
Abstract
The pathogenesis of MS is unknown. In our studies, we have demonstrated an important role for citrullinated myelin basic protein (MBP). The accompanying loss of positive charge compromises the ability of MBP to interact with the lipid bilayer. The conversion of arginine to citrulline in brain is carried out by an enzyme peptidyl arginine deiminase (PAD) 2. The amount of PAD 2 in brain was increased in MS normal-appearing white matter. The mechanism responsible for this increase involved hypomethylation of the promoter region in the PAD 2 gene in MS, but no change (compared to normal) was found in thymus tissue DNA from the same MS patients. In addition, no change was observed in other neurological diseases, including Alzheimer’s, Parkinson’s, and Huntington’s. We propose that citrullinated MBP, resulting from elevated levels of PAD 2 represents an important biochemical pathway in the pathogenesis of MS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Demyelinating diseases, such as multiple sclerosis (MS), the major demyelinating disease of humans affects not only young adults, but also children. It is the leading cause of neurological deficit in North America and Western Europe [1]. It is characterized by the patchy destruction of the myelin sheath around the axon and is, therefore, classified as a primary demyelinating disease [2]. The reason for this patchy destruction is not known. The MS plaque is characterized by a focal area of myelin destruction associated with astroglial scar formation. These lesions are scattered throughout the CNS with a predilection for optic nerves, brain stem, spinal cord, and periventricular white matter [3]. Perivascular lymphocytic infiltration with macrophages, microglial cells and astrocytes is a common feature. More recently, it has been recognized that extensive oligodendrocyte apoptosis and myelin macrophages are found in MS tissue, suggesting a primary neurodegenerative mechanism, followed by the autoimmune response as a secondary event [4]. In this type of apoptosis, the cardinal feature is damage to the most distal part of the oligodendiocyte process in the periaxonal region [5]. The chemical changes in myelin that precede this destruction are under intensive investigation. In the present overview, we suggest that enzymatic changes induced in myelin basic protein (MBP) may represent an important factor.
The cause of MS is unknown. Two major theories have emerged: an autoimmune or a neurodegenerative cause. The autoimmune etiology is considered to arise from sensitization of T-cells in the periphery by a mechanism of molecular mimicry, usually involving a viral antigen with peptide sequences similar to those found in myelin proteins, especially MBP, a candidate autoantigen in MS. The neurodegenerative hypothesis involves metabolic changes in the myelin constituents, which destabilize the membrane architecture, resulting in myelin degradation. Since the myelin proteins are primarily responsible for maintaining the structure of the membrane we focused our attention on myelin proteins, in particular, modifications that are found on MBP.
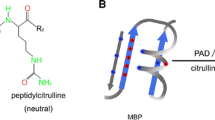
Myelin basic protein is an unusual protein. It is a single protein chain consisting of 170 amino acids. Since it does not contain cysteine, it cannot form disulfide bonds. It is a flexible structure, which adapts to environmental conditions, by changing conformation. It is a member of a group of proteins considered to be intrinsically disordered and, therefore, have considerable conformational flexibility (reviewed extensively in the contribution of Harauz and Musse in this issue). A further unusual feature of MBP is the large number and variety of post-translational modifications, such as N-terminal acylation, GTP- and ADP-ribose binding sites, deamidation, methylated arginine, methionine sulphoxide, phosphorylation, and deimination of arginyl residues. Citrullination occurs on six sites in the 18.5 kDa MBP from humans, while phosphorylation is found on ten sites at least. These are summarized in Fig. 1.

