
Abstract
Since the discovery of NMDA receptor (NMDAR) dependent long-term potentiation (LTP) in the hippocampus, many studies have demonstrated that NMDAR dependent LTP exists throughout central synapses, including those involved in sensory transmission and perception. NMDAR LTP has been reported in spinal cord dorsal horn synapses, anterior cingulate cortex and insular cortex. Behavioral, genetic and pharmacological studies show that inhibiting or reducing NMDAR LTP produced analgesic effects in animal models of chronic pain. Investigation of signalling mechanisms for NMDAR LTP may provide novel targets for future treatment of chronic pain.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hippocampal long-term potentiation (LTP) is a well investigated form of synaptic plasticity in the central nervous system. In the hippocampus, LTP is an activity-dependent form of synaptic plasticity in which increased synaptic transmission follows brief, high-frequency stimulation of input pathways. Three different major types of glutamate receptors have been found in the hippocampus: N-methyl-d-aspartic acid (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) and metabotropic glutamate receptors (mGluRs). While AMPA receptors (AMPARs) mediate normal synaptic transmission, activation of NMDA receptors (NMDARs) is required for the induction of LTP. Pre-treatment with the NMDAR antagonist d-2-amino-5-phosphonopentanoic acid (AP5) prevents the induction of LTP. Subsequent pharmacological and genetic studies have confirmed that NMDAR LTP in the hippocampus play important roles in spatial memory of the hippocampus [1]. New mechanism for the synaptic plasticity in the hippocampus has been cumulatively reported (for example, see [2]). In this article that contributes to the special issue for the discovery of NMDAR LTP by Prof. Graham Collingridge, we will review the roles of NMDAR LTP in pain systems, especially in the condition of chronic pain.
Basic Circuitry of Pain: From Periphery to the Cortex
Glutamate is the fast excitatory transmitter of first sensory synapses. Peripheral noxious stimuli activate nociceptive afferent fibers (Aδ and C fibers) and incoming action potentials trigger a release of glutamate in the spinal dorsal horn. In addition, some neuropeptides such as substance P (SP) and neurokinin A (NKA) are also released in the spinal dorsal horn. Glutamate and neuropeptides activate spinal dorsal horn neurons, including those that send projection terminals to supraspinal structures. Neurons in the thalamus play key roles in relaying these ascending inputs to different cortical area. Among them, anterior cingulate cortex (ACC) and insular cortex (IC) are believed to be critical the unpleasantness of pain. As protective responses to noxious stimuli, endogenous pain modulatory systems are also activated. Descending modulatory systems are biphasic, including descending inhibitory and facilitatory systems. LTP has been reported in synapses located in spinal cord dorsal horn and cortex that are related to pain.
NMDAR LTP in Spinal Dorsal Horn
Glutamate is a major transmitter between primary afferent fibers and spinal dorsal horn neurons, whereas neuropeptides (e.g., SP and NKA) mediate slow excitatory postsynaptic potentials (EPSPs) at synapses between small-myelinated Aδ and unmyelinated C fibers and dorsal horn neurons [3,4,5,6]. Fast excitatory synaptic transmission is mainly mediated by AMPAR and kainate receptor (KAR) [7]. In some synapses (or called silent synapses), synaptic responses can be mediated by pure NMDARs [4].
Unlike with the hippocampus, studies of spinal LTP are limited by the technical difficulty of spinal cord slices and the complexity of the spinal local neuronal network. Electrophysiological experiments using intracellular or whole-cell patch-clamp recordings from the spinal dorsal horn neurons generate some important findings related to spinal LTP. Strong tetanic stimulation (100 Hz, 1 s for three times at 10 s intervals) of the dorsal root induces long-lasting enhancement of synaptic responses to presynaptic stimulation [8, 9]. The enhancement is relatively long-lasting (ranging from 25 to 90 min) and input specific. Postsynaptic depolarization of dorsal horn neurons is critical for the induction of spinal LTP. Pairing postsynaptic depolarization with synaptic activity also induces long-lasting enhancement of synaptic responses. Interestingly, the level of postsynaptic depolarization may be important in determining whether synaptic transmission will be potentiated or depressed. In some experiments, synaptic depression can also be induced by pairing synaptic activity with modest postsynaptic depolarization. The induction of spinal LTP requires activation of NMDARs and/or the SP (NK1) receptors. Activation of NMDARs in spinal dorsal horn neurons leads to increases in intracellular Ca2+ [10]. Pre-treatment of spinal cord slices with an NMDAR antagonist AP5 prevents the induction of LTP. The contribution of neuropeptide SP to spinal LTP may act by enhancing NMDAR mediated currents in spinal dorsal projecting neurons [8]. The intracellular signal pathways of spinal LTP remain to be fully mapped. Evidence from other studies indirectly indicates that several protein kinases may be important for spinal LTP, such as phospholipid-dependent protein kinase C (PKC). Phorbol ester induces long-lasting facilitation of evoked EPSPs or EPSCs (excitatory postsynaptic currents) amplitude to stimulation of presynaptic fibers [11, 12]. One possible mechanism for PKC-dependent spinal LTP is through the recruitment of spinal silent synapses or the insertion of AMPARs. Brain-derived neurotrophic factor (BDNF) can induce NMDAR LTP in spinal dorsal horn via different signaling pathways [13].
