
Abstract
Purpose
Gliosarcoma is a histologic variant of glioblastoma (GBM), and like GBM carries a poor prognosis. Median survival is less than one (1) year with less than 5% of patients alive after 5 years. Although there is no cure, standard treatment includes surgery, radiation and chemotherapy. While very similar to GBM, gliosarcoma exhibits several distinct differences, morphologically and molecularly. Therefore, we report a comprehensive analysis of DNA copy number changes in gliosarcoma using a cytogenomic DNA copy number (CN) microarray (OncoScan®).
Methods
Cytogenomic DNA copy number microarray (OncoScan®) was performed on 18 cases of gliosarcoma. MetaCore™ enrichment was applied to the array results to detect associated molecular pathways.
Results
The most frequent alteration was copy number loss, comprising 57% of total copy number changes. The number of losses far exceeded the number of amplifications (***, < 0.001) and loss of heterozygosity events (***, < 0.001). Amplifications were infrequent (4.6%), particularly for EGFR. Chromosomes 9 and 10 had the highest number of losses; a large portion of which correlated to CDKN2A/B loss. Copy number gains were the second most common alteration (26.2%), with the majority occurring on chromosome 7. MetaCore™ enrichment detected notable pathways for copy number gains including: HOXA, Rho family of GTPases, and EGFR; copy number loss including: WNT, NF-kß, and CDKN2A; and copy number loss of heterozygosity including: WNT and p53.
Conclusions
The pathways and copy number alterations detected in this study may represent key drivers in gliosarcoma oncogenesis and may provide a starting point toward targeted oncologic analysis with therapeutic potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most aggressive primary malignant adult central nervous system tumor [1, 2]. Gliosarcoma, a histologic variant, accounts for 2% of all GBMs. Overall survival of gliosarcoma is similar to GBM except that systemic metastasis and skull invasion have been reported more frequently in gliosarcoma [3]. However, gliosarcoma exhibits several unique morphologic, immunohistochemical and molecular characteristics. Gliosarcoma is defined histologically as having biphasic neoplastic components; displaying both mesenchymal (fibroblastic, osseous, muscle or adipose differentiation) and glial differentiation [2]. The genomic alterations implicated in the malignant transformation of astrocytes are diverse, of which, IDH, TERT, EGFR, CDKN2A, TP53, PTEN, PDGFRA and NFKB1A are the most commonly reported [3, 4]. These genes are involved in oxidative decarboxylation, maintenance of telomeres, stimulating protein tyrosine kinase, tumor suppression, cell signaling and a variety of other cellular processes. Gain of 7p in combination with 10q loss is associated with EGFR amplification and is a frequent finding in IDH-wildtype GBM. Of GBMs with EGFR protein overexpression, 70–90% demonstrate EGFR gene amplification. Other mutations such as the EGFRvIII and missense mutations involving the extracellular domain are distinct from EGFR mutations in non-glial cancers [5]. Interestingly, EGFR alterations vary amongst GBM subtypes, being rare in IDH-mutated GBM, and more prevalent in IDH-wildtype GBM [6]. Similar to IDH-wildtype GBM, gliosarcomas contain PTEN, CDKN2A and TP53 alterations, but amplification of the EGFR (epidermal growth factor receptor) are uncommon (4–8%) [7,8,9]. Furthermore, the type of EGFR alterations reported in gliosarcoma are not usually seen in GBM; particularly EGFR point mutations have been detected in gliosarcomas (c.1831G > A) [10]. Amplification of EGFR is present in 35–45% of IDH-wildtype GBMs [2]. Of the detected EGFR amplifications in gliosarcoma, it is speculated that these results are derived from the glial component of the tumor rather than the sarcomatous component. Immunohistochemistry supports this, staining for EGFR is negative in the sarcomatous component and positive in the glial component [11]. Gains of chromosome 7 without EGFR amplification is frequent in gliosarcoma [9], leading one to believe the oncogenic driver for gliosarcomas may reside on chromosome 7, but not necessarily related to the EGFR pathway. The myriad of known candidate genes located on chromosome 7 (CDK6, PDGF-A, c-MET) may support this theory.
