
Abstract
Introduction
Medulloblastoma is a malignant embryonal tumor of the cerebellum that occurs predominantly in children. To find germline genetic variants associated with medulloblastoma risk, we conducted a genome-wide association study (GWAS) including 244 medulloblastoma cases and 247 control subjects from Sweden and Denmark.
Methods
Genotyping was performed using Illumina BeadChips, and untyped variants were imputed using IMPUTE2.
Results
Fifty-nine variants in 11 loci were associated with increased medulloblastoma risk (p < 1 × 10–5), but none were statistically significant after adjusting for multiple testing (p < 5 × 10–8). Thirteen of these variants were genotyped, whereas 46 were imputed. Genotyped variants were further investigated in a validation study comprising 249 medulloblastoma cases and 629 control subjects. In the validation study, rs78021424 (18p11.23, PTPRM) was associated with medulloblastoma risk with OR in the same direction as in the discovery cohort (ORT = 1.59, pvalidation = 0.02). We also selected seven medulloblastoma predisposition genes for investigation using a candidate gene approach: APC, BRCA2, PALB2, PTCH1, SUFU, TP53, and GPR161. The strongest evidence for association was found for rs201458864 (PALB2, ORT = 3.76, p = 3.2 × 10–4) and rs79036813 (PTCH1, ORA = 0.42, p = 2.6 × 10–3).
Conclusion
The results of this study, including a novel potential medulloblastoma risk loci at 18p11.23, are suggestive but need further validation in independent cohorts.
Similar content being viewed by others


Introduction
Medulloblastoma is the most common embryonal central nervous system malignancy in children. It is well known that a fraction of all cases is caused by germline mutations in TP53 (underlying Li-Fraumeni syndrome), APC (underlying Turcot syndrome), or PTCH1/PTCH2/SUFU (underlying basal cell nevus/Gorlin, syndrome) [1, 2]. A recent study including 1022 medulloblastoma patients found that 6% of all cases had a germline mutation in TP53, APC, PTCH1, SUFU, or in two additional genes with presumed tumor suppressor function: BRCA2 or PALB2 [3]. Another recent study reported a novel medulloblastoma predisposition gene in GPR161 [4]. The somatic genetic changes that occur in sporadic medulloblastoma tumors are also well-described, including alterations in CCND2, CTNNB1, DDX3X, GLI2, SMARCA4, MYC, MYCN, PTCH1, TP53, and KMT2D [5]. Although we know much about genetic aberrations in medulloblastoma tumors and the genetic syndromes that predispose to the disease, little is known about how common germline genetic variants (i.e. single nucleotide polymorphisms, SNPs) contribute to medulloblastoma susceptibility.
Prognosis for medulloblastoma patients is poor, with a ten-year survival rate of 63% [6]. As a consequence of the disease and intensive treatment, the children who survive have an increased risk of long-term neurocognitive dysfunction and secondary malignancies [7]. To improve treatment and prevention strategies for this devastating disease, a better understanding of medulloblastoma etiology is needed. We have conducted a genome-wide association study (GWAS) with the aim to identify genetic variants that are associated with medulloblastoma development in children and young adults. Identifying genetic variants that predispose to medulloblastoma development may provide new insights into the genetic pathways that contribute to the development of the disease and potential new targets for therapy.
Results
To find germline genetic variants associated with medulloblastoma risk, we conducted a genome-wide scan of 244 medulloblastoma cases and 247 control subjects from Sweden and Denmark that fulfilled the inclusion criteria (Figure S1; Table S1). Tests of association with medulloblastoma risk were performed for 1,288,472 SNPs that passed quality control. The Q–Q plot and inflation factor ʎ indicated no significant effect on the results by population stratification (Figure S2). Thirteen genetic variants in six genomic loci were associated with increased medulloblastoma risk (p < 1 × 10–5), but none were statistically significant when applying a conservative p-value threshold to adjust for multiple testing (p < 5 × 10–8; Table 1). We were able to analyze 12 of these variants in a validation cohort consisting of 249 cases and 629 controls (Table S1). In the validation cohort, one genetic variant, rs78021424 (18p11.23, PTPRM), was associated with medulloblastoma risk with an OR in the same direction as in the discovery cohort (Table 1).
