
Abstract
Glutamate transport (GluT) in brain is mediated chiefly by two transporters GLT and GLAST, both driven by ionic gradients generated by (Na+, K+)-dependent ATPase (Na+/K+-ATPase). GLAST is located in astrocytes and its function is regulated by translocations from cytoplasm to plasma membrane in the presence of GluT substrates. The phenomenon is blocked by a naturally occurring toxin rottlerin. We have recently suggested that rottlerin acts by inhibiting Na+/K+-ATPase. We now report that Na+/K+-ATPase inhibitors digoxin and ouabain also blocked the redistribution of GLAST in cultured astrocytes, however, neither of the compounds caused detectable inhibition of ATPase activity in cell-free astrocyte homogenates (rottlerin inhibited app. 80% of Pi production from ATP in the astrocyte homogenates, IC50 = 25 μM). Therefore, while we may not have established a direct link between GLAST regulation and Na+/K+-ATPase activity we have shown that both ouabain and digoxin can interfere with GluT transport and therefore should be considered potentially neurotoxic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
GLutamate and ASpartate Transporter (GLAST) is one of the two most important transporters handling acidic amino acids in brain tissue (the other being GLT; for reviews see [1–4]). Glutamate transport (GluT) is driven by free energy generated in the form of ionic gradients by (Na+, K+)-dependent ATPase (Na+/K+-ATPase). GLAST is located in actrocytes and its function appears to be regulated by rapid translocations between cytoplasm and the cell (plasma) membrane in response to the presence of substrates in the extracellular space [5–7]. The precise nature of the mechanism underlying these shifts is not understood but probably involves phosphorylation and changes in the activity of Na+/K+-ATPase [7–10].
Na+/K+-ATPase is a membrane-located enzyme which breaks down ATP and uses the resulting energy to maintain Na+ and K+ gradients across the cell membranes. It is found in virtually all cells and tissues but its activity and normal functioning would seem to be critically important in brain considering that the neuronal action potentials involve large increases in membrane permeability to both K+ and Na+ and, at the same time, the most important excitatory neurotransmitter and potential neurotoxin l-glutamate (for reviews see [11–13]) is cleared and inactivated by uptake into astrocytes via GluT, i.e. by a process driven by Na+/K+-ATPase-generated Na+ and K+ concentrations gradients [1, 2, 14]. Indeed, there may be not only functional but, also, close structural relationship between Na+/K+-ATPase and GluT [15–17].
Alterations in the activity of Na+/K+-ATPase have been noted in brain diseases, particularly those associated with elevated levels of extracellular glutamate. Rapport et al. [18] reported 60% decrease in Na+/K+-ATPase in the cerebral cortex of patients with epilepsy, global loss of Na+/K+-ATPase was found in a transgenic mouse model of amyotrophic lateral sclerosis [19] and the ionic movements occurring as a result of ischemia and hypoglycemia [20, 21] were consistent with a decrease in Na+/K+-ATPase activity. Inadequate Na+/K+-ATPase activity could, therefore, compromise normal function of GluT and lead to increases in extracellular glutamate resulting in neurotoxic events [3, 4, 22–24]. In fact, it has been known for some time that the Na+, K+-ATPase inhibitor ouabain is a powerful inhibitor of GluT in brain tissue [25, 26] and can act as a neurotoxin in vivo [27–29].
Recently, we have suggested that the disruption of GLAST traffic between cytoplasm and cell membrane caused by a protein kinase C-δ (PKC- δ) inhibitor and metabolic poison rottlerin could be explained by inhibition of Na+/K+-ATPase [10]. Such explanation raises a possibility that other Na+/K+-ATPase inhibitors could have similar effects on GLAST. In addition, inhibition of Na+/K+-ATPase activity accompanied by the loss of rapid regulation of GluT, effectively blocking an increase in GLAST activity in response to increased extracellular glutamate, could lead to pathologically high levels of l-glutamate around sensitive neurons. Therefore, naturally occurring Na+/K+-ATPase inhibitors (e.g. endogenous cardiac glycosides) and, in some cases such as digoxin, therapeutically used drugs, could be potentially highly neurotoxic if they entered into and/or, were allowed to build up to high concentrations in, the brain.