Post-translational modification of MBP. All post-translational modifications, except methylation of Arg107 (human sequence), decrease the positive charge. The known modifications are acylation of the N-terminus, GTP binding, ADP-ribosylation (if R25 is not citrullinated), deamidation, methionine sulphoxide, mono, and dimethylation of Arg107, phosphorylation at Ser/Thr residues and Tyr and deimination of arginyl residues (six sites in the 18.5 kDa MBP from normal brain). The structure, shown as a hairpin, occurs at the tri-proline region at the site of the bend. The figure was adapted and modified from Moscarello (1990, Myelin basic protein: a dynamically changing structure. In: Dynamic interactions of myelin proteins, George Hashim and Mario Moscarello (eds) Prog Clin Bio Res, vol 336)
One of the modifications, in which we are particularly interested, involves the conversion of peptide-bound arginine to peptide bound citrulline, an enzymatic reaction called deimination. The conversion of arginine to citrulline involves the release of ammonia. For each arginine converted to citrulline, one positive charge is lost from the protein. We first demonstrated that citrulline was present in a crude MBP fraction from myelin [6]. We then purified the citrulline containing MBP on CM52 cation exchange columns essentially as described earlier [7–9]. From these columns, we obtained a number of fractions, all of which were MBP. Fractionation of the various MBPs was shown to be dependent on the net charge of the highly modified MBP, resulting from phosphorylation, deamidation, C-terminal arginine loss, and others (Fig. 1). The citrullinated MBP was recovered in the flow-through volume and required further purification by high-performance liquid chromatography (HPLC). The purified protein was sequenced and the arginyl residues that were deiminated were identified. These were R25, R31, R122, R130, R159, and R169 [10]. This decreased positive charge affected the ability of the protein to interact with and organize lipid vesicles. Using liquid X-ray diffraction, we were able to show that the citrullinated MBP (termed “C-8” for convenience) was unable to organize lipid bilayers into compact multilayers, which was readily achieved by the non-citrullinated charge isomer (termed C-1, so-called because it bound most tightly to the CM52 column) [11]. At this time, we speculated that this failure to organize bilayers into compact multilayers, may contribute to myelin instability as seen in MS. These data demonstrated that the citrullinated MBP had a limited ability to organize and compact the lipid bilayers.
The citrullinated MBP was isolated from white matter of MS brain and compared to that isolated from normal brain. We reported that the relative amount of the citrullinated MBP was increased in MBP isolated from MS brain. Whereas, it accounted for 20% of the MBP isolated from normal brain, a threefold increase was found in chronic MS. Thus, the ratio of citrullinated MBP/non-citrullinated MBP was 0.82 in the normal and it was 2.45 in the MS MBP [12]. In a single case of Marburg’s disease (a fulminating form of MS), 80–90% of the MBP was citrullinated, with a citrullinated MBP/non-citrullinated MBP ratio of 6.7 [13, 14]. Therefore, MBP in Marburg’s type of MS was almost totally deiminated. Citrullination of MBP has been reported to diminish charge density, which diminishes its interaction with lipids, making it more vulnerable to proteolytic attack [15].
The effects of deimination on protein structure and protein-lipid interactions in myelin are diverse and have been thoroughly reviewed [16]. Briefly, deimination of MBP alters the 3D structure of MBP, producing a more open conformation. The more open conformation of MBP increases its susceptibility to proteases such as cathepsin D, a myelin associated protease. MBP containing six citrullinyl residues is digested four times faster than non-deiminated MBP. The MBP containing 18 citrullinyl residues was digested 45 times faster than MBP from normal brain. These data support the view that deimination of MBP not only decreases the positive charge on the molecule, but also creates a more open structure [17].
These perturbations in structure affect the interactions of the protein with the lipid bilayer and decrease the compaction of the multilayer structure of myelin. The deiminated protein had a reduced ability to bind and aggregate lipid vesicles and the highly deiminated form of MBP caused fragmentation of lipid vesicles [18], a process which may have implications for myelin breakdown in MS.
Electrostatic interactions between the negatively charged phospholipids and the positively charged amino acids (Arg and Lys) are the origin of attraction of MBP to the lipid. MBP is postulated to work as a glue for adjacent bilayers [19, 20] by binding to the polar head groups. This mechanism of interaction is interfered with by deimination of arginyl residues, since the resulting citrulline carries a neutral charge.
