
Abstract
Introduction
Glioblastoma multiforme (GBM) constitutes one of the deadliest tumors to afflict humans, although it is still considered an orphan disease. Despite testing multiple new and innovative therapies in ongoing clinical trials, the median survival for this type of malignancy is less than two years after initial diagnosis, regardless of therapy. One class of promising new therapies are chimeric antigen receptor T cells or CAR-T which have been shown to be very effective at treating refractory liquid tumors such as B-cell malignancies. However, CAR-T effectivity against solid tumors such as GBM has been limited thus far.
Methods
A Pubmed, Google Scholar, Directory of Open Access Journals, and Web of Science literature search using the terms chimeric antigen receptor or CAR-T, GBM, solid tumor immunotherapy, immunotherapy, and CAR-T combination was performed for publication dates between January 1987 and November 2021.
Results
In the current review, we present a comprehensive list of CAR-T cells developed to treat GBM, we describe new possible T-cell engineering strategies against GBM while presenting a short introductory history to the reader regarding the origin(s) of this cutting-edge therapy. We have also compiled a unique list of anti-GBM CAR-Ts with their specific protein sequences and their functions as well as an inventory of clinical trials involving CAR-T and GBM.
Conclusions
The aim of this review is to introduce the reader to the field of T-cell engineering using CAR-Ts to treat GBM and describe the obstacles that may need to be addressed in order to significantly delay the relentless growth of GBM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most common primary malignant brain tumor affecting up to 17 individuals per 500,000 adults per year. Despite decades of research, the prognosis is dismal with a median survival of less than 15 months with standard of care [1]. Current standard-of-care treatment regimens consist of tumor de-bulking followed by concomitant chemotherapy and radiation. Recent research has focused on therapies targeting the immune microenvironment of the tumors, as progression of GBM occurs concomitantly with high levels of immunosuppression. Many therapeutic strategies that have been successful with other cancers have failed in GBM as a result of its unique organ localization and immunosuppressive environment [2]. The three main immunotherapies used against GBM to date have been immune checkpoint inhibition, vaccination, and adoptive transfer of effector lymphocytes, with varying outcomes but none has consistently extended survival beyond 12 months. More recently, a newer immunotherapy using oncolytic viruses has shown significant improvement of mean survival [3]
One of the main obstacles conventional immunotherapies for GBM face is the low abundance of leukocytes in the brain under steady-state conditions. Although immune surveillance cells such as T cells and microglia exist, they are located in the choroid plexus stroma and the cerebrospinal fluid (CSF), which occupy the perivascular spaces. The central nervous system (CNS) has traditionally been regarded as immune privileged, and therefore excluded from the protection systemic immune surveillance affords other organs [4]. A distinct obstacle facing drugs and general oncolytic treatments is the presence of the blood brain barrier (BBB) or the blood-cerebrospinal fluid barrier that effectively blocks their entrance into the brain parenchyma where GBM-associated tumors are located. The absence of lymphatic vessels which normally drain antigen presenting cells further confirm the notion of the brain as an immune privileged site.
Single cell sequencing and flow cytometry has clarified the complexity of the cells that reside in the brain parenchyma. The most prominent immune cells in the brain are microglial cells, which serve as the first line of defense against pathogens [5]. Bone marrow-derived macrophages/monocytes are the primary immune cell in glioma and they may compose up to 30% of tumor mass. As such, there are two distinct populations: glioma bone marrow-derived macrophages and microglia.
During tumor progression, monocytes and T cells extravasate into the microenvironment through the compromised BBB. One of the most prominent T cell types in glioblastoma are CD8+ cells [6, 7]. These CD8+ cytotoxic T cells increase in glioblastoma due to an upsurge in chemoattractants such as CXCL9, CXCL10, and adhesion molecules such as ICAM. However, this massive infiltration eventually creates a heavily immunosuppressive tumor micro-environment (TME) by activation of tumor-associated macrophages (TAMs) and further recruitment of myeloid derived suppressor cells (MDSC) [8]. MDSCs can suppress cytotoxic CD8+ T cell proliferation and overall activation by—among others- increasing surface expression of IL-4Rα and the production of arginase and inducible nitric oxide synthase (iNOS) [9]. In response to inflammatory stimuli, such as tumor growth, brain stromal cells produce high levels of classic immunosuppressive cytokines such as transforming growth factor beta (TGF-β) and interleukin-10 (IL-10), which counteract the inflammatory cytokine signals to maintain homeostasis. Glioma cells also are known to produce huge amounts of indolamine 2,3 dioxygenase (IDO) which results in accumulation of regulatory T cells (Treg) which further suppress cytotoxic T cell activity. Combination therapy of patients with brain tumors has been focused on inhibiting the specific immunosuppressive factors, but targeting TGF-β and IDO has shown no clinical benefit so far, despite its success in animal models [10].
The ongoing success in the treatment of many other types of cancers and further progress in understanding of T cell immunotherapy, suggests further novel treatment strategies may be forthcoming for the treatment of brain cancers [11], including the up-and-coming new therapy of Chimeric Antigen Receptor T cells (CAR-Ts).
Introduction and brief history of CAR-T
Chimeric antigen receptor T cells or CAR-Ts are synthetic immune receptors that redirect cytotoxic T cells to specific targets through recognition of surface proteins expressed on targeted tumor cells. Thus, the principle of CAR-Ts is to genetically modify [12] existing T-cells from cancer patients in order to re-direct their immune response machinery toward a malignant target cell of interest. Theoretically, any cell surface molecule can be targeted through a CAR-T, thus over-riding the tolerance to self-antigens and the antigen recognition gaps in the T- cell repertoire that often limit their scope of reactivity. It should be noted that expression of CAR-Ts basically bypasses the HLA-restricted nature of T-cells, making them immune to evasion strategies such as MHC shedding by tumors. However, the CAR-T needs to be optimized in order to increase its binding and signaling properties. Interestingly, the persistence and strength of CAR-Ts can also be modulated by the design of the intracellular signaling domains.