Early Genetic Studies of NMDAR GluN2B (Also Known as NR2B) in Pain-Related Cortex
In addition to the spinal cord, early studies suggest that NMDAR in supraspinal structures may contribute to persistent or chronic pain. Direct evidence for the contribution of NMDARs in forebrains to behavioral nociceptive responses comes from genetic studies of NMDAR GluN2B transgenic mice. Tang et al. generated transgenic mice with forebrain-targeted GluN2B overexpression, and the normal developmental change in NMDAR kinetics was reversed [14]. GluN2B subunit expression was observed extensively throughout the cerebral cortex including the ACC and IC. In both the ACC and IC, GluN2B expression was significantly increased, and NMDAR mediated responses were enhanced [15]. Interestingly, while transgenic mice and wild-type mice were indistinguishable in tests of acute nociception, GluN2B transgenic mice exhibited enhanced behavioral responses after peripheral inflammation. These findings provide the first genetic evidence that forebrain NMDARs play a critical role in chronic pain.
NMDAR Dependent Postsynaptic LTP (Post-LTP) in the ACC
Stimulation Protocols
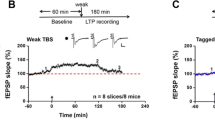
Among sensory-related cortical areas, LTP is well investigated in the ACC. In the ACC slices of adult animals, different stimulation protocols can be used to induce LTP using field recording and whole-cell patch-clamp recording techniques [16, 17]. For field recording from adult rat or mouse ACC slices, glutamatergic synapses in the ACC can undergo LTP in response to theta burst stimulation (TBS), a paradigm more closely to the activity of the ACC neurons. The potentiation lasted for at least 40–120 min [18]. Unlike the hippocampus, strong tetanic stimulation in the ACC did not cause reliable LTP. Whole-cell patch-clamp recordings allow better investigation of synaptic mechanisms for LTP in the ACC [16]. LTP can be induced using three different protocols, including the pairing training protocol, the spike-timing (spike-EPSPs) protocol, and TBS protocol [16, 19]. LTP induced by the pairing protocol is mainly triggered by the activation of NMDARs, but not through L-type voltage-gated calcium channels (L-VGCCs) [16].
NMDAR Dependent ACC LTP
In the ACC, NMDAR containing GluN2A or GluN2B subunits contribute to most NMDAR currents [16]. Bath application of a NMDAR GluN2A antagonist (NVP-AAM077) and GluN2B antagonist (ifenprodil/Ro compounds) produce almost completely blockade of NMDAR mediated EPSCs. Application of NMDAR GluN2A or GluN2B antagonist reduces ACC LTP, without complete abolishment of LTP. LTP is only abolished after the co-application of both inhibitors [16]. It is noted that LTP induced by spike-timing protocol seems to be more sensitive to NMDAR GluN2B blockade as compared with effects on LTP induced by pairing training protocol [16].