Given its poor prognosis, more research has been directed toward identifying specific mutations for targeted treatment of GBM. Several novel agents have been introduced that specifically target EGFR, but treatment with TKI’s and EGFR antibodies have not yielded successful clinical results. The hypothesis is that TKI’s work best for exon 19 and 21 mutations, which have not been detected in GBM [12]. The poor response of EGFR-targeted therapies raises the question of whether EGFR alterations truly represent key drivers in the genesis of glioblastomas. Therefore, we applied a whole-genome approach using the OncoScan® Assay to examine DNA copy number alterations, and identify any chromosome regions known to harbor oncogenic drivers.
Materials and methods
Tissue processing, histopathology and clinical history review
All tissue samples were obtained for diagnostic and research (under IRB approval) purposes at the time of surgical resection. A retrospective search within the institutional pathology database for “gliosarcoma” yielded 18 cases from which a OncoScan® was performed. Tissue processing consisted of fixation in 10% neutral buffered formalin and paraffin embedding (FFPE: formalin fixed paraffin embedded). Histopathologic tumor classification was rendered by multiple neuropathologists. Immunohistochemistry was performed following the manufacturers protocols using the Leica Bond Maxx III automated system for all primary antibodies: IDH1 (1:80, Dianova, Hamburg, Germany), GFAP (prediluted by manufacturer, DAKO, Carpinteria, CA). Positive and negative controls were stained alongside study materials.
Clinical data was retrieved from institutional electronic medical records and included: patient demographics, presenting symptoms, imaging characteristics, date of surgery, diagnosis, treatment, time to recurrence, length of follow-up, date of death. Less common IDH1 and IDH2 mutations were previously tested for clinical purposes using Emory University Hospital “SNaPshot” mutation panel, the results of which were reviewed for each case.
Copy number (CN)/single nucleotide polymorphism (SNP) microarray analysis
For 17 specimens, formalin-fixed paraffin embedded (FFPE) brain tumor tissue was obtained and processed on Thermo Fisher’s Oncoscan SNP DNA microarray. DNA was isolated using the QIAmp DNA FFPE Tissue kit. For quantification, DNA (80 ng) was prepared with the Qubit dsDNA Broad Range assay and detected with the Qubit Fluorometer 2.0. The assay was performed according to the manufacturer’s protocol using a standard gel electrophoresis system or the Lonza FlashGel System for DNA size assessments. The assay consisted of overnight annealing of approximately 240,000 molecular inversion copy number probes and 74 somatic mutation probes, single nucleotide integration for SNP detection, two rounds of PCR, HAEIII digestion, and overnight hybridization of the libraries to the arrays. For one case, fresh tumor tissue was processed on Thermo Fisher’s CytoScan HD SNP DNA microarray. DNA was isolated with the Qiagen QIAamp DNA mini kit with the Qiagen DNA Purification from Tissues protocol. The Nanodrop ND-2000 Spectrophotometer was used to quantify the DNA (250 ng). The assay was performed according to the manufacturer’s protocol. The arrays were scanned and CEL files were processed in Thermo Fisher’s Chromosome Analysis Suite (ChAS) software: NA33 workflow and hg19 genome Refs. [13,14,15,16,17,18].
Radiographic imaging
MRI images were manually inspected to characterize tumors according to criteria determined by the VASARI feature set [19] including: location, type of enhancement, margin definition, presence of hemorrhage or cysts, and invasion of adjacent structures such as the ependymal surfaces, pia, and cortex. The size of the T2 non-enhancing region, the T1 postcontrast enhancing region, and any areas of necrosis were recorded. Preoperative imaging (from our institution) on a comparable set of imaging from GBM patients (n = 20) from a similar time period (October 2014 and September 2015) was inspected using the same technique. Quantitative values, including measurements, were statistically compared using a two-sided t test assuming equal variance. Proportions between groups were compared using a Fischer exact test.