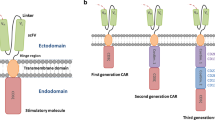
CAR-Ts have three main domains: the extracellular domain, which includes the antigen recognition domain, a transmembrane domain, and the intracellular domain, which is important for signal transmission (Fig. 1A). CAR-Ts are able to recognize antigen on any HLA background and can target tumor cells that have downregulated HLA expression or proteosomal antigen processing, mechanisms often attributed to tumor escape. A crude version of CAR-Ts was initially reported in 1987 by Yoshihisa Kuwana et al., [13] and almost concomitantly by Gideon Gross and Zelig Eshhar in 1989 [14]. While the first generation CAR-Ts were developed based on work from the Weiss lab demonstrating that inclusion of a CD3ζ domain could activate T-cells [15] (Fig. 1A), more versions followed quickly. Margo Roberts and Finney and colleagues during the 1990s contributed to the development of second generation CAR-Ts by incorporating co-stimulatory domains [16] such as CD28 which improved IL-2 production in Jurkat cells by 20-fold [17] (Fig. 1B). One of the advantages to CAR-Ts is their modular design properties that provide flexible domains which can be adjusted or swapped. Another seminal clinical improvement was the key finding of Rosenberg et al. in 1988, who demonstrated that that mild lympho-depletion in treated patients improved the proliferation of infused tumor-infiltrating lymphocytes (TILs) [18]. However, lympho-depletion is accompanied by very severe side-effects. All these crucial developments, and many others [19], paved the way for the eventual success of CAR-T cells clinically.

CAR-T design and current generations. A The basic CAR-T architecture consists of a target domain followed by a stem or hinge, a transmembrane domain and intracellular co-stimulatory motifs. Tags for easy identification can be placed in different key locations as shown. B Several types of co-stimulatory domain targets have been characterized to be used with CAR-T. C CAR-T have experienced a fast evolution, from the initial First Generation (First Gen) comprising only one stimulatory domain, all the way to second, third, fourth and next-generation CARs, comprising several combinations of co-stimulatory domain molecules as well as JAK/STAT signaling (Next-Gen)
However, unequivocal CAR-T results in a clinical setting were not demonstrated until two groups, one lead by Drs. June and Levine and funded by the Alliance for Cancer Gene Therapy or ACGT, and another by Rosenberg et al., from the NIH who showed remission in B-cell leukemia and lymphoma patients using a CAR-T approach [20, 21]. After this initial success combined with a little luck [22], more clinical trials followed [19, 23]. Experience with CD19-targeted CAR-Ts have had remarkable outcomes for patients with CD19-positive B cell malignancies which lead to FDA approval in 2017 [24, 25]. These CD19-targeted-CAR T therapies and the adoptive cellular therapy that followed in melanoma have also raised optimism for the treatment of CNS malignant tumors. For example, in patients with metastatic melanoma, Hong reported that 35% of patients achieved complete response in the brain metastases as well as extra-cranial disease [26].
As a result of the success of CAR-T cells on refractory liquid tumors, CAR-T technology has expanded substantially in recent years. Tagging the CAR-T at its amino (N)-terminus, upper or lower stem without losing functionality (Fig. 1A) has also been described when availability for detection is difficult [27]. Also of importance is the length of the CAR-T hinge [28], which ultimately can make a CAR-Ts effective in vitro but not in vivo [29]. This phenomenon is very unfortunate, because the vice-versa effect could also happen, i.e., where CAR-Ts don’t work in vitro but may work in vivo. Unfortunately such CAR-Ts would never be developed because very seldom do in vitro failures lead to in vivo testing.
Given the clinical success of CAR-Ts, development of co-stimulatory domains has been extensively characterized [30] as shown in Fig. 1B, as well as using nanobodies instead of antibodies as the targeting domain [31]. Nanobodies are smaller than conventional antibodies and are being explored in CAR-Ts in order to gain accessibility to difficult antigens [32]. Owing to the flexible nature of proteins, many more versions have been generated including third, fourth and next-generation CAR-Ts by incorporating JAK/Stat signaling (Fig. 1C).
While liquid tumors such as B-cell malignancies have experienced breathtaking success in achieving remission in up to 70–90% of treatment-refractory cancers, CAR-Ts designed to treat solid tumors have had considerably less success so far. Solid tumors pose greater challenges than liquid given the existence of a complex and heavily immuno-suppressed TME that surrounds the tumor and protects it from destruction [33]. The TME may also hamper the T-cell trafficking and infiltration necessary, resulting in T-cell exhaustion [34], and CAR-Ts that address the TME problem display enhanced anti-tumor efficacy in vivo [35]. Further, in some solid tumors, such as glioma, the TME is characterized by low nutrient regions and hypoxia [36]. However, given the versatile nature that CAR-T cell engineering offers, several improvements can be made to CAR-Ts in order to address the unique challenges that solid tumors present (Fig. 2). Other approaches to minimize TME effects have been; (1) designing CAR-Ts with dominant-negative TGFβrII receptors –dnTGFβII-, expression of mutant forms of FAS [37], (2) PD-1-CAR-T [38], dominant-negative PD-1 [39] or 3) as we describe below-secretion of key cytokines or heparanase [40].