The conversion of arginine→citrulline in proteins is carried out by the enzyme peptidyl arginine deiminase (PAD). Of the five known PAD enzymes, PAD 2 is the one involved in the deimination of MBP. Since our studies on the deiminated MBP in humans was obtained from autopsy material, the question of the origins of the citrullinated protein, as possibly artefactual, was addressed. To resolve this issue, we explored the temporal relationship between the appearance of the citrullinated protein and the up-regulation of the PAD 2 enzyme, which could only be done in a relevant animal model. Such a model was available to us. It is a transgenic model, containing 70 copies of the transgene for the myelin proteolipid protein DM20. This mouse is normal for the first 3 months of its life and then demyelinates spontaneously. We measured PAD enzyme and citrullinated MBP. We found that PAD 2 was up-regulated at 2 months of age and citrullinated MBP at 3 months of age, suggesting a precursor-product type relationship [21]. We postulated that up-regulation of PAD 2 was the primary response. To substantiate this hypothesis, a thorough study was carried out on PAD 2 regulation.
In this regard, we have reported that paclitaxel (Taxol) inhibited the activity of purified PAD 2 isolated from bovine brain in an in vitro assay [22]. In in vivo studies, we demonstrated that paclitaxel attenuated clinical disease in the spontaneously demyelinating mouse model described above [23]. In a recently submitted manuscript, we extended our Taxol studies further by demonstrating that the addition of the methyl donor, vitamin B12, suppressed both immune and non-immune demyelinating disease in mice (F. G. Mastronardi et al. 2006, submitted data), better than paclitaxel alone. These data suggest that drugs, which target PAD 2, in the presence of a methyl donor, such as vitamin B12, may be beneficial in demyelinating disease.
Another PAD enzyme, PAD 4, has been implicated in the pathogenesis of psoriasis [24], rheumatoid arthritis [25, 26] and MS (F. G. Mastronardi et al. 2006, submitted data). PAD 4 is different from other PAD enzymes, because the gene contains a nuclear localization signal (PPAKKKST) [27], not present in any of the other PAD enzymes. Translocation of the enzyme from the cytoplasm to the nucleus, has been shown to involve the cytokine tumor necrosis factor α (TNFα). The substrate for PAD 4 in the nucleus is histone (F. G. Mastronardi et al. 2006, submitted data). Deimination of arginyl residues of histones decreases the positive charge on histone, which compromises its ability to interact with DNA, possibly resulting in apoptosis of oligodendrocytes. On the other hand, deacetylation of histones would increase the positive charge on histone. Therefore, these two processes, the deimination of arginyl residues and the deacetylation of lysyl residues of histones affect the positive charge on histone in opposite ways, and these two processes may have a regulatory function.
Since we determined that up-regulation of PAD 2, preceded demyelination in the animal model, we searched for the mechanism responsible. An examination of the nucleotide sequence of the PAD 2 promoter revealed that it was high in CpG sequences. CpG sequences are important, because cytosine is a target for methylation by DNA methyltransferase, which produces 5-methylcytosine. Methylation of the promoter region of a gene silences the gene, whereas demethylation increases transcription.
Accordingly, we isolated DNA from white matter obtained from normal individuals and patients who died with MS. The DNA was treated with bisulphite to convert cytosine into uracil. The methylcytosine is not affected by bisulphite. We found that methylcytosine accounted for less than 4% of the cytosines in the PAD 2 promoter in MS (n = 17), whereas in the normal, the methylcytosine in the PAD 2 promoter was 12% (n = 8). Therefore, the PAD 2 promoter was hypomethylated in the DNA from MS white matter samples. DNA was also obtained from thymus of the same MS patients. The amount of methylcytosine was the same in both MS and normal samples. Therefore, the hypomethylation found in MS appeared to be tissue specific. When we examined other neurological diseases, including Alzheimer’s, Parkinson’s, and Huntington’s, the amounts of methylcytosine were the same as normal (Mastronardi et al. 2006 in preparation). Therefore, the hypomethylation of the PAD 2 promoter was unique to MS brain. This hypomethylation of the promoter of PAD 2 may account for the increase in PAD 2 in MS brain (summarized in Fig. 2).