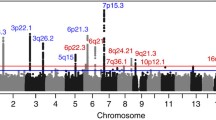
In a search for SNPs with even stronger associations at the 18p11.23 locus, and to find additional interesting regions, we imputed SNPs in the discovery dataset and performed association analyses of an additional 7,916,089 SNPs (Fig. 1). Forty-six imputed SNPs in eight genomic loci were associated with medulloblastoma risk (p < 1 × 10–5; Table S2). These associations were not, however, statistically significant after adjusting for multiple testing. The SNP with the strongest association in the 18p11.23 (PTPRM) locus was rs185966860 (ORper A allele = 4.01, 95% CI 2.43–6.63, p = 5.97 × 10–8).

Manhattan plot. P-values for the association between 9,204,561 genetic variants and medulloblastoma risk. Both genotyped and imputed SNPs are included. Solid line indicates genome-wide statistical significance (p = 5 × 10–8). Dashed line indicates p = 1 × 10–5
In addition to genome-wide analyses, we were specifically interested in seven genes, namely: APC, BRCA2, PALB2, PTCH1, SUFU, TP53, and GPR161 [3, 4]. Within these seven candidate genes, the strongest evidence for association was found for rs201458864, located within PALB2 (ORper T allele = 3.76, 95% CI 1.83–7.75, p = 3.2 × 10–4) and rs79036813, located within PTCH1 (ORper A allele = 0.42, 95% CI 0.24–0.74, p = 2.6 × 10–3) (Figure S3).
Discussion
In this first GWAS of medulloblastoma, we found a potential medulloblastoma risk locus at 18p11.23. Medulloblastoma is a rare disease, which makes it challenging to collect enough samples for adequate statistical power, especially for a GWAS. Compared to other epidemiologic studies of medulloblastoma, the number of cases included in this study is large. However, in relation to the number of statistical tests performed, the number of cases is still small, and our study was not powered to detect associations with a small effect size. Although GWAS of adult cancers usually report associations with small effect sizes, studies of early onset malignancies have reported associations with larger effects [8]. Analogous with this, for the majority of associations with p < 1 × 10–5 in this study, effect sizes were large, and carriers of the risk allele had a more than two-fold increased risk. Our findings were not statistically significant when using the p value threshold p < 5 × 10–8 to correct for multiple comparisons. Although a stringent p-value threshold is required in GWAS to reduce the presence of false positive findings, strict Bonferroni correction may be considered overly conservative due to linkage disequilibrium between many genetic variants. Twelve variants with evidence for associations in the initial analyses were investigated in an additional cohort. One of these SNPs, located in 18p11.23 (PTPRM) showed suggestive evidence for an association with medulloblastoma risk also in the validation cohort. The PTPRM gene product is a receptor-type protein tyrosine phosphatase that mediates cell–cell adhesion. Altered expression, mutations, or aberrant methylation of PTPRM have been described in different malignancies, including glioblastoma [9]. The role of PTPRM in medulloblastoma is, to our knowledge, unknown, but it is interesting to note that the PTPRM protein has been shown to interact with beta-catenin [10]. Beta-catenin is a central part of the Wnt signaling pathway and is encoded by the gene CTNNB1, which is frequently mutated in WNT medulloblastoma [5]. However, only about 10% of all medulloblastoma tumors belong to the WNT subgroup [5], and this subgroup is therefore represented by few patients in the study cohort. Investigation of imputed variants across the genome indicated the presence of additional variants associated with medulloblastoma risk in the 18p11.23 locus and variants in five additional genetic regions that remain to be validated in an independent cohort.