Types of CAR-T developed. Several different strategies have been developed involving CAR-Ts owing to their flexible nature. A Bi-specific CAR, targeting two antigens independently. B iCAR, targeting two independent antigens leads to inhibition (thus sparing healthy tissues). C Intra-CAR-T can be designed to target antigens that are located intracellularly but expressed via MHC. D Syn-Notch CAR-T where a secondary-targeting antigen is strongly controlled by binding of a primary one. E Split CAR-T are created by targeting two independent antigens with the co-stimulatory domains split. Thus, only when both antigens are present is the CAR-T fully activated. F Supra-CAR: the CAR-T construct is split allowing for one co-stimulatory domain (B-zip) to bind several targeting domains (A-zip). G Tan-CAR-T are achieved by fusing two antigen-targeting domains into one. H Trivalent CAR-T, where three independent antigen-targeting CAR-T are expressed independently within the same cell. I Uni-CAR-T consist of an antigen-binding domain that bind not an endogenous target but a soluble one administered exogenously acting as a bridge (purple)
Methods
Electronic databases, Pubmed, Google Scholar, Directory of Open Access Journals, and Web of Science, were searched from January 1987 to August 2021. Database searches included the following key words: ‘glioblastoma or glioma’, ‘solid tumor immunotherapy’, ‘CAR-T’, ‘immunotherapy’, and ‘CAR-T combination’. We further manually screened references within the related articles to expand the search range. Two researchers (AKF and DS) extracted the relevant information and validated their inclusion in the current review.
CAR-T versus GBM
Several CAR-Ts have been developed to treat GBM and other solid tumors, but to-date none has led to long-term remission [11]. Despite these initial failures, the use of CAR-Ts against GBM is ongoing with multiple designs undergoing clinical trials (Table 1). The flexibility that T-cell genetic engineering offers is a very strong impetus to improve tumor targeting in a way no other treatment allows: i.e., using the plethora of cell-killing mechanisms that the immune system offers. CAR-Ts have been successfully used to treat several liquid (blood) cancers owing to the existence of very specific tumor targets. Thus, the rationale behind using CAR-Ts to treat GBM is that sooner or later a way to target multiple highly-specific tumor targets will be discovered. Indeed, multiple GBM-specific targets have already been identified, the most notable including EGFRvIII [41,42,43], HER2 [44], EphA2 [45] or IL-13Rα2-CAR [46, 47] among many others identified in Fig. 3 and Supplementary Table 1. Novel designs include an IL-13Rα2-CAR developed by Brown et al. [48], using a peptide as the targeting-domain (zetakine) or by Pituch et al., using an antibody [47]. Both approaches showed promise in murine studies targeting the IL-13Rα2 receptor in glioma cells and the zetakine version induced remission in a Phase I human GBM trial [46]. However, in this clinical trial the remission was relatively short-lived, since under the IL-13Rα2-CAR pressure, the tumor is suspected to have undergone antigenic loss, an acknowledged major obstacle in combating GBM with CAR-T therapy, since it represents the disappearance of the primary CAR-T target, which renders CAR-Ts ineffective. To compound this problem, antigenic loss can be the result of the heterogeneous cellular nature of GBM, with existence of GBM IL-13Rα2neg cells that expand to bypass CAR-T or instead IL-13Rα2+ cells that somehow undergo gene loss of IL-13Rα2. However, a clinical trial conducted by Brown et al., demonstrated that despite a heavily immunosuppressive milieu, CAR-Ts were able to eradicate IL-13Rα2+ cells. More recently, a T cell receptor fusion construct (TRuC) against IL-13Rα2 has been developed and tested in a U251 NG1 murine model of GBM, showing superior reactivity and safety profiles compared to conventional CAR-T cells [49].

List of anti-GBM CAR-T amino acid sequences. A comprehensive list of several CAR-T targeting different antigens on GBM is shown based on an extensive literature search. Within each CAR-T, different colors on amino acid sequences identify their nature and functionality. TMD transmembrane domain, CLTX chlorotoxin peptide
CAR-T cells have also been developed against the EGFRvIII antigen as mentioned above. The interest in the EGFRvIII variant stems from it being a mutated form of EGFR present in about 52% of glioma cells, but not healthy tissues [50]. In an EGFRvIII clinical study, the patients’ tumors also underwent antigenic loss, rendering the tumor CAR-T resistant [41]. It is interesting to note that despite knowledge regarding this phenomenon, it is not discouraging other groups from developing additional EGFRvIII-targeted CAR-Ts using novel higher affinity antibodies as recently as 2021 [51, 52]. Others have developed CAR-T secreting Bi specific T cell Engagers or BiTEs targeting EGFRvIII [53].
On another front, in order to improve CAR-T control even further, recent work by Dr. Wendel Kim, building on the development of Syn-Notch receptors [54], has developed a sigmoidal Syn-Notch that can discriminate tumor antigens very accurately [55]. Even more recently, another take on Syn-Notch by their creators has been developed using a two-strike trivalent CAR-T against EGFRvIII and EphA2/IL-13Rα+ TanCAR in a GBM murine model [56]. However, Syn-Notch circuits are exogenous and could lead to allergic reactions, as mentioned in the original publication [57].