The chief aim of the present study has been to test whether ouabain and digoxin could alter the d-aspartate induced redistribution of GLAST in glial cells. As an experimental model we have used, as in previous similar studies, cultured rat astrocytes in which GLAST transporter was immunofluorescently labelled by a custom made antibody [30] and the subcellular location of the immunofluorescence was studied using deconvolution microscopy [9, 10].
Experimental Procedure
Cultured Astrocytes
Cultured astrocytes were prepared from brains of neonatal Sprague–Dawley rats as described elsewhere [9]. Cells were first grown in Dulbecco modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS) in 25 cm2 flasks, then passaged onto coverslips (13 mm in diameter) and maintained until they reached even distribution.
Immunocytochemistry
The methodology was described in recent publications [9, 10]. The coverslips with astrocytes were washed and incubated in the presence of d-aspartate and/or drugs dissolved in 500 μl of serum-free DMEM (sfDMEM). Incubations (30 min) were carried out at 37°C in 5% CO2. Drug-free sfDMEM was used as a control.
Following the exposure to d-aspartate and/or rottlerin, the astrocytes were double-labelled with antibodies against glial fibrillary acidic protein (GFAP, marker for astrocytes, mouse monoclonal antibodies) and antibodies against the glutamate transporter GLAST (polyclonal antibody raised in rabbit; [30]). The antibodies were diluted in phosphate buffered saline (PBS) containing 1% bovine serum albumin (BSA) and 0.05% saponin. The coverslips were first washed in 2 ml of PBS for 5 min, then fixed with paraformaldehyde (2% in PBS) for 10 min and again washed with 2 ml of PBS for 5 min. The preparation was then “blocked” with the BSA-containing PBS (BSA/PBS) for 30 min and exposed to 200 μl of solutions containing the antibodies at dilutions of 1:4,000 (GLAST) and 1:400 (GFAP). The incubations (2 h) were carried out at room temperature in a humidified environment. The coverslips were then washed three times for 5 min with 2 ml of the BSA/PBS solution and exposed to secondary antibodies.
Goat-generated anti-mouse IgG conjugated to Alexa Fluor 488 (AF 488) was used to visualize GFAP while goat-generated anti-rabbit IgG conjugated to Alexa Fluor 594 (AF 594) was used to label the anti-GLAST antibody. Both secondary antibodies were diluted in the BSA/PBS solution; incubations were for 1 h at room temperature and washed as described above for the primary antibodies; three times for 5 min with 2 ml of BSA/PBS.
After washing, the coverslips were blotted and mounted upside down, using 50% solution of glycerol in PBS, on slides and secured along the edges with nail polish.
Deconvolution Microscopy and Image Analysis
Deconvolution microscope (Axioplan 2, Zeiss) was used for image acquisition [9, 10]. AF 488 and AF 594 were excited at 499 nm (emission at 520 nm) and 590 (emission at 618 nm) respectively. The images were optically sectioned at 0.513 μm intervals and subjected to deconvolution using an inverse filter algorithm to remove out-of-focus (background) signals. Sections from the midplane of the stacks (usually five to seven sections) were used for the image analyses. Examples of such sections are shown in Fig. 1. Each image represented a randomly selected single cell.