Deimination pathway in the pathogenesis of demyelinating disease. Normal myelin is shown on the left. The promoter of the PAD 2 gene contains methylated cytosines (closed lollipops) along its CpG island, which regulates its transcription. Some MBPcit is made under normal conditions by the deimination of arginyl residues “C” on MBP. The amount of MBPcit is not enough to destabilize the bilayer. During demyelination, the promoter of PAD 2 is demethylated (open lollipops) at cytosines in the CpG island. This demethylated promoter increases transcription of PAD 2, which increases the amount of MBPcit in myelin with subsequent destabilization of the myelin. Degradation of myelin protein fragments, in particular MBPcit fragments, become available to sensitize T-cells in the periphery, initiating the immune response
The removal of methyl groups from methylcytosine in CpG sequences is carried out by a DNA demethylase. We proposed that up-regulation of the demethylase may account for the hypomethylation state of the PAD 2 promoter in MS. When we measured the total demethylase activity in brain homogenates from normal and MS white matter, using a synthetic methylcytosine-guanosine (CpG) substrate [28] we demonstrated that the enzyme in NAWM from MS brain was increased twofold over normal, suggesting the DNA demethylase may have an important role in the hypomethylation of the PAD 2 promoter. Therefore, the hypomethylated state of the PAD 2 promoter may involve up-regulation of the DNA demethylase at least in part.
As a result of the work summarized in this overview, we postulate the following sequence of events during the pathogenesis of MS. The citrullinated MBP, because of its decreased positive charge, prevents proper compaction of the bilayer, so that the multilamellar structure is less stable. Since PAD 2 has been shown to be present in myelin by immunogold electron microscopy (unpublished results), the conversion of non-citrullinated to citrullinated MBP can occur locally in myelin. Preliminary results have shown that immunogold labeling of PAD 2 in mouse optic nerve was not random, but was found in clusters in the axon itself and in the myelin sheath. In the latter, the clusters were found mainly at the junction of the myelin sheath and the axon (periaxonal) with some gold particles distributed in the myelin sheath. The presence of the PAD 2 enzyme at these sites suggests that the citrullinated MBP would be formed in these clusters in myelin, eventually resulting in plaques.
When we examined the extent of labeling in the optic nerve of the spontaneously demyelinating transgenic mouse, we found a threefold increase in the number of gold particles. This increase in PAD 2 labeling demonstrated that increased amounts of PAD 2 were present in the transgenic mouse, accounting for an increase in the citrullinated MBP. Since citrullinated MBP is present in the normal myelin sheath (20% of the total MBP), citrullinated MBP can be tolerated to this extent. However, increased amounts as found in chronic MS (threefold), and in fulminating MS (six to sevenfold), cannot be accommodated in a compact myelin sheath, resulting in destabilization.
Peptidyl arginine deiminase 2 has been reported recently to be elevated in primary open-angle glaucoma, where it was considered responsible for the deimination of several optic nerve proteins, in addition to MBP. Concomitant with this protein deimination, a decrease in arginine methylation was also observed [29]. Therefore, the PAD 2 enzymes have important roles in several nervous system diseases.