Statistics, pathway analysis, literature review
The genes within CNV regions were obtained by using BEDTools (v2.26.0) intersect that overlapped hg19 human reference genome and CNV regions. Kaplan–Meier Survival curves were created using CASAS tool [20]. DNA copy number results were analyzed using MetaCore™ to generate molecular maps/pathways (confined to p < 0.05 for gains, losses, and LOH). Amplification events were too low in number to generate statistically significant ontology data. Literature review was attained via a Scopus® search for the keywords “gliosarcoma and EGFR,” from 1 January 1995 to 31 July 2018.
Results
Clinical characteristics, radiographic features and histopathology
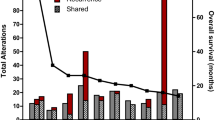
The cohort included 18 specimens (17 patients; 2 specimens were from the same patient) with a diagnosis of gliosarcoma. Twelve patients were male (70.6%), and 5 patients were female (29.4%) with a male to female ratio of 2.4:1. The median age at diagnosis was 61 years, with a range of 33–75 years. The median survival (Fig. 1) was 313 days (10.3 months) compared to 9 months (range 3–44 months) for a similar cohort of 20 IDH-wildtype GBM cases from our database (resected between 2014 and 2015) [21].

Kaplan Meier survival curves for 17 patients with gliosarcoma. The median survival was 0.9 years (313 days) (95% CI 0.3, 2.2) versus 9 months for a similar cohort of 20 IDH-wildtype GBM cases (9 months). Bottom graphic: number of patient’s surviving (considered to be “at risk”) after each time period
Fifteen (15) patients had preoperative imaging available for review (Fig. 2). Gliosarcomas typically presented as aggressive appearing masses with extensive edema (diameter 84 mm), marked enhancement (100%), solid or thick enhancement (87%), cortical involvement (87%), pial invasion (53%), and ependymal invasion (40%). All had low or mixed average diffusion coefficient values (100%). These findings were compared to a control cohort of 20 IDH-wildtype GBM cases (from our institution) taken from patient’s who underwent surgical resection or biopsy between October 2014 and September 2015. The only distinct feature of gliosarcomas (compared to GBM) was a slightly larger area of edema (84 vs. 71 mm, p = 0.03 for a comparable size enhancing mass (48 vs. 43 mm, p = 0.26). Gliosarcomas were less likely to involve the occipital lobe (6% vs. 35%, p = 0.1) and have a thin rim of enhancement (13% vs. 40%, p = 0.13). Both GBM and gliosarcomas invaded adjacent structures including the cerebral cortex, ependymal surfaces, and pia, although only one (1) gliosarcoma had invasion of the adjacent calvarium, a feature which was not seen in the glioblastoma set.

Typical MRI features of gliosarcomas. a FLAIR image demonstrating a centrally isointense mass (black arrow) with extensive surrounding edema extending into the basal ganglia (white arrow). b ADC image showing low diffusion values in the central enhancing portion of the mass (black arrow). c GRE image demonstrating central areas of susceptibility compatible with internal hemorrhage. d–f Postcontrast T1-weighted images showing a thick rim of enhancement (d black arrow) with central necrosis (d white arrow), pial invasion (e black arrows), and ependymal invasion (f black arrow)
All cases were diagnosed as gliosarcoma, the glial component of which, exhibited confirmatory GFAP immunohistochemical positivity. A sarcomatous morphology was present in all 18 cases, but in 3 cases the mesenchymal portion consisted of adipocytic differentiation, osteoid differentiation, and myxoid/metaplastic morphology. All cases were IDH-negative in both the glial and sarcomatous components by immunohistochemistry and IDH-wildtype by molecular testing. The approximate proportion of sarcomatous components in each case ranged from 10 to 60%.