Distribution of GLAST immunoreactivity (IR) in cultured astrocytes. Green fluorescence marks glial fibrillary acidic protein, red corresponds to GLAST IR. Top two panels are controls (a and b); absence of added glutamate transport substrates, bottom two panels (c and d) are cells in the presence of 500 μM d-aspartate. The left two panels (a and c) show combination of GLAST and GFAP IR, the panels on the right (b and d) show GLAST IR only. Scale bar = 10 μm. For interpretation of the references to color in this figure legend, the reader is referred to the online version of this article
Mean fluorescence density (MFD) was determined in both the cytoplasm and membrane (cMFD and mMFD) and the membrane/cytoplasm ratio of fluorescent intensity (RFI = mMFD/cMFD) was used as an index of the distribution of GLAST between membrane and cytoplasm. Examples of such distributions in fluorescence density are shown in Fig. 2.

Example of GLAST IR density distribution across cells displayed as XY plots and histograms. The fluorescence intensity (optical density of the fluorescence produced by AF 594, see Experimental Procedure for details) is in units provided by the deconvolution microscope, the distance units are arbitrary. The top two panels are controls, bottom two panels show the distribution in the presence of 500 μM d-aspartate. The panels on the left show examples of lines across osingle cells while those on the right show mean fluorescence density inside cells (“In”, i.e. in the cytoplasm, cMFD) or at the perimeter, close to, or at, the plasma membrane (“Out”, mMFD), determined as described in detail in [9, 10]. RFI is the ratio of mMFD to cMFD [9] and, when calculated from the data shown in the panels on the right (means ± SEM from five controls, six in the presence of d-aspartate), it was found to be significantly higher (at P < 0.001 by unpaired t-test) in the presence of d-aspartate when compared to controls
Activity of Na+, K+-Dependent ATPase in Prisms of Adult Cerebral Cortex
We have used a technique that employs uptake of Rb+ as a measure of the activity of (Na+, K+)-dependent ATPase in rat brain tissue. The methodology is similar to that used to study uptake of radiolabelled amino acid [25] and has been described in detail earlier [26]. Prisms of adult (3–6 months) rat cerebral cortex (0.1 × 0.1 × thickness of cortex) were suspended at 25 mg/10 ml of incubation medium (phosphate buffered Krebs–Ringer; [31]) and allowed 15 min for recovery in a shaking water bath at 37°C. Uptake of Rb+ was started by adding 500 μl of the medium in which 5 mM KCl was replaced by 5 mM RbCl. Incubation was terminated 10 min later by rapid filtration (assisted by negative-pressure at ~20 psi) through Whatman No. 1 filters (2.5 cm in diameter) and washing the filters twice with 2 ml of RbCl-free medium. The filters were then extracted overnight with 1.5 ml of deionized water and the concentration of Rb+ in the extracts estimated using acetylene-flame atomic absorption spectroscopy (AAS).
ATPase Activity Studied by Production of Pi (Inorganic Phosphate)
Confluent rat cultured astrocytes were mechanically detached from the cultured flasks and homogenized in ice-cold homogenization medium (containing 250 mM sucrose, 5 mM EDTA, 20 mM imidazole, pH 7.4) at 3% (w/v) using a glass-glass homogenizer and frozen at app. −20°C. The homogenates were later thawed and first preincubated (aliquots of 150 μl) for 10 min at 37°C in the assay medium (containing 30 mM histidine, 4 mM MgCl2, 128 mM NaCl and 20 mM KCl, pH 7. 4) with or without the drugs.
Estimation of ATPase activity followed an established method [32]: Following the preincubation, 3 mM ATP was added to the samples to initiate the activity of ATPase(s) and the incubation was continued for 10 min. The reaction was terminated by the addition of an equal volume of ice-cold 0.8 N perchloric acid. The samples were centrifuged at 1,200g at 2°C for 15 min in a Hettich Universal 16R centrifuge. Aliquots of 250 μl from the centrifuged solution were transferred into cuvettes and diluted with 250 μl of distilled water. A volume (500 μl) of the colour reagent was then added. The colour reagent was prepared by dissolving 4 g of FeSO4·7H2O in 100 ml of a 1% ammonium molybdate ((NH4)6Mo7O24·4H2O) solution in 3.3 ml of 98% H2SO4, and was used within 2 h after the preparation, was added to the cuvettes. The absorbance of the cuvettes was read after 10 min of the resulting colour at 750 nm by a UV-1601 UV–Visible spectrophotometer Shimadzu, Tokyo, Japan. The standard used for inorganic phosphate was from a stock solution of 1 mM KH2PO4.