Germline mutations in APC, BRCA2, PALB2, PTCH1, SUFU, and TP53 occur in up to 6% of all medulloblastoma cases [3]. Another potential medulloblastoma predisposing mutation has been reported in the gene GPR161 [4]. In candidate gene analysis restricted to these seven genes, we observed associations between genetic variants in PALB2 and PTCH1 and medulloblastoma risk. Pathogenic genetic variants in PALB2 have been associated with increased risk of medulloblastoma as well as breast cancer [3, 11]. Genetic testing of PALB2 has been suggested for clinical testing in breast cancer families and in specific subgroups of medulloblastoma based on clinical and molecular tumor characteristics [3, 12]. Germline mutations in PTCH1 give rise to basal cell nevus (Gorlin) syndrome, which comes with an increased risk of different malignancies, including basal cell carcinoma and medulloblastoma. In the present study, we investigated common germline genetic variants (minor allele frequency > 1%), and we could not assess the rare germline mutations in PALB2 and PTCH1 reported by Waszak et al. [3].
Medulloblastoma tumors comprise four or more molecular subgroups [5]. The cases in our discovery sets were diagnosed during a period when these molecular subgroups of medulloblastoma were not established. In the present study, tissue samples are not possible to obtain, and molecular subgroups cannot be taken into consideration in the analyses, which is a limitation of the study. In GWAS of glioma, which is also a heterogeneous group of brain tumors, we and others have shown that many established risk loci are specific for certain subtypes [13, 14]. However, even in early GWAS of glioma, in which all glioma were included, we found several genetic variants that were associated with an increased risk of all glioma, irrespective of molecular subtype [15]. Another potential limitation of the study is the inclusion of study subjects from six different countries in the validation phase, whereas patients and control subjects in the discovery phase were born in either Sweden or Denmark.
An advantage of the study is that, although cases were retrospectively identified, their blood samples were collected prior to disease diagnosis. With this study design, we avoided survival bias, which can be a problem in a case–control study of an aggressive disease, where mortal cases would be underrepresented. On the contrary, we may have an underrepresentation of less aggressive medulloblastoma, since a subset of surviving cases chose not to participate in the study.
In summary, we have identified 11 loci that may be associated with medulloblastoma development in children and young adults, including the 18p11.23 (PTPRM) loci that was validated in a separate cohort. None of the observed associations were, however, statistically significant after conservative correction for multiple testing, and to know the relevance of these loci in medulloblastoma etiology, replication in independent cohorts is needed. If these associations proves robust in independent validations, it is a step towards enhanced understanding of medulloblastoma etiology, which in turn may enable development of improved treatment and prevention strategies. For sufficient power of future studies of genetic variants in medulloblastoma, broad international collaborations are required.
Materials and methods
Study subjects
Medulloblastoma cases diagnosed between 1975 and 2008, under the age of 25, were identified from the national cancer registries in Sweden (n = 136) and Denmark (n = 128) [16] (Table S1). Dried blood spot samples were collected from the Swedish Phenylketonuria Screening Registry [17] and the Danish Newborn Screening Biobank, which are national biobanks containing dried blood spot samples from newborns. For each medulloblastoma case, one control subject was identified among samples that were physically located close to the case sample in the biobanks. Control subjects were matched by date of birth (Swedish and Danish controls) and sex (Danish controls only).
In Sweden, the study was approved by the Data Inspection Board and the Regional Ethical Review Board. All living Swedish subjects provided informed consent. The Regional Ethical Review Board approved the use of samples from deceased Swedish cases without informed consent from close relatives. In Denmark, the study was approved by the Research Ethics committee of the Capital Region (Copenhagen), the Danish Data Protection Agency, and by the Danish Newborn Screening Biobank Steering Committee. According to Danish law, the regional Ethics Committee can grant exemption from obtaining informed consent for research projects on biobank samples under certain circumstances [18]. For this study, such an exemption was granted.
The validation study included 249 cases and 629 controls originally recruited to four different studies: (1) Studies at Children’s Hospital Los Angeles and the USC Keck School of Medicine (CA, USA) [19], (2) a study conducted at Baylor College of Medicine in Houston (TX, USA), (3) a study conducted at the University of Medical Sciences in Poznan, Poland, and (4) the CEFALO study conducted in Denmark, Sweden, Norway, and Switzerland [20] (Table S1). Ethical approval and informed consent from validation study subjects were obtained at the respective study site.