DNA copy number microarray analysis
Total copy number abnormalities including amplification, loss of heterozygosity, gains, and losses for 18 gliosarcoma specimens was 305 (Table 1). Per specimen, the copy number changes ranged from 6 to 52 (median = 13). Amplifications were infrequent (4.6%); were present in 6 cases and included regions on: chromosome 3p, 7q, 9p, 12q, 4q, and 7p. One case exhibited amplification of EGFR (7p11.2). However, gain of EGFR occurred in 13 cases (72%) (Fig. 3). The most frequent type of copy number alteration was loss (n = 175), comprising 57% of the total copy number changes. The number of losses far exceeded the number of amplifications (***, < 0.001) and loss of heterozygosity events (***, < 0.001). Chromosomes 9 and 10 had the highest number of losses. Particularly, chromosome 10 harbored 13 (33%) of whole chromosome losses. A large number of losses were from chromosome 9 (n = 17), mostly correlating to loss of CDKN2A/B (Table 1, Fig. 3). DNA copy number gains were the second most common change, totaling 80 events (26.2%). The majority of gains occurred on chromosome 7, which harbored 20 total gain events. Copy neutral loss of heterozygosity (LOH) events were infrequent, consisting of 39 events. Chromosome 17 exhibited the highest number of LOH (Table 2) in the gliosarcoma cohort. One case exhibited tetraploidy, but all other cases were referenced in diploid.

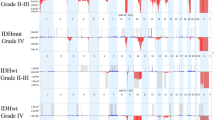
Whole genome view illustrating Oncoscan (CN/SNP DNA microarray) results. 18 gliosarcoma specimens (top) and 580 glioblastoma cases (bottom, taken from the TCGA). Focal amplification of EGFR (7p11.2) is evident in the GBM TCGA dataset (arrow), but gains of chromosome 7 (*) without EGFR (7p11.2) amplification are present in the gliosarcoma cases. Loss of chromosome 10 (+) and loss of 9p containing CDKN2A/B (arrowheads) are present in both GBM and gliosarcoma (+). X axis: each chromosome. Y axis: SGOL score, which represents segments of gains (blue) and losses (red) across chromosomes. Both graphics created with Copy Number Explorer. https://arraycgh.shinyapps.io/Copy_Number_Explorer/ [52]. Data from bottom graphic provided by GBM TCGA Dec 2014
Pathway enrichment (MetaCore™)
Pathways associated with copy number loss (Table 3) in the gliosarcoma cohort included WNT signaling, NF-kß, and CDKN2A. Several regions that showed chromosome loss were areas that code for WNT pathway proteins (Tcf (Lef), WNT, Sirtuin1, beta-TrCP, BMI-1, TCF7L2 (TCF4), DKK1). In addition, pathway enrichment revealed copy number loss in areas coding for components of the OX40L/OX40 pathway (NF-kB2 (p52), IKK-alpha, NF-kB2 (p100), Calcineurin A (catalytic), PKC-theta, Perforin, NF-kB). Copy number loss of the regions containing CDKN2A/p16INK4 (9p21) was present in several pathway maps. The NF-kß protein complex was also repeatedly represented.
Pathways associated with copy number gains included HOXA, EGFR, actin, adenylate cyclase, PKA-reg, IBP3, cytochrome c, Rac1, G-protein alpha-12 family, F-actin cytoskeleton, MRLC, IL-6. Gain of EGFR occurred in 13 of 18 cases (72%). Gain of regions that code for adenylate cyclase, MRLC (myosin regulatory light chains) and PKA-reg were frequent and routed to pathway maps related to myogenesis, and regulation of smooth muscle tone. The pathways with the highest statistical significance involved gains of areas containing HOXA genes, specifically centered around demethylation and methylation of histone H3 at lysine 27 (H3K27), and their role in stem cell differentiation. Rac1, a key member of the Rho family of GTPases, was a frequently identified network object related to pathways including cytoskeletal remodeling through kinase effectors of Rho GTPases. Chromosomal gains in the region coding for cytochrome c was also present, and linked to multiple maps related to apoptosis regulation and cell survival. IL-6 was frequently represented as a network object for copy number gains [22, 23].
Pathways associated with copy number loss of heterozygosity (LOH) included p53, ephrin-B, PLD2, PI3K, MEK4, CRK, and Dsh. Chromosome 17 exhibited the highest number of LOH events. This included the region encoding p53 (17p13.1), and mapped to pathways citing inhibiting of apoptosis. Interestingly, there was also WNT pathway involvement through LOH of Dsh (Dishevelled), PLD2, MEK4, and CRK; mapping to canonical WNT signaling in colorectal cancer, hepatocellular cancer, and lung cancer; frequently in the same maps associated with p53. LOH of regions encoding for several proteins related to angiogenesis was present (ephrin-B, PDF, TWEAK).