NMDAR Dependent Calcium Influx
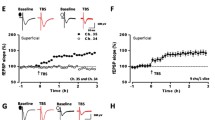
Calcium (Ca2+) signaling is critical for the induction of NMDAR LTP [16, 20]. Using the two-photon imaging method, it has been shown that NMDARs contribute to postsynaptic Ca2+ signal increases induced by different synaptic stimulation in ACC pyramidal neurons. Furthermore, LTP inducing protocols also triggered postsynaptic Ca2+ influx, which were NMDAR dependent [19]. These studies provide the first direct study of Ca2+ signals in the ACC and demonstrate that NMDARs play important roles in postsynaptic Ca2+ signals (see Fig. 1). As a result of postsynaptic increase of Ca2+, Ca2+ binds to calmodulin (CaM) and leads to activation of Ca2+-stimulated signaling pathways [21]. In support of the role of Ca2+ in LTP, postsynaptic injection of 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) completely blocked the induction of LTP [16]. Furthermore, a study using electroporation of mutant CaM in the ACC suggests that Ca2+ binding sites of CaM are critical for the induction of cingulate LTP [21].

NMDAR dependent LTP in the ACC. A Neurons in the ACC receive sensory afferents from the thalamus. Synaptic LTP is believed to be the key cellular mechanism for chronic pain in the ACC. LTP can be blocked by NMDAR antagonist AP5. B NMDAR mediated postsynaptic calcium signals in the ACC neurons. Representative calcium transient waveforms and average traces of fluorescence changes (ΔF/F) in responsive spines evoked by puff-application of glutamate in the control artificial cerebro-spinal fluid (ACSF), presence of CNQX (20 µM), and AP5 (50 µM) in the ACC, respectively. C ACC microinjection NMDAR antagonists reduced chronic pain. The NR2B receptor antagonists Ro 25-6981 and Ro 63-1908 microinjected into the ACC significantly reduced mechanical allodynia in the CFA injection 3 days mice.
Downstream Intracellular Signaling Pathways
Cyclic AMP (cAMP) signaling pathways are important signaling pathways in biological systems. Among more than 10 adenylyl cyclase (AC) subunits, AC subtype 1 (AC1) and subtype 8 (AC8) are two AC subtypes that respond positively to Ca2+–CaM [22]. As compared with AC8, AC1 is more sensitive to Ca2+ increase. In the ACC, AC1 is highly expressed in cingulate neurons located in most layers [23]. AC1 is selective for plastic changes and gene deletion of AC1 does not affect basal glutamate transmission in the ACC. By contrast, LTP induced by TBS or pairing stimulation are abolished in cingulate pyramidal cells [24]. Whole-cell patch-clamp recording also revealed that AC1 activity is required for the induction of LTP in ACC pyramidal cells. By using chemical design and biochemical screening, several selective inhibitors of AC1 have been identified. Consistently, pharmacological inhibition of AC1 in the ACC neurons abolished LTP induced by pairing training [25, 26].
Expression of ACC LTP
At least four possible mechanisms may contribute to the expression of LTP: (1) presynaptic enhancement of glutamate release; (2) postsynaptic enhancement of glutamate receptor mediated responses; (3) recruitment of previously ‘silent’ synapses or synaptic trafficking or insertion of AMPA receptors; (4) structural changes. Under in vitro brain slice conditions, it appears that the LTP mechanism may depend on the induction protocol in certain cases. Paired-pulse facilitation (PPF) was not altered after the induction of cingulate LTP [16]. However, we do not rule out the possibility of presynaptic changes in the ACC during other physiological/pathological conditions. Among these possibilities, we have recently investigated the roles of AMPAR GluA1 and GluA2/3 using genetic and pharmacological approaches. We found that GluA1 subunit C-terminal peptide analog, Pep1-TGL, blocked the induction of cingulate LTP [27]. Thus, in the ACC, the interaction between the C-terminus of GluA1 and postsynaptic density-95/Discs large/zona occludens-1 (PDZ) domain proteins is required for the induction of LTP. Synaptic delivery of the GluA1 subunit from extrasynaptic sites is the key mechanism underlying synaptic plasticity [28, 29] and GluA1–PDZ interactions are a critical intermediate in this plasticity. Our pharmacological experiments show that the application of philanthotoxin-74 (PhTx) 5 min after paired training reduced to synaptic potentiation, while PhTx had no effect on basal responses. Therefore, we believe that Ca2+-permeable GluA2-lacking receptors contribute to the maintenance of LTP and are necessary for subsequent LTP stabilization. These findings indicate that it is likely that selective contribution of AMPARs to cingulate LTP. Recently, we found that the expression of LTP and network LTP was significantly impaired in the GluA1 knock-in mutation mice at the protein kinase A (PKA) phosphorylation site serine 845 (s845A), but not in the CaMKII/PKC phosphorylation site serine 831 (s831A) mice. These results provide strong evidence that PKA phosphorylation of GluA1 is important for the network LTP expression in the ACC [30].