Several approaches have been developed to address the obstacle of antigenic loss (Fig. 2). Most notably, a novel CAR-T employing the scorpion toxin peptide Chlorotoxin (CLTX) has been developed recently to target CAR-Ts towards GBM antigens [58]. This CLTX-CAR-T induced long-term remission in murine studies [58]. Since CLTX is known to recognize 100% glioma cells through at least three tumor-associated proteins while not affecting healthy tissue [59], the clever rational behind this approach is that a CLTX-CAR-T could potentially reduce antigenic loss once and for all. However, through their very elegant set of studies, Wang et al., identified expression of the surface protein MMP-2 on glioma cells as necessary to mediate the CLTX binding. Loss of MMP2 in GBM -which has been described [60]- may hamper CLTX targeting. Current clinical trials are underway (NCT04214392) that will determine whether CLTX-CAR-Ts can improve GBM prognosis in patients. It is interesting to note that a CLTX-antibody was developed in 2012 but its clinical application remains unknown [61]. We have also compiled updated lists of current trials in Table 1 and completed trials in Table 2 adapted from reviews by Land C.A. et al. [62] and Maggs L. et al. [63]
Other CAR-T approaches to treat gliomas have been to include helper genes. As such, a recent advancement has been reported from Huang et al., who have engineered a CD70-CAR [64]. Over-expression of CD70 on glioma cells has been known to induce apoptosis on T-cells via CD27 [65] and Huang et al., cleverly replaced the intracellular portion of CD27 by 41BB and CD3ζ, thus basically turning CD27 into a CD70 targeting CAR-T. The authors showed that this type of CAR could kill CD70+ glioma cells in vitro and in vivo, although remission was only achieved using 100 × 106 CAR-T cells [64]. Then, in a follow-up study, they co-expressed CXCR2 expression using a Furin-V5-P2A sequence [66] with their CD70-CAR in order to guide the modified T-cells towards IL-8 producing glioma cells [67]. Since IL-8 expression is known to promote tumor resistance and invasion [68] besides being one of the predominantly expressed chemokines in GBM [69], the authors rationale is that it would help T-cells hone in on, and infiltrate tumors more efficiently. Using this approach, the authors needed only 2 × 106 CARs to achieve remission in vivo in preclinical modeling after 150 days [67]. Interestingly, since the authors were able to treat not just U87 murine xenographs but also pancreatic and ovarian-type cell lines, it seems co-expression of CXCR2 could improve treatment of solid tumors overall. However, it is unknown whether CD70+ glioma cells can undergo CD70 loss under long-term CAR-T pressure, so reproducibility and clinical feasibility of these studies needs further investigation. However, CD70 CAR-T clinical trials involving solid tumors such as pancreatic, renal and breast cancer were recently suspended after enrolling only two patients (NCT02830724). CD70 has also been combined with targeting B7-H3 (CD276) with a TanCAR strategy (Fig. 2G) that showed enhanced anti-tumor functionality against gliomas and many other solid tumors [70].
Another helper gene recently used has been IL-15 [71]. IL-15 is a cytokine that has been demonstrated to enhance survival of T-cells. However, use of this cytokine in vivo is complex, since it needs to be expressed in trans with its receptor in order to extend IL-15 half-life and thus function effectively in vivo [72].
In order to address the glioma antigenic escape problem, a daring Trivalent CAR strategy was developed by Bielamowicz et al. [73], using a single lentiviral construct expressing three individual CARs directed against IL-13Rα2, HER and EphA2. The authors reported almost 100% success using two different patient-derived cell line xenographs in five mice during a 60-day period. However, several limitations appear to surround this approach. Requirements for producing high enough viral titers for such long constructs in a clinical setting remains to be seen [74] and the inter- and intra-patient GBM variability appears to be wider than what the authors describe a Universal CAR or UCAR can achieve [75]. However, if such strategy can be proven effective at containing the growth of GBM, surely the manufacturing of such large constructs would find a way to translate clinically.
Owing to the versatility that transmembrane proteins confer, several other types and combinations of CARs have been developed and are listed in Supplementary Table 1, however, their viability and utilization against GBM remains to be seen.
However, solving the challenge of antigenic loss may not be the only obstacle in order to achieve long-term remission of gliomas. The TME and the alleged detrimental effects it exerts on T-cells such as T-cell exhaustion and poor tumor trafficking, are major obstacles that need to be account for in any new strategies [76,77,78]. As Wang et al., suggest in their studies, some CLTX-CARs that failed to achieve durable remission in murine models may be due to T-cell exhaustion [58].
Combined CAR-T use with other therapies
Recently, CAR-T therapeutics aimed at GBM have been used in combination with existing anti-tumor agents, such as temozolamide [79] or immune checkpoint inhibitors (ICI) PD1/PD-L1 against refractory diffuse large B-cell lymphoma [80]. However, the clinical efficacy of ICI against GBM may be moot with open questions regarding their usefulness against GBM [81] after usage of Nivolumab –a PD1/PD-L1 inhibitor- failed to show any clinical benefit to patients with recurrent GBM [82]. Owing to the plastic nature of CAR-Ts, work has also been published using CAR-Ts of many carriers of oncolytic viruses [83] or oncolytic viruses targeting IL-13Rα2 [84]. Additionally, modified CAR-NK cells have been used to eliminate MDSC before CAR-T administration in murine models [85] as well as co-expressing CXCR4 and EGFRvIII in NK cells to improve immunotherapy against CXCL12-secreteing GBM [86].
Special details of CAR-T modules
Since the development of additional co-stimulatory modules to CAR-Ts in order to improve their in vivo potency and lasting effect (Fig. 1B), several additional modifications to certain modules have been reported or are de facto known and used in the CAR-T community. One of these is a key modification on the CD28 co-stimulatory domain where a single amino acid change involving an asparagine to a phenylalanine (N193F) improves long-term survival and exhaustion of T-cells [87]. Interesting too is the recently reported non-canonical CD3e motif -RKxQRxxY- that has also been described to provide improved function [88]. Improvements on the stem/hinge section of CAR-Ts have also been reported. For example, a mutant with two amino acid changes -L235E and N297Q- thus coined EQ on the IgG4 domain is reported to improve T cell persistence and anti-tumor efficacy while avoiding Fc receptor binding in CD19-CAR-Ts [89]. One should also note that several modifications of co-stimulatory domains or hinges are not reported in the literature, but are described in filed patents, such as the change of two leucines to glycines in the CD28 intracellular domain (L186G and L187G) designated LLmGG [90] and recent patent WO2017066481A1. Schönfeld et al., have also filed a patent reporting that a serine to cysteine change in the CD8 hinge regions (S164C) improves functionality and expression of CAR-T [91] and patent EP3115373A1. Finally, a double mutant CD3ζ domain with mutation Q65K and deletion Q101 has been reported from Mackall’s group, albeit no functional difference from the wt sequence was noted (Dr. Benjamin Salzer, personal communication and present in patent WO/2020/118076).
Untested approaches in gliomas
Several novel therapeutic approaches involving T cell engineering have been developed, but none have been tested against gliomas or GBM. For example, CD40 is known to be upregulated in 40% of all GBM [92] and constitutive expression of its ligand CD40L on CAR-T has been shown to enhance IL-12 secretion, extend survival of T cells, and increase cytotoxicity against tumors [93]. Other groups have engineered heat-controlled CAR-T that display enhanced intra-tumoral activity [94].