Statistical Analyses
The data were analysed by using the Prism Software (GraphPad, San Diego Ca, USA). Lines were fitted by linear regression. Values are shown as mean ± SEM. Statistical comparisons were evaluated by using one way analysis of variance (ANOVA), Tukey’s multiple comparison test or unpaired t-test.
Chemicals
Antibodies against GFAP were obtained from SIGMA Chemical Company (St. Louis, Mo, USA), the AF secondary antibodies were purchased from Bioscientific Pty Ltd. (Sydney, Australia).
Rottlerin was purchased from Calbiochem (La Jolla, CA, USA), ouabain, digoxin, ATP (adenosine triphosphate), EDTA (ethylenediaminotetraacetic acid), DMSO (dimethyl sulfoxide), imidazole and all tissue culture supplies were from Sigma Chemical Co (St. Louis, MO., USA). d-Aspartate was a gift from Prof. Graham Johnston (Department of Pharmacology, The University of Sydney). All other chemicals were of analytical grade and were purchased from commercial suppliers. Rottlerin was solubilized using DMSO [10] while digoxin was solubilized by ethanol (final contribution from ethanol was 0.7% and this concentration was either included in the control or it was established in separate experiments that it had no effect on the reaction studied).
Results
Effects of Na+/K+-ATPase Inhibitors on the Distribution of GLAST in Astrocytes
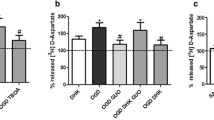
In the absence of drugs the fluorescence-labelled GLAST immunoreactivity (GLAST-IR) was distributed approximately equally between cytoplasm and the areas at, or in the immediate vicinity of, the plasma membrane. Even though the distribution of GLAST-IR within the cytoplasm was not even, the values of RFI (ratio of mean fluorescence density in cytoplasm to mean fluorescence density at or near the plasma membrane, mMFD/cMFD) were close to one (Fig. 3). In the presence of d-aspartate RFI increased and reached values of about 1.5–1.8 (significantly different from controls at P < 0.001; Figs. 2 and 3). Neither digoxin nor ouabain (at 100 μM) alone had any effect on the GLAST-IR distribution, however, both Na+/K+-ATPase inhibitors prevented the d-aspartate induced translocation of GLAST-IR towards the plasma membranes (significantly different from d-aspartate exposed cells at P < 0.01, not different from controls at P > 0.05; Fig. 3).

Relative fluorescence intensity (RFI, see legend of Fig. 2 and [9]) for GLAST IR in the presence of d-aspartate and the cardiac glycosides ouabain and digoxin (both at 100 μM; values are mean ± S.E.M. obtained from the number of cells shown in parenthesis). **Statistically different from the values obtained in the presence of d-aspartate at P < 0.01 by ANOVA, using Tukey’s multiple comparison test
Studies of Na+/K+-ATPase Activity in Cortical Prisms and Astrocyte Homogenates
Neither 1 mM ouabain nor 100 μM digoxin caused significant inhibitions of Pi production from ATP by homogenates of astrocyte membranes. This could have been caused not only by low activities of ouabain- and digoxin-sensitive Na+/K+-ATPase in the homogenates but also by low signal-to-noise ratios resulting from small amounts of tissue (limited by the availability of cultured astrocytes). The mean values of controls varied from experiment to experiment between about 16 and 26 μg Pi/mg protein/hour. Rottlerin, however, produced, under similar experimental conditions, statistically significant inhibitions of Pi production, with maximum inhibition (Imax) computed at about 80% (Fig. 4). Additionally, we tested digoxin against Rb+ uptake by prisms of rat cerebral cortex to estimate its activity as an inhibitor of Na+/K+-ATPase in adult brain tissue. We obtained a significant dose-dependent effect (Fig. 5). Uptake of Rb+ by tissue prisms prepared from adult rat cerebral cortex has been used as a quantitative measure of Na+/K+-ATPase activity previously [10, 26].