Genotyping and imputation
DNA extraction and genotyping have previously been described in detail [16]. In brief, DNA was extracted using the Extract-N-amp kit (Sigma-Aldrich) [21,22,23] and was whole-genome-amplified using the REPLIg kit (QIAGEN; Danish subjects) or the GenomePlex Single Cell Whole Genome Amplification kit (Sigma-Aldrich; Swedish subjects). Genotyping was performed using a high-density SNP-array (HumanOmni2.5–8 BeadChip, Illumina). Subjects were excluded if their call-rate was less than 97% or if technical issues were identified, for example conflicting information on reported sex versus X chromosome genotypes or the presence of unexpected duplicate samples. We also excluded subjects identified as outliers using principal component analysis [24, 25] (Figure S2). Based on these criteria, 20 cases and 17 controls were excluded (Figure S1). All subjects included in the association analyses were unrelated (PI-HAT < 0.2). SNPs were excluded based on call-rate (< 95%), minor allele frequency (MAF) (< 1%), and Hardy–Weinberg test (p < 1 × 10–4). We also excluded any A/T and C/G SNPs. Quality control was performed using PLINK (version 1.07, https://zzz.bwh.harvard.edu/plink/) [26]. Imputation was based on 1,288,472 SNPs that passed quality control in the Swedish and Danish datasets and was performed using IMPUTE2 and SHAPEIT2 software and data from the 1000 Genomes Project as reference [27,28,29,30]. Imputed SNPs with MAF < 1% or imputation info score < 0.8 were excluded from all subsequent analyses.
In the validation phase of the study, we used the Sequenom iPLEX Gold platform when genotyping all subjects, except for control subjects from the study conducted at Children’s Hospital Los Angeles and the USC Keck School of Medicine. These subjects were genotyped using Illumina BeadChips, and SNPs that were not represented on the arrays were imputed using MACH v1.0 and the HapMap phase 2 release 21 consensus CEU or CEU + ASN haplotypes as reference.
Selection of SNPs
The genes APC, BRCA2, PALB2, PTCH1, SUFU, TP53, and GPR161 were selected for investigation using a candidate gene approach. The selection was based on two recent studies that found germline mutations in one of these genes in 6% of all medulloblastoma cases [3, 4]. The 1446 SNPs located within these genes include directly genotyped as well as imputed SNPs. We have previously reported the association between genotyped variants in PTCH1 and TP53 and medulloblastoma risk based on the same study population [16].
Statistical methods
Association between genetic variants and medulloblastoma risk was assessed using a frequentist test under an additive model and the score method using SNPTEST v2.5.2 [31]. Analyses were adjusted for sex and five principal components. Principal component analyses were conducted using EIGENSOFT version 6.1.4 (https://www.hsph.harvard.edu/alkes-price/software/) [24, 25].
In the validation phase of the study, logistic regression analysis was preformed separately in two subsets of validation study subjects. Subset 1 included subjects from Children’s Hospital Los Angeles and the USC Keck School of Medicine, and subset 2 included all other validation study subjects. The results from these two subsets were then combined using fixed-effect model meta-analysis.
In genome-wide (agnostic) analyses, p < 5 × 10–8 was considered statistically significant. For candidate gene analyses, p < 0.007 was considered statistically significant, corresponding to Bonferroni correction for testing seven independent loci.
Data availability
The datasets generated and/or analysed during the current study are not publicly available due to legal restrictions but are available from the corresponding author on reasonable request for researchers who meet the criteria for access to confidential data.