Last year a novel monomorphic MHC class I-related protein, MR1, was described to be expressed in virtually all types of cancers but not healthy cells [95]. Authors showed this particular surface MR-1 molecule could be targeted by a TCR that recognizes vitamin B-related metabolites present in malignant cells but not healthy ones. Although MR1-restricted mRNA has been detected in glioma cell lines such as U-373 [96], its expression in another commonly used glioma cell line U87 or patient-derived GSC is unknown. Another promising approach has been the development of Supra-CARs, i.e., inter-changeable CAR-Ts that can be modulated by soluble adaptors [97]. Protein logic, i.e., combinatorial antigens- targeting HER2+ cells, have also been developed to achieve precise discriminatory effector functions targeting antigens present in tumor, but not healthy, cells [98].
Another promising approach to developing CARs targeting difficult tumor antigens was used by Rafiq et al., in 2017, creating a CAR with the ability to target intracellular Tumor-Associated Antigens (TAA) such as Wilms tumor 1 (WT1), known to be up-regulated in gliomas [99]. Using this approach, the CAR is guided by an antibody portion that recognizes a surface MHC molecule loaded with peptide fragments of WT1 [99], which otherwise cannot be accessible to T-cells as it is ordinarily intracellular (Fig. 2I). WT1 is an oncogenic, zinc-finger transcription factor involved in differentiation, proliferation and apoptosis among other functions [100]. WT1 is known to be over-expressed in many malignancies, including GBM [101], offering broad tumor therapeutic potential. Expanding on this strategy to target MHC-presented peptides from key intracellular oncoproteins, Yamarkovich M. et al. have recently created a peptide-centric CAR (PC-CAR) that can target neuroblastoma dependency gene and intracellular transcriptional regulator PHOX2B and induce remission in a murine model of neuroblastoma [102].
In order to increase local potency of CAR-Ts, Fourth Generation CAR-Ts—“T cells redirected for antigen-unrestricted cytokine-initiated killing” (TRUCKS) have been tested in murine models [103]. As shown in Fig. 2, CAR-T along with NFAT-controlled cytokines IL-12 or IL-18 can increase potency against tumors only after the primary CAR-T is engaged and T cells activate [104, 105]. However, although IL-12 TRUCKs showed efficacy in murine models, they provided little therapeutic effects in a Phase 1 clinical trial for metastatic seminal vesicle cancer, although no adverse side effects were reported [106]. An additional detrimental effect of this approach is that NFAT-controlled cytokines might activate independently of the primary CAR-T engagement if the T-cell is activated by alternative mechanisms. This hindrance may be solved using Syn-Notch receptors as introduced above. Other cytokines that have been constitutively co-expressed along with CAR-Ts include IL-7 and CLL19, which promote survival via decreased T-cell exhaustion- and tumor infiltration respectively [107]. More recently, CAR-Ts constitutively expressing IL-9 have also been described as having a superior tumor-fighting phenotype against liquid and solid tumors, displaying central memory phenotype, decreased exhaustion markers and robust proliferative capacity [108].
Delivering the gene cargo into CAR-Ts has also been a logistical problem, and its difficulty in clinical translation of bench developments have been mentioned. Buchholz et al., have developed a lentivirus capable of creating CD19-CAR-T cells in vivo without the requirement for cell expansion [109]. Utilization of this approach versus solid tumors remains to be explored.
Many CAR-T improvements have been developed using combinatorial antigens [110] or AvidCARs [57] to improve selective tumor eradication. It has also been demonstrated that it is possible to restrict CAR-T expression to anoxic conditions similar to the hypoxic environment, a hallmark of GBM, by C-terminally attaching a HIF motif into the CAR that grants it oxygen-sensing features [111]. Scientists have also developed adaptor molecules that can recognize specific antigens while engaging the TCR machinery, thus minimizing tonic signaling [112]. One such approach is called T cell antigen coupler or TAC and has demonstrated efficacy in murine models against HER+ cells [113], another known surface antigen over-expressed in GBM. Other approaches have been even more daring such as directly combining antibody-based targeting domains with the TCR-like activation machinery itself [114]. In this approach, Yue Liu et al., showed that synthetic TCR and antigen receptor or STAR receptors, offer greater functionality compared to conventional CAR-Ts.
However, besides the clinical feasibility of these groundbreaking studies, a major obstacle that plagues most CAR-T or T cell engineering studies is the virtual impossibility to exactly reproduce CAR-T or similar expression experiments. Most often published reports do not include the exact protein sequence of the constructs used, or they do so in a convoluted manner that makes reproducibility very difficult and time consuming. Obtaining the original plasmids used by the authors from which the constructs are expressed is even more difficult—even though it is encouraged in published work. Sometimes, the only recourse is to reverse engineer the constructs or obtain the sequences from other source documentation such as patent applications. This adds another level of ambiguity, where one can never be completely sure whether the construct is the actual one used or not in the published work. This observation is compounded by the inflexible nature of proteins, where a single amino acid change can be very influential. In order to help readers regarding this issue, we have compiled a list in Fig. 3 and accompanying Supplementary Table 1 describing the constructs used in each study for which we were able to find the precise amino acid sequences. Hopefully, this will aid researchers in dissemination of vital information necessary to propel research forward and advance potential treatments for this devastating disease.
CAR safety, efficacy and caveats in GBM
Safety concerns are a major issue when using CAR-Ts [115]. Targeting tumor cells while ensuring normal tissue remains undamaged needs to be extensively assessed using in vitro and preclinical in vivo testing prior to testing their clinical efficacy in human patients. One of the main toxicities associated with CAR-T therapy is induction of cytokine release syndrome (CRS) as well as poorly understood neurological sequelae. Fortunately CRS can be managed with the use of anti-IL-6 [116] and corticosteroids, but it can reduce the benefits of CAR-T as a result. For example, it was recently mentioned in an online conference-CelliCon Valley 2021- by Dr. Carl June, that a subgroup of neurons express very low levels of CD19 which might make them a target for the CD19-CAR-T approved clinically. However, the physiodynamics underlying CRS are extensive and could well fill a separate review [25, 115, 117,118,119].