Inhibition of ATPase (Pi production from ATP) in astrocyte homogenates by rottlerin. Values relative to control (100) are plotted against Log of rottlerin concentration (μM). For details how the lines and constants (mean ± S.E.M.) were computed see [35]. “n” corresponds to the number of points (as circles in the figure)

Discussion
As in previous studies [9, 10], the present experiments using immunocytochemistry have indicated that, in cultured astrocytes, the glutamate transporter GLAST translocates from cytoplasm to plasma membrane in response to the presence of d-aspartate. The most parsimonious interpretation of earlier data [6, 7, 33] would be that the presence of GluT substrates in the vicinity of GLAST-expressing astrocytes triggers a process resulting in translocation of additional GLAST molecules to the plasma membrane. Accordingly, greater availability of substrate(s) would cause higher activity of GluT and this would lead to the recruitment of additional transporter (GLAST) molecules at the surface of the cell. Therefore, the most straightforward explanation of our main findings would be that d-aspartate did not induce the GLAST translocation when ouabain and digoxin were present because GluT activity could not increase while Na+/K+-ATPase was inhibited.
However, the inhibition of GLAST traffic by ouabain and digoxin may not be simply caused by an overall decrease in free energy supply resulting from a nonselective inhibition of Na+/K+-ATPase. This is underscored by the lack of an observable effect of digoxin and ouabain on the Pi production by ATPase activity in the cell-free preparations (though some degree of Na+/K+-ATPase inhibition by ouabain in intact cell cultures has been observed; [10]). More likely, ouabain and digoxin would have a strong effect on only a small portion of the total Na+/K+-ATPase activity, perhaps mediated by an isoform of the enzyme which is specifically susceptible to the inhibition by ouabain and digoxin. Inhibition of this fraction of the enzyme would then be enough to stop the translocation and activation of GLAST.
In a previous study, ouabain, when tested in rat cerebral cortical tissue in vitro, produced strong inhibition of GluT (IC50 < 1 μM) but was at least an order of magnitude weaker (IC50 app. 17 μM) as an inhibitor of Na+/K+-ATPase activity [23]. In other words, almost all GluT in those experiments was inhibited at ouabain concentrations which had only marginal effect on the overall activity of Na+/K+-ATPase in the tissue [26]. Such findings, too, could be explained by postulating a link between a highly ouabain-sensitive form of Na+/K+-ATPase (which would account for only a small fraction of the total Na+/K+-ATPase and glutamate transport, in agreement with conclusions based on other studies [34]. It has also been pointed out that administration of ouabain into neonatal rat brains in vivo can produce excitotoxic events, putatively caused by increased extracellular levels of l-glutamate, even when no major disruption of energy metabolism could be detected [29]. That observation, too, is consistent with the notion that only a small but strategically positioned fraction of Na+/K+-ATPase is of crucial importance for the normal function of GluT.
Regardless of whether ouabain and digoxin act primarily by disrupting of a close functional link between GluT (GLAST) and a subset of Na+/K+-ATPase molecules or by directly interfering with the GLAST translocation to the plasma membrane, both compounds should be viewed as potentially serious neurotoxins.
In summary, the present study has demonstrated that (Na+, K+)-dependent ATPase inhibitors ouabain and digoxin can inhibit the cytoplasm-to-membrane translocation of glutamate transporter GLAST. The mechanism does not appear to depend on an overall inhibition of (Na+, K+)-dependent ATPase activity but an involvement of a small fraction of (Na+, K+)-dependent ATPase linked directly to GluT cannot be ruled out. The findings have implications for the mechanisms underlying neurotoxicity of ouabain and digoxin.