GluA2/3 subunits may continually replace synaptic GluA2/3 subunits in an activity-independent manner that maintains constant synaptic transmission [31,32,33,34]. We also examined the role of these peptides in synaptic potentiation in the ACC and found that the GluA2/3–PDZ interaction had no effect on cingulate LTP. We did find that the same interfering peptides inhibited cingulate long-term depression (LTD) [35]. These findings suggest that GluA1 and GluA2/3 play different roles in cingulate LTP vs LTD.
LTP in the IC
Both the ACC and IC are key cortical areas for pain perception, in addition to other key brain functions [20, 26, 36, 37]. Excitatory synapses in the ACC are highly plastic, and LTP of excitatory transmission can be induced experimentally in brain slice preparation and in vivo conditions by periphery injury. Recent studies characterized LTP in the IC using 64-channel recording system [38]. TBS induced a prolonged potentiation lasting for at least 3 h. The induction of IC LTP is NMDAR dependent, and both NMDAR GluN2A and GluN2B receptors are required for the induction of LTP [38]. Using in vivo recording techniques, it has bene reported that IC LTP induced by tetanic stimulation applied to the basolateral nucleus of the amygdala is blocked by NMDAR antagonist 3-(R-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) in adult rats [39]. Using in vivo optical imaging methods, Mizoguchi et al. has reported that tetanic stimulation caused LTP in the IC of adult rats [40].
IC LTP can also be induced by the LTP pairing protocol using whole-cell patch-clamp recording. We found that, using a pairing training protocol, LTP was induced in pyramidal neurons in the IC slices. NMDAR is important for the induction of LTP, since application of AP5 blocked the induction of LTP. NMDAR GluN2B is important and Ro 25-6981, a selective GluN2B containing NMDAR antagonist, also significantly reduced IC LTP [41]. We also found that postsynaptic calcium is important for the induction of post-LTP, since the postsynaptic application of BAPTA completely blocked the induction of LTP. Ca2+-activated AC1 is required for potentiation. By contrast, AC8 is not required. Inhibition of Ca2+ permeable AMPA receptors (CP-AMPARs) or protein kinase M zeta (PKMζ) reduced the expression of LTP in the IC [42].
During IC LTP, the paired-pulse ratio is not affected at 1 and 3 h after LTP induction, suggesting that the potentiation is likely not purely expressed presynaptically. Application of 1-naphthyl acetyl spermine (NASPM) (50 µM), a potent CP-AMPARs blocker, reversed the potentiation at 3 h after LTP induction, suggesting postsynaptic recruitment of CP-AMPARs is a necessary step for IC L-LTP expression [43, 44].
AC1 is the major Ca2+/calmodulin-stimulated AC isoform among the cAMP signaling pathway [45, 46]. Using knockout mice lacking AC1 (AC1−/− mice), we found that the amount of synaptic GluA1 and its phosphorylation at the Ser845 site remained unchanged in the IC after injury. Furthermore, no upregulation of A-kinase anchoring protein (AKAP)79/150, PKA catalytic subunit α (Cα), or PKA regulatory subunit IIβ (RIIβ) were detected from the AC1−/−mice with nerve ligation. Taken together, these results indicate that AC1 is essential for the translocation of AKAP79/150 and PKA to the synaptic site, and then for the enhancement of synaptic GluA1 in the IC.