Oligodendrocyte apoptosis has been reported in MS white matter in the absence of lymphocytes or myelin phagocytes [4]. The major feature of this type of injury is damage to the most distal part of the oligodendrocyte process (periaxonal), which results in clumping of proteins [5], followed by apoptosis. The possible role of the PAD 2 enzyme in this process has not yet been demonstrated. If it can be established that protein deimination at sites of PAD 2 clusters does occur, it may suggest that demyelination begins in the periaxonal myelin sheath and subsequently moves outward. Although this hypothesis is speculative, it warrants investigation. The increased amount of PAD 2 in MS white matter probably reflects a greater production of the enzyme. Since the PAD 2 promoter is rich in CpG islands, we showed that the promoter was hypomethylated. These studies are ongoing in our laboratory. They suggest immediately that the reversal of the hypomethylated state may be a therapeutic target in MS.
The importance of methylation in a therapeutic strategy of MS has been suggested by us recently [(F. G. Mastronardi et al. 2006, submitted data), 30]. In these studies, we demonstrated that vitamin B12 in combination with either interferon β or paclitaxel, effectively ameliorated demyelinating disease in both EAE and non-immune demyelinating disease. In a detailed mass spectrophotometric study of the various components of MBP, we showed arginine methylation was decreased in most of the components of the MBPs from MS material [31], supporting the view that defects in methylation may involve protein methylation as well. The methylation process requires Vitamin B12, which transfers its methyl group to homocyteine synthesizing methionine, which is then converted to S-adenosytmethionine, the methyl donor in all biological methylation reactions, including methylation of cytosine in DNA to methylcytosine. These studies with the preliminary studies reported above suggest an important role for methylation/demethylation reactions in the pathogenesis of MS.
References
McFarlin DE, McFarlin HF (1982) Multiple sclerosis (second of two parts). N Engl J Med 307:1246–1251
Volmer TL, Waxman SG (1991) Multiple sclerosis and other demyelinating disorders. Raven Press, New York
Adams CW (1977) Pathology of multiple sclerosis: progression of the lesion. Br Med Bull 32:15–20
Barnett MH, Prineas JW (2004) Relapsing and remitting multiple sclerosis. Pathology of the newly forming lesion. Ann Neurol 55:458–468
Stadlemann G, Ludwin S, Tabira T, Guseo A, Lucchinetti CF, Leel-Ossy L, Ordinario A, Bruck W, Lassmann H (2005) Tissue preconditioning may explain concentric sclerosis in Balo’s type of multiple sclerosis. Brain 128:979–987
Finch PR, Wood DD, Moscarello MA (1971) The presence of citrulline in a myelin protein fraction. FEBS Lett 15:145–148
Martenson RE, Diebler GE, Kies MW (1971) Microhetero geneity and species related differences among myelin basic proteins. Res Publ Assoc Res Nerv Ment Dis 49:76–94
Deibler GE, Martenson RE, Kies MW (1992) Large Scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prep Biochem 2:139–165
Moscarello MA, Brady GW, Fein DB, Wood DD, Cruz TF (1986) The role of charge microheterogeneity of protein in the formation and maintenance of multilayered structure of myelin: a possible role in multiple sclerosis. J Neurosci Res 15:87–99
Wood DD, Moscarello MA (1989) The isolation, characterization and lipid aggregating properties of a citrulline-containing myelin basic protein. J Biol Chem 264:5121–5127
Brady GW, Murthy NS, Fein DB, Wood DD, Moscarello MA (1981) The effect of basic myelin protein on multilayer membrane formation. Biophys J 34:345–350
Moscarello MA, Wood DD, Ackerley C, Boulias C (1994) Myelin in multiple sclerosis is developmentally immature. J Clin Invest 94:146–154