References
Hottinger AF, Khakoo Y (2009) Neurooncology of familial cancer syndromes. J Child Neurol 24:1526–1535. https://doi.org/10.1177/0883073809337539
Smith MJ, Beetz C, Williams SG et al (2014) Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32:4155–4161. https://doi.org/10.1200/JCO.2014.58.2569
Waszak SM, Northcott PA, Buchhalter I et al (2018) Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19:785–798. https://doi.org/10.1016/S1470-2045(18)30242-0
Begemann M, Waszak SM, Robinson GW et al (2020) Germline GPR161 mutations predispose to pediatric medulloblastoma. J Clin Oncol 38:43–50. https://doi.org/10.1200/JCO.19.00577
Northcott PA, Korshunov A, Pfister SM, Taylor MD (2012) The clinical implications of medulloblastoma subgroups. Nat Rev Neurol 8:340–351. https://doi.org/10.1038/nrneurol.2012.78
Weil AG, Wang AC, Westwick HJ et al (2017) Survival in pediatric medulloblastoma: a population-based observational study to improve prognostication. J Neurooncol 132:99–107. https://doi.org/10.1007/s11060-016-2341-4
De Braganca KC, Packer RJ (2013) Treatment options for medulloblastoma and CNS primitive neuroectodermal tumor (PNET). Curr Treat Options Neurol 15:593–606. https://doi.org/10.1007/s11940-013-0255-4
Raynor LA, Pankratz N, Spector LG (2013) An analysis of measures of effect size by age of onset in cancer genomewide association studies. Genes Chromosom Cancer 52:855–859. https://doi.org/10.1002/gcc.22081
Craig SE, Brady-Kalnay SM (2015) Regulation of development and cancer by the R2B subfamily of RPTPs and the implications of proteolysis. Semin Cell Dev Biol 37:108–118. https://doi.org/10.1016/j.semcdb.2014.09.004
Brady-Kalnay SM, Rimm DL, Tonks NK (1995) Receptor protein tyrosine phosphatase PTPmu associates with cadherins and catenins in vivo. J Cell Biol 130:977–986. https://doi.org/10.1083/jcb.130.4.977
Couch FJ, Shimelis H, Hu C et al (2017) Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 3:1190–1196. https://doi.org/10.1001/jamaoncol.2017.0424
Manahan ER, Kuerer HM, Sebastian M et al (2019) Consensus guidelines on genetic` testing for hereditary breast cancer from the American Society of Breast Surgeons. Ann Surg Oncol 26:3025–3031. https://doi.org/10.1245/s10434-019-07549-8
Melin BS, Barnholtz-Sloan JS, Wrensch MR et al (2017) Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat Genet 49:789–794. https://doi.org/10.1038/ng.3823
Labreche K, Kinnersley B, Berzero G et al (2018) Diffuse gliomas classified by 1p/19q co-deletion, TERT promoter and IDH mutation status are associated with specific genetic risk loci. Acta Neuropathol 135:743–755. https://doi.org/10.1007/s00401-018-1825-z
Shete S, Hosking FJ, Robertson LB et al (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41:899–904. https://doi.org/10.1038/ng.407
Dahlin AM, Hollegaard MV, Wibom C et al (2015) CCND2, CTNNB1, DDX3X, GLI2, SMARCA4, MYC, MYCN, PTCH1, TP53, and MLL2 gene variants and risk of childhood medulloblastoma. J Neurooncol 125:75–78. https://doi.org/10.1007/s11060-015-1891-1
Hannelius U, Lindgren CM, Melen E et al (2005) Phenylketonuria screening registry as a resource for population genetic studies. J Med Genet 42:e60. https://doi.org/10.1136/jmg.2005.032987
Norgaard-Pedersen B, Hougaard DM (2007) Storage policies and use of the Danish Newborn Screening Biobank. J Inherit Metab Dis 30:530–536. https://doi.org/10.1007/s10545-007-0631-x
Torgerson DG, Ampleford EJ, Chiu GY et al (2011) Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet 43:887–892. https://doi.org/10.1038/ng.888
Aydin D, Feychting M, Schuz J et al (2011) Mobile phone use and brain tumors in children and adolescents: a multicenter case-control study. J Natl Cancer Inst 103:1264–1276. https://doi.org/10.1093/jnci/djr244
Hollegaard MV, Grauholm J, Borglum A et al (2009) Genome-wide scans using archived neonatal dried blood spot samples. BMC Genomics 10:297. https://doi.org/10.1186/1471-2164-10-297
Hollegaard MV, Grove J, Grauholm J et al (2011) Robustness of genome-wide scanning using archived dried blood spot samples as a DNA source. BMC Genet 12:58. https://doi.org/10.1186/1471-2156-12-58