Functional Implications of NMDAR in Chronic Pain
Spinal Cord
It is well known that NMDARs play important roles in spinal pain processes. For example, responses of sensory dorsal horn neurons to noxious and non-noxious stimuli are enhanced by application of NMDA, indicating that the NMDAR may contribute to both hyperalgesia and allodynia [47]. Activation of NMDARs in the spinal cord by glutamate or NMDA produces nociception or behavioral hyperalgesia and the effects are blocked by an NMDARs antagonist. NMDA excites spinal dorsal horn neurons (including identified spinothalamic tract cells) and enhances their response to peripheral sensory stimuli. Blockage of NMDARs in the spinal cord by different antagonists reduces thermal hyperalgesia, mechanical hyperalgesia/allodynia, and spontaneous nociceptive behavior or autotomy (a behavioral model of neuropathic pain) in rats with different types of persistent pain [47]. NMDAR antagonists applied systemically or intrathecally significantly reduce pain sensation in patients with different kinds of persistent pain [48]. For example, Kristensen et al. reported that intrathecal administration of a NMDAR antagonist relieved pain sensation to mechanical and thermal stimuli in a patient with nerve injury, while the allodynia response, as well as deep pain sensation, were still present [49]. Lagraize et al. found that intrathecal (i.t.) injection the NMDAR channel blocker MK-801 blocked complete Freund’s adjuvant (CFA) induced thermal hyperalgesia [50]. These results suggest that NMDAR dependent synaptic plasticity may actually play an important role in the induction and/or maintenance of persistent pain. Future studies to directly address the relationship between spinal LTP and persistent pain are needed.
Cortex
Does genetic overexpression of NMDAR GluN2B mimic physiological or pathological conditions? Our study provides the first evidence that the up-regulation of NMDAR GluN2B in the ACC contributes to inflammation-related persistent pain. After persistent inflammation, the expression of NMDAR GluN2B in the ACC was up-regulated, thereby increasing the GluN2B component in NMDAR mediated responses [51]. Consistently, microinjection into the ACC and systemic administration of GluN2B receptor selective antagonists inhibited behavioral responses to peripheral inflammation. These results are in good accordance with our previous report, showing that GluN2B forebrain overexpression selectively enhanced inflammation-related persistent pain in transgenic mice [15]. Furthermore, we believe that these findings provide critical evidence that NMDAR GluN2B receptors undergo long-term plastic changes in the brain after injury. It should be noted that neurons of the ACC have been implicated in other brain functions, and our present results do not rule out roles for NMDAR GluN2B receptors in other ACC-related physiological functions.
We believe that this GluN2B receptor up-regulation is likely to be reliant on activity-dependent mechanisms. Several lines of evidence support this prediction: (1) the molecular motor protein KIF 17 has been shown to be involved in the active transport of GluN2B [52,53,54]; (2) GluN2B, mRNA and protein, is highly expressed in ACC neurons [15] and (3) GluN2B contains a CREB (cAMP response element-binding protein) binding domain which may couple increases in intracellular calcium with the increase in GluN2B expression. Since NMDARs play an important role in activity-dependent plasticity in the ACC, we suggest that GluN2B may be regulated through NMDA–calcium–CaM-dependent signaling pathways. The activation of NMDARs trigger postsynaptic calcium, leading to the activation of calcium-stimulated CREB in the ACC after injury [23]. Since GluN2B contains a CREB binding domain, it is likely that GluN2B may be activated downstream from the CREB signaling pathway through the activation of NMDARs. A recent study showed that persistent pain induced by tissue inflammation or nerve injury were significantly reduced in PDZ-93 knockout mice, in part due to the lower level of GluN2B expression at the spinal and cortical levels in knockout mice [55]. Yang et al. reported that Caveolin-1 directly binding with GluN2B subunit and promotion of GluN2B surface levels in the ACC contributed to modulation of chronic neuropathic pain. Disrupting the interaction of Cav-1 and GluN2B through microinjection of a short peptide derived from the C-terminal of GluN2B into the ACC exhibited a significant antinociceptive effect associated with decrease of surface GluN2B expression [56].
Behavioral and pharmacological experiments consistently demonstrate that inhibiting IC LTP produce analgesic effects in animal models of chronic pain. In an animal model of neuropathic pain, microinjection of AMPAR/KAR antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) bilaterally into the IC significantly reduced injury related behavioral sensitization. Similar analgesic effects have been found with microinjections of NMDAR antagonist AP5 or a selective GluN2B-NMDARs antagonist Ro 25-6981 bilaterally into the IC [41]. PKMζ activity has been known to be important for maintaining potentiation in different parts of the brain, including the hippocampus and the ACC [57, 58]. Although its role in IC potentiation remain to be examined, microinjection of zeta-pseudosubstrate inhibitory peptide (ZIP), an inhibitor of PKMζ and other PKC isoforms, into the IC produced analgesic effects in rats with nerve injury [59]. Interestingly, the critical roles of IC PKMζ in the conditioned taste aversion (CTA) have been reported. While inhibiting PKMζ produced erasure of long-term memory in the CTA test [60], overexpression of PKMζ in the IC can enhance consolidated long-term memory [61].