References
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Balcar VJ (2002) Molecular pharmacology of the Na+-dependent transport of acidic amino acids I the mammalian central nervous system. Biol Phar Bull 25:291–301
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Internat 51:333–355
Beart PM, O’Shea RD (2007) Transporters for l-glutamate: an update on their molecular pharmacology and pathological involvement. Brit J Pharmacol 150:5–17
Duan S, Anderson CM, Stein BA et al (1999) Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci 19(23):10193–10200
Robinson MB (2002) Regulated trafficking of neurotransmitter transporters: common notes but different melodies. J Neurochem 80:1–11
Susarla BTS, Seal RP, Zelenaia O et al (2004) Differential regulation of GLAST immunoreactivity and activity by protein kinase C: evidence for modification of amino and carboxyl termini. J Neurochem 91:1151–1163
Susarla BTS, Robinson MB (2003) Rottlerin, an inhibitor of protein kinase C-δ (PKC-δ), inhibits astrocytic glutamate transport activity and reduces GLAST immunoreactivity by a mechanism that appears to be PKC–δ–independent. J Neurochem 86:635–645
Shin JW, Nguyen KTD, Pow DW et al (2009) Distribution of glutamate transporter GLAST in membranes of cultured astrocytes in the presence of glutamate transport substrates and ATP. Neurochem Res 34:1758–1766
Nguyen KTD, Shin JW, Rae C et al (2009) Rottlerin inhibits (Na+, K+)-dependent ATPase activity in brain tissue and alters d-aspartate dependent redistribution of glutamate transporter GLAST in cultured astrocytes. Neurochem Res 34:1767–1774
Fonnum F (1984) Glutamate: A neurotransmitter in mammalian brain. J Neurochem 42:1–11
Bennett MR, Balcar VJ (1999) Forty years of amino acid transmission in brain. Neurochem Int 35:269–280
Watkins JC, Jane DE (2006) The glutamate story. Brit J Pharmacol 147(S1):S100–S108
Abe K, Saito H (2000) Involvement of Na+-K+ pump in l-glutamate clearance by cultured rat cortical astrocytes. Biol Pharm Bull 23:1051–1054
Cholet N, Pellerin L, Magistretti PJ, Hamel E (2002) Similar perisynaptic glial localizationfor the Na+, K+-ATPase alpha-2 subunit and the glutamate transporters GLAST and GLT in the rat somatosensory cortex. Cereb Cortex 12:515–525
Gegelashvili M, Rodriguez-Kern A, Sung L, Shimamoto K, Gegelashvili G (2007) Glutamate transporter GLAST/EAAT1 directs cell surface expression of FXYD2/γ subunit of Na+, K+-ATPase in human fetal astrocytes. Neurochem Int 50:916–920
Rose EM, Koo JCP, Antflick JE, Ahmed SM, Angers S, Hampson DR (2009) Glutamate transporter coupling to Na, K-ATPase. J Neurosci 29:8143–8155
Rapport RL 2nd, Harris AB, Friel PN (1975) Human epileptic brain Na+, K+-ATPase activity and phenytoin concentrations. Arch Neurol 32:549–554
Ellis DZ, Rabe J, Sweadner KJ (2003) Global loss of Na+/K+-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 23:43–51
Hansen AJ (1985) Effects of anoxia on ion distribution in the brain. Physiol Rev 65:101–141
Wieloch T, Harris RJ, Symon L et al (1984) Influence of severe hypoglycemia on brain extracellular calcium and potassium activities, energy and phospholipid metabolism. J Neurochem 43:160–168
Vandenberg RJ (1998) Molecular pharmacology and physiology of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 25:393–400
Maragakis NJ, Rothstein JD (2004) Glutamate transporters: animal models to neurologic disease. Neurobiol. Disease 15:461–473
O’Shea RD (2002) Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 29:1018–1023
Balcar VJ, Johnston GAR (1972) The structural specificity of the high affinity uptake of l-glutamate and l-aspartate by rat brain slices. J Neurochem 19:2657–2666
Nanitsos EK, Acosta GB, Saihara Y et al (2004) Effects of glutamate transport substrates and glutamate receptors ligands on the ativity of (Na+, K+)-dependent ATPase in brain tissue in vitro. Clin Exp Pharmacol Physiol 31:762–769