Hollegaard MV, Thorsen P, Norgaard-Pedersen B, Hougaard DM (2009) Genotyping whole-genome-amplified DNA from 3- to 25-year-old neonatal dried blood spot samples with reference to fresh genomic DNA. Electrophoresis 30:2532–2535. https://doi.org/10.1002/elps.200800655
Price AL, Patterson NJ, Plenge RM et al (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909. https://doi.org/10.1038/ng1847
Patterson N, Price AL, Reich D (2006) Population structure and eigenanalysis. PLoS Genet 2:e190. https://doi.org/10.1371/journal.pgen.0020190
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955–959. https://doi.org/10.1038/ng.2354
Howie B, Marchini J, Stephens M (2011) Genotype imputation with thousands of genomes. G3 (Bethesda) 1: 457–470. https://doi.org/10.1534/g3.111.001198
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529. https://doi.org/10.1371/journal.pgen.1000529
Delaneau O, Marchini J, Zagury JF (2011) A linear complexity phasing method for thousands of genomes. Nat Methods 9:179–181. https://doi.org/10.1038/nmeth.1785
Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39:906–913. https://doi.org/10.1038/ng2088
Acknowledgements
Open access funding provided by Umeå University. We acknowledge the role of the late Dr. Mads V. Hollegaard in this study. Dr. Hollegaard was included in all parts of the study, with most important contributions in the conceptualization and formal analyses. The Cefalo Study Group includes Michaela Prochazka, Maral Adel Fahmideh, Birgitta Lannering, Lisbeth S. Schmidt, Christoffer Johansen, Astrid Sehested, Claudia Kuehni, Michael Grotzer, Tore Tynes, Tone Eggen, and Lars Klæboe. We are grateful for the support early in the study by Dr. Bent Nørgaard-Pedersen at the Danish Neonatal Screening Biobank and the assistance in sample collection by Dr. Ulrika von Döbeln at the Swedish phenylketonuria screening registry. This research was funded by the Swedish Childhood Cancer Foundation (Grant Nos. NCS2009-0001, PR2017-0157, NC2011-0004, and TJ2015-0044); the Acta Oncologica Foundation through The Royal Swedish Academy of Science; the Swedish Cancer Foundation (Grant No. CAN 2018/390); the Swedish Research Council (Grant No. 2016-01159_3) ; the Cancer Research Foundation in Northern Sweden (Grant Nos. LP 14-2044, LP 10-1842); Umeå University Hospital (Cutting Edge Grant) (Grant Nos. 7002485, 7002994); and NIH-NIEHS grant, P30ES007033, R01CA116724 and R03CA106011. This research has been conducted using the Danish National Biobank resource, supported by the Novo Nordisk Foundation. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization, BM, UH, and AMD; methodology, ID, CW and AMD; validation, MES, CCL, RMC, RJK, LTH, JY, ASM, JBT, WJG, MFe, JS, MR, KK, DJL, MFi, JN, SSN, and SA; formal analysis, AMD, and UA; resources, DMH; data curation, ID, AMD, and JBG; writing—original draft preparation, AMD; writing—review and editing, all authors; visualization, AMD; supervision, BM; project administration, UA, and LM; funding acquisition, BM, and AMD.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments.
Informed consent
Informed consent was obtained from all Swedish living individual participants included in the discovery phase of the study. According to Danish law, the regional Ethics Committee can grant exemption from obtaining informed consent for research projects on biobank samples under certain circumstances. For the Danish arm of the discovery phase of this study, such an exemption was granted. Informed consent was obtained at the respective study site from participants in the validation phase of the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11060_2020_3424_MOESM1_ESM.pdf
Figure S1, Subject Inclusion; Figure S2, PCA and Q-Q plots; Figure S3, Candidate Genes; Table S1, Study Subjects; Table S2, Genotyped and imputed genetic variants associated with medulloblastoma risk (p < 1 × 10−5) (PDF 921 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dahlin, A.M., Wibom, C., Andersson, U. et al. A genome-wide association study on medulloblastoma. J Neurooncol 147, 309–315 (2020). https://doi.org/10.1007/s11060-020-03424-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-020-03424-9