Conclusion and Future Directions
Discovery of the basic mechanism of NMDAR dependent LTP has greatly improved our understanding of brain plasticity. These forms of plasticity not only contribute to various key physiological functions such as learning and memory, but also contribute to the development of pathological conditions that are still difficult to be treated, such as chronic pain and drug addiction. The involvement of NMDAR dependent LTP in pain modulation is better supported by pharmacological inhibitors that are reducing or blocking the expression of LTP without affecting the function of NMDARs such as ZIP.
The complaints of poor translation from animal studies to clinical treatment are not just that these basic mechanisms do not exist in human brains. Instead, poor funding and government regulation have supported less translational works as compared with basic researches. Furthermore, the side effects of various chemicals targeted at NMDARs, including various subtypes, have prevented this translational discovery into human treatment.
One possible solution is selectively targeting downstream proteins from NMDARs, for example, Ca2+ stimulation AC1. AC1 serves as a key downstream signaling enzyme in sensory neurons, from spinal dorsal horn to cortical neurons. While genetic deletion of NMDARs caused developmental defects and impairment of cognitive functions, gene deletion of AC1 produce no or minor behavior defects. One possible explanation is that other forms of ACs, as well as downstream protein kinases, may compensate or take over key physiological functions. We hope that fully mapping the signaling pathways of LTP in sensory and cortical neurons may help us to identify new targets, and eventually help to treat patients with different illness such as chronic pain and drug addiction.
Abbreviations
- AC:
-
Adenylyl cyclase
- ACC:
-
Anterior cingulate cortex
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid
- AP5:
-
d-2-Amino-5-phosphonopentanoic acid
- BAPTA:
-
1,2-Bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- BDNF:
-
Brain-derived neurotrophic factor
- CaM:
-
Calmodulin
- cAMP:
-
Cyclic AMP (cAMP)
- CNQX:
-
6-Cyano-7-nitroquinoxaline-2,3-dione
- CP-AMPARs:
-
Ca2+ permeable AMPA receptors
- CPP:
-
3-(R-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid
- CTA:
-
Conditioned taste aversion
- EPSCs:
-
Excitatory postsynaptic currents
- EPSPs:
-
Excitatory postsynaptic potentials
- IC:
-
Insular cortex
- KAR:
-
Kainate receptor
- LTP:
-
Long-term potentiation
- mGluRs:
-
Metabotropic glutamate receptors
- NAPSM:
-
1-Naphthyl acetyl spermine
- NKA:
-
Neurokinin A
- NMDA:
-
N-Methyl-d-aspartate
- PDZ:
-
Postsynaptic density-95/Discs large/zona occludens-1
- PKC:
-
Protein kinase C
- PKMζ:
-
Protein kinase Mζ
- PPF:
-
Paired-pulse facilitation
- SP:
-
Substance P
- ZIP:
-
ζ-Pseudosubstrate inhibitory peptide
References
Bliss TV, Collingridge GL (2013) Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol Brain 6:5
Wang J, Lv X, Wu Y, Xu T, Jiao M, Yang R, Li X, Chen M, Yan Y, Chen C, Dong W, Yang W, Zhuo M, Chen T, Luo J, Qiu S (2018) Postsynaptic RIM1 modulates synaptic function by facilitating membrane delivery of recycling NMDARs in hippocampal neurons. Nat Commun 9:2267
Li P, Zhuo M (2001) Substance P and neurokinin A mediate sensory synaptic transmission in young rat dorsal horn neurons. Brain Res Bull 55:521–531
Li P, Zhuo M (1998) Silent glutamatergic synapses and nociception in mammalian spinal cord. Nature 393:695–698
Li P, Calejesan AA, Zhuo M (1998) ATP P2x receptors and sensory synaptic transmission between primary afferent fibers and spinal dorsal horn neurons in rats. J Neurophysiol 80:3356–3360
Yoshimura M, Jessell T (1990) Amino acid-mediated EPSPs at primary afferent synapses with substantia gelatinosa neurones in the rat spinal cord. J Physiol 430:315–335
Li P, Wilding TJ, Kim SJ, Calejesan AA, Huettner JE, Zhuo M (1999) Kainate-receptor-mediated sensory synaptic transmission in mammalian spinal cord. Nature 397:161–164
Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J (2003) Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 299:1237–1240