Since existing patient T-cells need to be engineered in order to express the CAR-T, isolation, extraction and expansion of functional peripheral T-cells in a Glioma patient is required. This can be a challenge itself as GBM patients are heavily immunosuppressed and their T-cells are documented to have a wide range of T-cell dysfunction including, senescence, anergy, tolerance and exhaustion [120]. On top of this, the GBM median survival of 15–17 months after diagnosis can limit the timing of isolation, expansion and re-infusion of modified T-cells back into patients, since production alone of CAR-T can generally take approximately 2–4 weeks or more depending on the patient’s clinical status and chemotherapy usage [121, 122]. In order to shorten this step, off-the-shelf CAR-T are under investigation but their clinical usefulness has not been assessed yet [123].
Conclusions
GBM tumors are among the most devastating types of cancers to afflict humans. As such, it is highly unlikely any single conventional or unconventional treatment that worked against other tumor types will decisively reverse unstoppable growth or adaptability and escape of GBM tumors. In this regard, CAR-T therapies have proven to be no exception despite their decisive role in reversing the prognosis of patients with liquid tumor malignancies. However, the advantage CAR-Ts offer versus other therapies is the unlimited potential for improvement via refined T cell engineering. Taking advantage of the flexible nature of DNA and delivery systems, it is possible that effective CAR-T combinations versus solid tumors such as GBM will be developed in the foreseeable future. In order to achieve this goal, a holistic approach should be undertaken. One in which several immunologic obstacles are addressed simultaneously including the challenge of antigenic loss, protecting CAR-T from the TME, T cell exhaustion, improving tumor homing, and making T cells resistant to immunosuppression. There is also room for improvement on safety profiles in order to reduce cytokine release syndrome, neurotoxicity and long-term control through iCasp9 [124], HSV-TK [125] gene or a suicide epitope [126]. However, the future is very promising, with novel and very innovative improvements being reported almost weekly. As such, the possibilities of CAR-T and T cell engineering has never looked more promising in designing potential therapeutics for treatment of solid tumors and specifically GBM, the meanest of them all.
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
Alexander BM, Cloughesy TF (2017) Adult glioblastoma. J Clin Oncol 35(21):2402–2409
Kipnis J (2016) Multifaceted interactions between adaptive immunity and the central nervous system. Science 353(6301):766–771
Desjardins A, Gromeier M, Herndon JE 2nd et al (2018) Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med 379(2):150–161
Quail DF, Joyce JA (2017) The microenvironmental landscape of brain tumors. Cancer Cell 31(3):326–341
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10(12):1538–1543
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C et al (2008) Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res 14(16):5166–5172
Kmiecik J, Poli A, Brons NH et al (2013) Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol 264(1–2):71–83
Wang Q, Hu B, Hu X et al (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(1):42–56
Wei J, Wu A, Kong LY et al (2011) Hypoxia potentiates glioma-mediated immunosuppression. PLoS One 6(1):e16195
Wainwright DA, Dey M, Chang A, Lesniak MS (2013) Targeting tregs in malignant brain cancer: overcoming IDO. Front Immunol 4:116
Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE (2019) CAR T cells for brain tumors: lessons learned and road ahead. Immunol Rev 290(1):60–84
Lim WA, June CH (2017) The principles of engineering immune cells to treat cancer. Cell 168(4):724–740
Kuwana Y, Asakura Y, Utsunomiya N et al (1987) Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun 149(3):960–968
Gross G, Gorochov G, Waks T, Eshhar Z (1989) Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transpl Proc 21(1 Pt 1):127–130
Irving BA, Weiss A (1991) The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 64(5):891–901
AC Tran, D Zhang, R Byrn, MR Roberts (1995) Chimeric zeta-receptors direct human natural killer (NK) effector function to permit killing of NK-resistant tumor cells and HIV-infected T lymphocytes. J Immunol 155(2):1000–1009
Finney HM, Lawson AD, Bebbington CR, Weir AN (1998) Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol 161(6):2791–2797
Dudley ME, Wunderlich JR, Robbins PF et al (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298(5594):850–854
Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348(6230):62–68
Kalos M, Levine BL, Porter DL et al (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3002842
Kochenderfer JN, Wilson WH, Janik JE et al (2010) Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116(20):4099–4102
Rosenbaum L (2017) Tragedy, perseverance, and chance—the story of CAR-T therapy. N Engl J Med 377(14):1313–1315
Gill S, June CH (2015) Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263(1):68–89
Maude SL, Laetsch TW, Buechner J et al (2018) Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 378(5):439–448
Neelapu SS, Locke FL, Bartlett NL et al (2017) Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med 377(26):2531–2544
Hong JJ, Rosenberg SA, Dudley ME et al (2010) Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res 16(19):4892–4898
Berahovich R, Xu S, Zhou H et al (2017) FLAG-tagged CD19-specific CAR-T cells eliminate CD19-bearing solid tumor cells in vitro and in vivo. Front Biosci 22:1644–1654
Guedan S, Calderon H, Posey AD Jr, Maus MV (2019) Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev 12:145–156
Hudecek M, Sommermeyer D, Kosasih PL et al (2015) The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3(2):125–135
Labanieh L, Majzner RG, Mackall CL (2018) Programming CAR-T cells to kill cancer. Nat Biomed Eng 2(6):377–391
Xie YJ, Dougan M, Jailkhani N et al (2019) Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci USA 116(16):7624–7631
Yang EY, Shah K (2020) Nanobodies: next generation of cancer diagnostics and therapeutics. Front Oncol 10:1182
Barsoum IB, Smallwood CA, Siemens DR, Graham CH (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 74(3):665–674
Berger TR, Maus MV (2021) Mechanisms of response and resistance to CAR T cell therapies. Curr Opin Immunol 69:56–64
Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ (2017) Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep 7(1):10541
Howie D, Waldmann H, Cobbold S (2014) Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol 5:409
Yamamoto TN, Lee PH, Vodnala SK et al (2019) T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest 129(4):1551–1565
Liu X, Ranganathan R, Jiang S et al (2016) A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res 76(6):1578–1590
Chen N, Morello A, Tano Z, Adusumilli PS (2017) CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology 6(2):e1273302
Caruana I, Savoldo B, Hoyos V et al (2015) Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 21(5):524–529
Sampson JH, Choi BD, Sanchez-Perez L et al (2014) EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res 20(4):972–984
Morgan RA, Johnson LA, Davis JL et al (2012) Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther 23(10):1043–1053
O’Rourke DM, Nasrallah MP, Desai A et al (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaa0984
Ahmed N, Salsman VS, Kew Y et al (2010) HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res 16(2):474–485
Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G (2018) Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev 9:70–80
Brown CE, Alizadeh D, Starr R et al (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 375(26):2561–2569
Pituch KC, Miska J, Krenciute G et al (2018) Adoptive transfer of IL13Ralpha2-specific chimeric antigen receptor T cells creates a pro-inflammatory environment in glioblastoma. Mol Ther 26(4):986–995
Brown CE, Starr R, Aguilar B et al (2012) Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res 18(8):2199–2209
Baeuerle PA, Ding J, Patel E et al (2019) Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat Commun 10(1):2087
Wikstrand CJ, Hale LP, Batra SK et al (1995) Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 55(14):3140–3148
Abbott RC, Verdon DJ, Gracey FM et al (2021) Novel high-affinity EGFRvIII-specific chimeric antigen receptor T cells effectively eliminate human glioblastoma. Clin Transl Immunol 10(5):e1283
Chuntova P, Hou Y, Naka R et al (2021) Novel EGFRvIII-CAR transgenic mice for rigorous preclinical studies in syngeneic mice. Neuro-Oncology. https://doi.org/10.1093/neuonc/noab182
Choi BD, Yu X, Castano AP et al (2019) CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 37(9):1049–1058
Roybal KT, Williams JZ, Morsut L et al (2016) Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167(2):419–432
Hernandez-Lopez RA, Yu W, Cabral KA et al (2021) T cell circuits that sense antigen density with an ultrasensitive threshold. Science 371(6534):1166–1171
Choe JH, Watchmaker PB, Simic MS et al (2021) SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abe7378
Salzer B, Schueller CM, Zajc CU et al (2020) Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat Commun 11(1):4166
Wang D, Starr R, Chang WC et al (2020) Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaw2672
Cohen-Inbar O, Zaaroor M (2016) Glioblastoma multiforme targeted therapy: the chlorotoxin story. J Clin Neurosci 33:52–58
Ramachandran RK, Sorensen MD, Aaberg-Jessen C, Hermansen SK, Kristensen BW (2017) Expression and prognostic impact of matrix metalloproteinase-2 (MMP-2) in astrocytomas. PLoS One 12(2):e0172234
Kasai T, Nakamura K, Vaidyanath A et al (2012) Chlorotoxin fused to IgG-Fc inhibits glioblastoma cell motility via receptor-mediated endocytosis. J Drug Deliv 2012:975763
Land CA, Musich PR, Haydar D, Krenciute G, Xie Q (2020) Chimeric antigen receptor T-cell therapy in glioblastoma: charging the T cells to fight. J Transl Med 18(1):428
Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S (2021) CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci 15:662064
Jin L, Ge H, Long Y et al (2018) CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol 20(1):55–65
Wischhusen J, Jung G, Radovanovic I et al (2002) Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res 62(9):2592–2599
Yang S, Cohen CJ, Peng PD et al (2008) Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther 15(21):1411–1423
Jin L, Tao H, Karachi A et al (2019) CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun 10(1):4016
David JM, Dominguez C, Hamilton DH, Palena C (2016) The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines 4(3):22
Sharma I, Singh A, Sharma K, Saxena S (2017) Gene expression profiling of chemokines and their receptors in low and high grade astrocytoma. Asian Pac J Cancer Prev 18(5):1307–1313
Yang M, Tang X, Zhang Z et al (2020) Tandem CAR-T cells targeting CD70 and B7–H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 10(17):7622–7634
Krenciute G, Prinzing BL, Yi Z et al (2017) Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol Res 5(7):571–581
Han KP, Zhu X, Liu B et al (2011) IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 56(3):804–810
Bielamowicz K, Fousek K, Byrd TT et al (2018) Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol 20(4):506–518
Ponterio E, De Maria R, Haas TL (2020) Identification of targets to redirect CAR T cells in glioblastoma and colorectal cancer: an arduous venture. Front Immunol 11:565631
Caruso H, Heimberger AB (2018) Comment on “trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma.” Neuro Oncol 20(7):1003–1004
Henke E, Nandigama R, Ergun S (2019) Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci 6:160
Liu G, Rui W, Zhao X, Lin X (2021) Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol 18(5):1085–1095
Anderson KG, Stromnes IM, Greenberg PD (2017) Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell 31(3):311–325
Suryadevara CM, Desai R, Abel ML et al (2018) Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology 7(6):e1434464
Chong EA, Melenhorst JJ, Lacey SF et al (2017) PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129(8):1039–1041
Khasraw M, Reardon DA, Weller M, Sampson JH (2020) PD-1 Inhibitors: do they have a future in the treatment of glioblastoma? Clin Cancer Res 26(20):5287–5296
Filley AC, Henriquez M, Dey M (2017) Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 8(53):91779–91794
VanSeggelen H, Tantalo DG, Afsahi A, Hammill JA, Bramson JL (2015) Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol Ther Oncolytics 2:15014
Kim JW, Young JS, Solomaha E, Kanojia D, Lesniak MS, Balyasnikova IV (2015) A novel single-chain antibody redirects adenovirus to IL13Ralpha2-expressing brain tumors. Sci Rep 5:18133