Comparison to published data
A Scopus® search for “gliosarcoma and EGFR,” from 1 January 1995 to 31 July 2018 yielded 58 articles, 9 of which included EGFR testing (Table 3). Actor et al. analyzed the largest number of cases (n = 38) and detected 3 gliosarcoma cases with EGFR amplification using a combination of comparative genomic hybridization, single-strand conformation polymorphism/heteroduplex analysis, duplex PCR, and southern blot analysis. Three (3) studies [24,25,26] used florescent in situ hybridization (FISH) to detect amplification of EGFR. Two (2) studies used next-generation sequencing (NGS), which detected EGFR mutations, c.1831G > A and G719D [10, 27]. Although EGFR immunohistochemistry was performed in 2 studies [11, 28], the ability of immunohistochemistry to differentiated EGFR gain from amplification is uncertain. OncoScan® (DNA copy number analysis) was not performed in any of the 9 articles reviewed.
Discussion
Overall, our data showed that DNA copy number losses were frequent and amplifications were infrequent in gliosarcoma. The majority of copy number loss occurred on chromosomes 9 and 10; localizing to regions containing CDKN2A and CDKN2B genes. The CDKN2A gene encodes for proteins p16 and p14arf, tumor suppressors that regulate the p53 and RB1 cell cycle [29]. The CDKN2B gene encodes for the p15ink4b protein, a member of the p16ink4 (CDKN2A) family, and a cell growth regulator that inhibits G1 progression [30]. Homozygous loss of CDKN2A is common in GBM (35–50% [2]) and loss of this locus occurred in 14 of 18 gliosarcoma specimens [31]. Other potential pathways involved in gliosarcoma include activation of the OX40L/OX40 pathway, which has been shown to induce strong immunity and antitumor effects in GBM [32]. DNA copy number loss was present in areas coding for several protein components of the OX40L/OX40 pathway (NF-kB2 (p52), IKK-alpha, NF-kB2 (p100), Calcineurin A (catalytic), PKC-theta, Perforin, NF-kB). Conversely, several regions that showed chromosome loss were areas that coded for WNT pathway proteins (Tcf (Lef), WNT, Sirtuin1, beta-TrCP, BMI-1, TCF7L2 (TCF4), DKK1). In the activated or overexpressed state, most of these WNT pathway proteins (with the exception of DKK1) promote cell proliferation and cell survival [33]. However, DKK1 (Dickkopf-1) is considered to be a negative regulator of WNT pathway, and has been implicated as a candidate gene that is epigenetically silenced in medulloblastoma [34], loss of which, may provide an avenue for WNT pathway activation with subsequent cell proliferation and survival. One study showed that DKK1 expression led to glioma cell sensitivity to chemotherapy-induced apoptosis [35]. Another protein with repetitive representation after pathway enrichment was NF-kß, a protein complex that controls DNA transcription, but can induce cell proliferation and anti-apoptosis if misregulated or constitutively activated. Aberrant activation of NF-kß in glioblastoma, leading to cell invasive capabilities, resistance to radiotherapy, and even promotion of mesenchymal phenotype has been reported [36]. Studies to evaluate the therapeutic effect of inhibition of NF-kß, based on these mechanisms have been published [37]. However, due to the multifactorial role NF-kß plays in a diverse number of biological processes (cell proliferation, survival, motility, DNA repair, inflammation), a direct pathway to neoplasia in GBM is unclear. Our data shows that there is copy number loss in the region encoding for NF-kß in gliosarcoma, implying that activation of the NF-kß pathway does not play a role in gliosarcoma genesis. However, it is possible that loss of NF-kß could lead to loss of DNA repair mechanisms, resulting in neoplasia.