Randic M, Jiang MC, Cerne R (1993) Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci 13:5228–5241
MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL (1986) NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 321:519–522
Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M (1999) AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci 2:972–977
Hori Y, Endo K, Takahashi T (1996) Long-lasting synaptic facilitation induced by serotonin in superficial dorsal horn neurones of the rat spinal cord. J Physiol 492(Pt 3):867–876
Zhou LJ, Zhong Y, Ren WJ, Li YY, Zhang T, Liu XG (2008) BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Exp Neurol 212:507–514
Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ (1999) Genetic enhancement of learning and memory in mice. Nature 401:63–69
Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M (2001) Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci 4:164–169
Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M (2005) Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron 47:859–872
Wei F, Li P, Zhuo M (1999) Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. The J Neurosci 19:9346–9354
Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M (2002) Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci 5:573–579
Li XH, Song Q, Chen T, Zhuo M (2017) Characterization of postsynaptic calcium signals in the pyramidal neurons of anterior cingulate cortex. Mol Pain 13:1744806917719847
Zhuo M (2008) Cortical excitation and chronic pain. Trends Neurosci 31:199–207
Wei F, Xia XM, Tang J, Ao H, Ko S, Liauw J, Qiu CS, Zhuo M (2003) Calmodulin regulates synaptic plasticity in the anterior cingulate cortex and behavioral responses: a microelectroporation study in adult rodents. J Neurosci 23:8402–8409
Xia M, Sreedharan SP, Bolin DR, Gaufo GO, Goetzl EJ (1997) Novel cyclic peptide agonist of high potency and selectivity for the type II vasoactive intestinal peptide receptor. J Pharmacol Exp Ther 281:629–633
Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M (2002) Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 36:713–726
Liauw J, Wu LJ, Zhuo M (2005) Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J Neurophysiol 94:878–882
Chen T, O’Den G, Song Q, Koga K, Zhang MM, Zhuo M (2014) Adenylyl cyclase subtype 1 is essential for late-phase long term potentiation and spatial propagation of synaptic responses in the anterior cingulate cortex of adult mice. Mol Pain 10:65
Bliss TV, Collingridge GL, Kaang BK, Zhuo M (2016) Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci 17:485–496
Toyoda H, Wu LJ, Zhao MG, Xu H, Zhuo M (2007) Time-dependent postsynaptic AMPA GluR1 receptor recruitment in the cingulate synaptic potentiation. Dev Neurobiol 67:498–509
Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287:2262–2267
Passafaro M, Piech V, Sheng M (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4:917–926
Song Q, Zheng HW, Li XH, Huganir RL, Kuner T, Zhuo M, Chen T (2017) Selective phosphorylation of AMPA receptor contributes to the network of long-term potentiation in the anterior cingulate cortex. J Neurosci 37:8534–8548
Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40:361–379
Song I, Huganir RL (2002) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25:578–588
Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126
Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Muller T, Pin JP, Prezeau L (2001) BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol 59:965–973
Toyoda H, Zhao MG, Zhuo M (2006) NMDA receptor-dependent long-term depression in the anterior cingulate cortex. Rev Neurosci 17:403–413
Zhuo M (2016) Contribution of synaptic plasticity in the insular cortex to chronic pain. Neuroscience 338:220–229
Zhuo M (2014) Long-term potentiation in the anterior cingulate cortex and chronic pain. Philos Trans R Soc Lond B 369:20130146
Liu MG, Kang SJ, Shi TY, Koga K, Zhang MM, Collingridge GL, Kaang BK, Zhuo M (2013) Long-term potentiation of synaptic transmission in the adult mouse insular cortex: multielectrode array recordings. J Neurophysiol 110:505–521