Parihar R, Rivas C, Huynh M et al (2019) NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res 7(3):363–375
Muller N, Michen S, Tietze S et al (2015) Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother 38(5):197–210
Guedan S, Madar A, Casado-Medrano V et al (2020) Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest 130(6):3087–3097
Hartl FA, Beck-Garcia E, Woessner NM et al (2020) Noncanonical binding of Lck to CD3epsilon promotes TCR signaling and CAR function. Nat Immunol 21(8):902–913
Jonnalagadda M, Mardiros A, Urak R et al (2015) Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther 23(4):757–768
Barish M, Brown CB, Forman SJ (2016) Chimeric antigen receptors containing a chlorotoxin domain 2016, patent: WO 2017/066481 Al
Kurt Schönfeld CS, Winfried W (2017) Chimeric antigen receptors with an optimized hinge region, patent: EP3115373A1
Seimiya M, O-Wang J, Bahar R et al (2003) Stage-specific expression of Clast6/E3/LAPTM5 during B cell differentiation: elevated expression in human B lymphomas. Int J Oncol 22(2):301–304
Curran KJ, Seinstra BA, Nikhamin Y et al (2015) Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther 23(4):769–778
Miller IC, Zamat A, Sun LK et al (2021) Enhanced intratumoural activity of CAR T cells engineered to produce immunomodulators under photothermal control. Nat Biomed Eng 5:1348–1359
Crowther MD, Dolton G, Legut M et al (2020) Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol 21(2):178–185
Riegert P, Wanner V, Bahram S (1998) Genomics, isoforms, expression, and phylogeny of the MHC class I-related MR1 gene. J Immunol 161(8):4066–4077
Cho JH, Collins JJ, Wong WW (2018) Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 73(6):1426–1438
Lajoie MJ, Boyken SE, Salter AI et al (2020) Designed protein logic to target cells with precise combinations of surface antigens. Science 369(6511):1637–1643
Rafiq S, Purdon TJ, Daniyan AF et al (2017) Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia 31(8):1788–1797
Hohenstein P, Hastie ND (2006) The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet 15:R196–R201
Sugiyama H (2010) WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol 40(5):377–387
Yarmarkovich M, Marshall QF, Warrington JM et al (2021) Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature. https://doi.org/10.1038/s41586-021-04061-6
Chmielewski M, Abken H (2015) TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther 15(8):1145–1154
Chmielewski M, Abken H (2017) CAR T cells releasing IL-18 convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep 21(11):3205–3219
Hu B, Ren J, Luo Y et al (2017) Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep 20(13):3025–3033
You F, Jiang L, Zhang B et al (2016) Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci 59(4):386–397
Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K (2018) IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol 36(4):346–351
Liu L, Bi E, Ma X et al (2020) Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat Commun 11(1):5902
Frank AM, Braun AH, Scheib L et al (2020) Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv 4(22):5702–5715
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M (2013) Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31(1):71–75
Juillerat A, Marechal A, Filhol JM et al (2017) An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep 7:39833
Long AH, Haso WM, Shern JF et al (2015) 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21(6):581–590
Helsen CW, Hammill JA, Lau VWC et al (2018) The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun 9(1):3049
Liu Y, Liu G, Wang J et al (2021) Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abb5191
Greenbaum U, Kebriaei P, Srour SA et al (2021) Chimeric antigen receptor T-cell therapy toxicities. Br J Clin Pharmacol 87(6):2414–2424
Lee DW, Gardner R, Porter DL et al (2014) Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124(2):188–195
Wei J, Liu Y, Wang C et al (2020) The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct Target Ther 5(1):134
Abramson JS, Gordon LI, Palomba ML, Lunning MA, Arnason JE, Foreroorres A, Wang M, Maloney DG, Sehgal A, Andreadis C, Purev E, Solomon SR, Ghosh N, Albertson TM, Xie B, Garcia J, Siddiqi T (2018) Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol 36:7505–7750
Neelapu SS, Tummala S, Kebriaei P et al (2018) Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat Rev Clin Oncol 15(1):47–62
Woroniecka K, Fecci PE (2018) T-cell exhaustion in glioblastoma. Oncotarget 9(82):35287–35288
Fesnak AD, June CH, Levine BL (2016) Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 16(9):566–581
Levine BL, Miskin J, Wonnacott K, Keir C (2017) Global Manufacturing of CAR T Cell Therapy. Mol Ther Methods Clin Dev 4:92–101
Martinez Bedoya D, Dutoit V, Migliorini D (2021) Allogeneic CAR T cells: an alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front Immunol 12:642
Diaconu I, Ballard B, Zhang M et al (2017) Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther 25(3):580–592
Bonini C, Ferrari G, Verzeletti S et al (1997) HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 276(5319):1719–1724
Paszkiewicz PJ, Frassle SP, Srivastava S et al (2016) Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest 126(11):4262–4272
Funding
DCS, AK-F and AES were partially supported by the Peter D. Cristal Chair, the Kimble Family Foundation, the Gerry Kaufman Jr. Foundation, and the Ferry Family Foundation.
Author information
Authors and Affiliations
Contributions
All authors contributed to the analysis and interpretation of reviewed data, conception and design of this manuscript. The first draft of the manuscript was written by DCS and AK-F and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. Figures 1–3 were illustrated by DCS.
Corresponding author
Ethics declarations
Conflict of interest
Author DCS declares that he has no conflict of interest. Author AK-F declares that she has no conflict of interest. Author TSM declares that he has no conflict of interest. Author AES declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Soler, D.C., Kerstetter-Fogle, A., McCormick, T.S. et al. Using chimeric antigen receptor T-cell therapy to fight glioblastoma multiforme: past, present and future developments. J Neurooncol 156, 81–96 (2022). https://doi.org/10.1007/s11060-021-03902-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-021-03902-8