kittner H, Franke H et al (2001) Stimulation of P2 receptors in the ventral tegmental area enhances dopaminergic mechanisms in vivo. Neuropharmacology 40:1084–1093
Krügel U, Kittner H, Illes P (2001) Mechanisms of adenosine 5′-triphosphate-induced dopamine release in the rat nucleus accumbens in vivo. Synapse 39:222–232
Gan M, Moussaud S, Jiang P et al (2015) Extracellular ATP induces intracellular alpha-synuclein accumulation via P2X1 receptor-mediated lysosomal dysfunction. Neurobiol Aging 36:1209–1220
Marcellino D, Suarez-Boomgaard D, Sanchez-Reina MD et al (2010) On the role of P2X7 receptors in dopamine nerve cell degeneration in a rat model of Parkinson’s disease: studies with the P2X7 receptor antagonist A-438079. J Neural Transm 117:681–687
Carmo MR, Menezes AP, Nunes AC et al (2014) The P2X7 receptor antagonist Brilliant Blue G attenuates contralateral rotations in a rat model of Parkinsonism through a combined control of synaptotoxicity, neurotoxicity and gliosis. Neuropharmacology 81:142–152
Hracskó Z, Baranyi M, Csölle C et al (2011) Lack of neuroprotection in the absence of P2X7 receptors in toxin-induced animal models of Parkinson’s disease. Mol Neurodegener 6:28
Wilkaniec A, Gassowska M, Czapski GA et al (2017) P2X7 receptor-pennexin 1 interaction mediates extracellular alpha-synuclein-induced ATP release in neuroblastoma SH-SY5Y cells. Purinergic Signal 13:347–361
Jiang T, Hoekstra J, Heng X et al (2015) P2X7 receptor is critical in α-synuclein-mediated microglial NADPH oxidase activation. Neurobiol Aging 36:2304–2318
Coppi E, Pedata F, Gibb AJ (2012) P2Y1 receptor modulation of Ca2+-activated K+ currents in medium-sized neurons from neonatal rat striatal slices. J Neurophysiol 107:1009–1021
Wang Y, Chang CF, Morales M et al (2013) Diadenosine tetraphosphate protects against injuries induced by ischaemia and 6-hydroxidopamine in rat brain. J Neurosci 23:7958–7965
Yang X, Lou Y, Liu G et al (2017) Microglia P2Y6 receptor is related to Parkinson’s disease through neuroinflammatory process. J Neuroinflamm 14:38
Qian Y, Xu S, Yang X et al (2017) Purinergic receptor P2Y6 contributes to 1-methyl-4-phenylpyridinium-induced oxidative stress and cell death in neuronal SH-SY5Y cells. J Neurosci Res 96:253–264
Neher JJ, Neniskyte U, Hornik T et al (2014) Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 62:1463–1465
Dunkel P, Chai CL, Sperlágh B et al (2012) Clinical utility of neuroprotective agents in neurodegenerative diseases: current status of drug development for Alzheimer’s, Parkinson’s and Huntington’s diseases, and amyotrophic lateral sclerosis. Expert Opin Investig Drugs 21:1267–1308
Acknowledgements
Open access funding provided by MTA Institute of Experimental Medicine (MTA KOKI). This study was supported by Research Grants from Hungarian Research and Development Fund (Grant K116654 to BS), Hungarian Brain Research Program (2017-1.2.1.-NKP-2017-00002 to BS) and the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie Grant Agreement No. 766124.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors are pleased to be part of the SI dedicated to Professor Vera Adam-Vizi and grateful for the great inspiration and collaboration.
Special Issue of Neurochemical Research: In honour of Professor Vera Adam-Vizi.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tóth, A., Antal, Z., Bereczki, D. et al. Purinergic Signalling in Parkinson’s Disease: A Multi-target System to Combat Neurodegeneration. Neurochem Res 44, 2413–2422 (2019). https://doi.org/10.1007/s11064-019-02798-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02798-1