Chromosome gains were the second most common copy number abnormality. Gains in regions coding for signaling molecules known to promote cell proliferation (F-actin cytoskeleton, actin cytoskeletal) and cytoskeleton remodeling (G-protein alpha-12 family, F-actin cytoskeleton) were present [38]. Interestingly, the Rho GTPase family, including rac1has been implicated in the modulation of glioma cell migration through cytoskeletal rearrangement [39]. Homeobox (HOX) genes are responsible for regulation of transcription factors during embryonic development, the differential expression of which, have been tied to many different cancers, including glioblastoma [40]. Specifically, high expression of HOXA9 and HOXA10 have been reported in human glioma cell lines [40]. The expression of HOXA13 has been proposed as an activator of WNT and TGF-ß-induced epithelial to mesenchymal transition in glioma progression [41]. Resistance to radiation and chemotherapy through the activation of HOXA, thereby inducing increased proliferation and decreased apoptosis in cultured glioblastoma cells has also been reported [42]. Our microarray data showed gains in the regions encoding HOXA1, HOXA3, HOXA7, HOXA11, and HOXA13. Interestingly, several studies have demonstrated overexpression of HOX genes in Ewing sarcoma [43, 44] and undifferentiated small round blue cell sarcoma [45]; perhaps raising the question of whether HOX genes play a role in mesenchymal phenotype in gliosarcoma.
Copy neutral loss of heterozygosity (LOH) events were less frequent. OncoScan™ detected loss of the region encoding p53 (17p13.1), and while p53 alterations have been well documented in gliomas, some studies suggests that LOH of a single cell cycle regulator, such as p53 may be insufficient for development of gliomas [46]. In the context of this study, concurrent loss of p16 with LOH of p53 may play a large role in the genesis of gliosarcoma. Another pathway that was well represented across LOH, gains and losses after MetaCore™ enrichment was chemotaxis lysophosphatidic acid signaling. Lysophosphatidic acid (LPA) is a phospholipid that binds to G protein-coupled receptors (GPCRs) leading to chemotaxis, cell proliferation, cell growth and cell survival through a complex network of signaling cascades [38]. LPA was shown to be increased in GBM stem cells from the subventricular zone via LPA/Rho signaling cascades and proposed as a mechanism for GBM invasion and angiogenesis, a possible therapeutic target using LPAR antagonist and LPA synthesis inhibitors [47, 48]. Another region with copy neutral LOH encoded for the network object Ephrin-B, a protein ligand known to promote angiogenesis, as well as in developmental processes such as axon guidance, cell migration, and maturation of cortical dendrites. The Eph receptor tyrosine kinases and ephrin ligands have been implicated in both the inhibition and promotion of neoplasia and may play a key role in glioma genesis [49].
Epidermal growth factor receptor (EGFR) is mutated or amplified in 35–45% of IDH-wildtype glioblastomas [2] and implicated as a key driver [50]. Gliosarcoma, however, does not exhibit amplification or mutations of EGFR at the same frequency, suggesting there may be additional/alternate mechanisms propelling tumorigenesis and potentially mesenchymal transition into a sarcoma phenotype. Our data supports other studies that show a very low prevalence of EGFR amplification in gliosarcoma, but did show frequent gain of chromosome 7 (72%) containing EGFR locus. It is important to note that amplification or mutation of EGFR is not necessarily required for EGFR activation. For example, copy number gain of SYNJ2 in breast cancer leads to EGFR activation by altered trafficking pathways [50]. Therefore, even though EGFR amplification is not common in gliosarcoma, EGFR pathway activation may still be present. Gain as oppose to amplification of EGFR may be sufficient for EGFR pathway activation in of itself. In contrast, some believe copy number gain (as oppose to amplification) of EGFR reflects chromosomal instability in cancer cells and has no biological significance [51]. Thus, it is unclear whether EGFR pathway activation is present in gliosarcoma; perhaps it is present through indirect mechanisms and not necessarily through overexpression of EGFR due to gene amplification. Our study confirms that EGFR amplification is uncommon in gliosarcoma and provides whole-genome evidence of possible driver pathways from a DNA copy number perspective. Further analysis to pinpoint specific loci within the altered copy number regions is needed.