Escobar ML, Alcocer I, Chao V (1998) The NMDA receptor antagonist CPP impairs conditioned taste aversion and insular cortex long-term potentiation in vivo. Brain Res 812:246–251
Mizoguchi N, Fujita S, Koshikawa N, Kobayashi M (2011) Spatiotemporal dynamics of long-term potentiation in rat insular cortex revealed by optical imaging. Neurobiol Learn Mem 96:468–478
Qiu S, Chen T, Koga K, Guo YY, Xu H, Song Q, Wang JJ, Descalzi G, Kaang BK, Luo JH, Zhuo M, Zhao MG (2013) An increase in synaptic NMDA receptors in the insular cortex contributes to neuropathic pain. Sci Signal 6:ra34
Yamanaka M, Matsuura T, Pan H, Zhuo M (2017) Calcium-stimulated adenylyl cyclase subtype 1 (AC1) contributes to LTP in the insular cortex of adult mice. Heliyon 3:e00338
Clem RL, Huganir RL (2010) Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330:1108–1112
Lu W, Chen H, Xue CJ, Wolf ME (1997) Repeated amphetamine administration alters the expression of mRNA for AMPA receptor subunits in rat nucleus accumbens and prefrontal cortex. Synapse 26:269–280
Cooper DM, Crossthwaite AJ (2006) Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol Sci 27:426–431
Cooper DM, Karpen JW, Fagan KA, Mons NE (1998) Ca(2+)-sensitive adenylyl cyclases. Adv Second Messenger Phosphoprot Res 32:23–51
Willis WD (2002) Long-term potentiation in spinothalamic neurons. Brain Res Rev 40:202–214
Robinson DA, Zhuo M (2002) Glutamatergic synapses serve as potential targets for controlling persistent pain. Curr Anaesth Crit Care 13:321–327
Kristensen JD, Svensson B, Gordh T Jr (1992) The NMDA-receptor antagonist CPP abolishes neurogenic ‘wind-up pain’ after intrathecal administration in humans. Pain 51:249–253
Lagraize SC, Guo W, Yang K, Wei F, Ren K, Dubner R (2010) Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience 171:1341–1356
Wu LJ, Toyoda H, Zhao MG, Lee YS, Tang J, Ko SW, Jia YH, Shum FW, Zerbinatti CV, Bu G, Wei F, Xu TL, Muglia LJ, Chen ZF, Auberson YP, Kaang BK, Zhuo M (2005) Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J Neurosci 25:11107–11116
Guillaud L, Setou M, Hirokawa N (2003) KIF17 dynamics and regulation of NR2B trafficking in hippocampal neurons. J Neurosci 23:131–140
Wong RW, Setou M, Teng J, Takei Y, Hirokawa N (2002) Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc Natl Acad Sci USA 99:14500–14505
Setou M, Nakagawa T, Seog DH, Hirokawa N (2000) Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science 288:1796–1802
Tao YX, Rumbaugh G, Wang GD, Petralia RS, Zhao C, Kauer FW, Tao F, Zhuo M, Wenthold RJ, Raja SN, Huganir RL, Bredt DS, Johns RA (2003) Impaired NMDA receptor-mediated postsynaptic function and blunted NMDA receptor-dependent persistent pain in mice lacking postsynaptic density-93 protein. J Neurosci 23:6703–6712
Yang JX, Hua L, Li YQ, Jiang YY, Han D, Liu H, Tang QQ, Yang XN, Yin C, Hao LY, Yu L, Wu P, Shao CJ, Ding HL, Zhang YM, Cao JL (2015) Caveolin-1 in the anterior cingulate cortex modulates chronic neuropathic pain via regulation of NMDA receptor 2B subunit. J Neurosci 35:36–52
Sacktor TC (2012) Memory maintenance by PKMzeta—an evolutionary perspective. Mol Brain 5:31
Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, Shim J, Lee K, Collingridge GL, Kaang BK, Zhuo M (2010) Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science 330:1400–1404
Han J, Kwon M, Cha M, Tanioka M, Hong SK, Bai SJ, Lee BH (2015) Plasticity-related PKMzeta signaling in the insular cortex is involved in the modulation of neuropathic pain after nerve injury. Neural Plast 2015:601767
Shema R, Sacktor TC, Dudai Y (2007) Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science 317:951–953
Shema R, Haramati S, Ron S, Hazvi S, Chen A, Sacktor TC, Dudai Y (2011) Enhancement of consolidated long-term memory by overexpression of protein kinase Mzeta in the neocortex. Science 331:1207–1210
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, XH., Miao, HH. & Zhuo, M. NMDA Receptor Dependent Long-term Potentiation in Chronic Pain. Neurochem Res 44, 531–538 (2019). https://doi.org/10.1007/s11064-018-2614-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2614-8