Lees GJ, Leong W (1994) Brain lesions induced by nonspecific inhibitors of sodium-potassium ATPase. Brain Res 649:225–233
Lees GJ, Leong W (1995) The sodium-potassium inhibitor ouabain is neurotoxic in the rat substantia nigra and striatum. Neurosci Lett 188:113–116
Veldhuis WB, van der Stelt M, Delmas F et al (2003) In vivo excitotoxicity induced by ouabain, a Na+/K+-ATPase inhibitor. J Cereb Blood Flow Metabolism 23:62–74
Scott HL, Pow DV, Tanneberg AE et al (2002) Aberrant expression of glutamate transporter EAAT1 in Alzheimer’s disease. J Neurosci 22(RC206):1–5
Rae C, Lawrance ML, Dias LS et al (2000) Strategies for studies of neurotoxic mechanisms involving deficient transport of l-glutamate: antisense knockout in rat brain in vivo and changes in the neurotransmitter metabolism following inhibition of glutamate transport in guinea pig brain slice. Brain Res Bull 53:373–381
Turner N, Haga KL, Else PL et al (2006) Scaling of Na+, K+-ATPase molecular activity and membrane fatty acid composition in mammalian and avian hearts. Physiol Pharmacol Zool 79:522–533
Poitry-Yamate CL, Vutskits L, Rauen T (2002) Neuronal-induced and glutamate-dependent activation of glial glutamate transporter function. J Neurochem 82:987–997
Pellerin L, Magistretti PJ (1997) Glutamate uptake stimulates Na+, K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J Neurochem 69:2132–2137
Takamoto A, Quiggin LB, Lieb I et al (2002) Differences between d–and l-aspartate binding to the Na+-dependent binding sites on glutamate transporters in frozen sections of rat brain. Life Sci 70:991–1001
Acknowledgments
Thanks are expressed to Dr. Louise Cole of Bosch Institute Microscopy Facility for her assistance with the image analysis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The present study reflects my very long-lasting interest in amino acid transport, going back to my PhD years in early 1970s in Canberra. There was not much to read on amino acid transport in brain at the time, except, of course, for the pioneering studies of Abel Lajtha and his group. Abel’s papers became a continuing source of inspiration for me as well as the benchmark for the standard of research in the area. Some years later, as a postdoctoral fellow in Strasbourg, France, I ventured to submit my first manuscript to Neurochemical Research. It came back with several pages of referees’ comments and objections, essentially asking me to rewrite and change the conclusion. I haven’t got the details of my response anymore but I do remember that it ended rather curtly; stating that this was our study, our data and it will be our conclusion. Of course, I expected the Editor-in-Chief, Dr. Abel Lajtha to reject the paper but, to my great astonishment, he sided with us and the paper was published! I always enjoyed meeting Abel either in his Institute in New York or having him to listen patiently to my opinions on AA transport at international meetings, sometimes even while sharing airport taxis in strange cities. Abel introduced me to the International Society for Neurochemistry, backed my application for membership in 1978 and I have been a beneficiary of his tolerance, kindness and generosity on quite a few more occasions since then (VJB).
Special issue article in honor of Dr. Abel Lajtha.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nguyen, K.T.D., Buljan, V., Else, P.L. et al. Cardiac Glycosides Ouabain and Digoxin Interfere with the Regulation of Glutamate Transporter GLAST in Astrocytes Cultured from Neonatal Rat Brain. Neurochem Res 35, 2062–2069 (2010). https://doi.org/10.1007/s11064-010-0274-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-010-0274-4