Wood DD, Bilbao JM, O’Connors P, Moscarello MA (1996) Acute multiple sclerosis (Marburg type) is associated with developmentally immature myelin basic protein. Ann Neurol 40:18–24
Whitaker JM, Mitchell GW (1996) A possible role for myelin basic protein in multiple sclerosis. Ann Neurol 40:3–4
Shanshiashvili LV, Suknidze CN, Machidze GG, Mideladze DG, Ramsden JJ (2003) Adhesion and clustering of charge isomers of myelin basic protein at model myelin membranes. Arch Biochem Biophys 419:170–177
Harauz G, Ishiyama N, Hill CMD, Bates IR, Libich DS, Fares C (2004) Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron 35:503–542
Pritzker LB, Joshi S, Gowan JJ, Harauz G, Moscarello MA (2000) Deimination of myelin basic protein I. Effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathespsin D. Biochemistry 39:5324–5381
Boggs JM, Rangaraj G, Koshy KM, Ackerley C, Wood DD, Moscarello MA (1999) Highly deiminated isoform of myelin basic protein from multiple sclerosis brain causes fragmentation of lipid vesicles. J Neurosci Res 57:529–535
Hu Y, Douvedski I, Wood DD, Moscarello MA, Husted C, Genain C, Zasadzinski JA, Israelchivili J (2004) Synergistic interactions of lipids and myelin basic protein. Proc Natl Acad Sci 101:13466–13471
Cristofolini L, Fontana MP, Serra F, Fasano A, Riccio P, Konovalov O (2005) Microstructural analysis of the effects of incorporation of myelin basic protein in phospholipid layers. Eur Biophys J 34:1041–1048
Moscarello MA, Pritzker L, Mastronardi FG, Wood DD (2002) Peptidyl arginine deiminase: a candidate factor in demyelinating disease. J Neurochem 81:385–343
Pritzker LB, Moscarello MA (1998) A novel microtubuler independent effect of paclitaxel: the inhibition of peptidylarginine deiminase from bovine brain. Biochim Biophys Acta 1388:154–160
Moscarello MA, Mak B, Nguyen TA, Wood DD, Mastronardi FG, Ludwin SK (2002) Paclitaxel (Taxol) attenuates clinical disease in a spontaneously demyelinating transgenic mouse and induces remyelination. Mult Scler 8:130–138
Ishida-Yamamoto A et al (2000) Decreased deiminated keratin K1 in psoriatic hyperproliferative epidermis. J Investig Dermatol 114:701–705
Masson-Bessiere D et al (2001) The major synovial targets of the rheumatoid arthritis specific antifilaggrin antibodies are deiminated forms of the 2 and 3 chains of fibrin. J Immunol 166:4177–4184
Van Boekel MA, Vossenaar ER, Van Den Hoogen FH, Van Venrooj WJ (2002) Antibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res 4:87–93
Nakashima K, Hagiwara T, Yamada M (2002) Nuclear localization of peptidyal arginine deiminase V and histone deimination in granulocytes. J Biol Chem 277:49562–49568
Szyf M, Battacharya SK (2002) Measuring DNA demethylase activity in vitro. Methods Mol Biol 200:155–161
Bhattacharya SK, Crabb JS, Bonilha VL, Gu X, Takahara H, Crabb JW (2006) Proteomics implicates peptidyl arginine deiminase 2 and optic nerve citrullination in glaucoma pathogenesis. Invest Ophthalmol Vis Sci 47:1–7
Mastronardi FG, Min W, Wang H, Winer S, Dosch M, Boggs JM, Moscarello MA (2004) Attenuation of experimental autoimmune encephalomyelitis and non-immune demyelination by IFN-beta plus vitamin B12. Treatment to modify notch-1/sonic hedgehog balance. J Immunol 172:6418–6426
Kim JK, Mastronardi FG, Wood DD, Lubman DM, Zand R, Moscarello MA (2003) Multiple Sclerosis, an important role for post translational modifications of myelin basic protein in pathogenesis. Mol Cell Proteomics 2:453–462
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue dedicated to Anthony Campagnoni.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moscarello, M.A., Mastronardi, F.G. & Wood, D.D. The Role of Citrullinated Proteins Suggests a Novel Mechanism in the Pathogenesis of Multiple Sclerosis. Neurochem Res 32, 251–256 (2007). https://doi.org/10.1007/s11064-006-9144-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9144-5