The strengths of this study are that it comprises the largest cohort of gliosarcoma cases with DNA copy number analysis by OncoScan™. However, there were a few limitations. We intended this to represent an initial overview of copy number changes in a large cohort of gliosarcoma cases, and did not include OncoScan™ data from GBM. In the future, it would be ideal to compare our gliosarcoma cohort to regional-matched GBM cases. Also, no general effort was made to separate sarcomatous from glioblastoma components for OncoScan™ analysis. An interesting next step would be to separate the glioblastoma from sarcoma components microscopically, and re-analyze the separate components.
Conclusions
Gliosarcoma, much like GBM, is a fatal diagnosis with no cure. We present a comprehensive whole-genome copy number analysis of gliosarcoma performed in an effort to identify chromosome regions that may represent plausible drivers for gliosarcoma genesis. This study may provide a starting point to direct more targeted oncologic analysis and discover genetic alterations that lead to pathways with therapeutic potential.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- GBM:
-
Glioblastoma
- GFAP:
-
Glial fibrillary acid protein
- EGFR:
-
Epidermal growth factor receptor
- FFPE:
-
Formalin fixed paraffin embedded
- H&E:
-
Hematoxylin & eosin
- ChAS:
-
Chromosome analysis suite
- LOH:
-
Loss of heterozygosity
- Hmz:
-
Homozygous (LOH)
- WC:
-
Whole chromosome
- TCGA:
-
The Cancer Genome Atlas
References
Walid M (2008) Prognostic factors for long-term survival after glioblastoma. Perm J. https://doi.org/10.7812/tpp/08-027
International Agency for Research on Cancer (2016) WHO Classification of Tumours of the Central Nervous System (IARC WHO Classification of Tumours). http://publications.iarc.fr/Book-And-Report-Series/Who-Iarc-Classification-Of-Tumours/Who-Classification-Of-Tumours-Of-The-Central-Nervous-System-2016%0Ahttp://link.springer.com/10.1007/s00401-016-1545-1
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(2016):1–18. https://doi.org/10.1007/s00401-016-1545-1
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R (2014) The somatic genomic landscape of glioblastoma. Cell 157(3):753
Aldape K, Gelareh Z, Von Deimling A (2015) Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:809–827. https://doi.org/10.1007/s00401-015-1424-1
Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, Ohgaki H (1996) Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 6:217–223. https://doi.org/10.1097/00005072-199605000-00017 discussion 23-4
Biernat W, Huang H, Yokoo H, Kleihues P, Ohgaki H (2004) Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol 14:131–136. https://doi.org/10.1111/j.1750-3639.2004.tb00045.x
Han SJ, Yang I, Tihan T, Prados MD, Parsa AT (2010) Primary gliosarcoma: key clinical and pathologic distinctions from glioblastoma with implications as a unique oncologic entity. J Neurooncol 96:313–320. https://doi.org/10.1007/s11060-009-9973-6
Reis RM, Konu-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H (2000) Genetic profile of gliosarcomas. Am J Pathol 178:2496–2503. https://doi.org/10.1016/j.ajpath.2011.02.031
Cachia D, Kamiya-Matsuoka C, Mandel JJ, Olar A, Cykowski MD, Armstrong TS, Fuller GN, Gilbert MR, De Groot JF (2015) Primary and secondary gliosarcomas: clinical, molecular and survival characteristics. J Neurooncol 125:401–410. https://doi.org/10.1007/s11060-015-1930-y
Actor B, Ludwig Cobbers JMJ, Büschges R, Wolter M, Knobbe CB, Reifenberger G, Weber RG (2002) Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components, Genes Chromosom. Cancer 34:416–427. https://doi.org/10.1002/gcc.10087
Padfield E, Ellis HP, Kurian KM (2015) Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol 5:5. https://doi.org/10.3389/fonc.2015.00005
Affymetrix Inc, Oncoscan FFPE Assay kit Manual (2013)
Affymetrix Inc, CytoScan Assay User Manual (n.d.)
Qiagen, Qiagen QIAamp DNA mini kit and Blood Mini Handbook, 5th Ed. (n.d.). https://www.qiagen.com/us/shop/sample-technologies/dna/genomic-dna/qiaamp-dna-mini-kit/#resources. Accessed 7 July 7 2018
Invitrogen, Qubit 2.0 Fluorometer Assay Manual, (n.d.)
Invitrogen, Qubit 2.0 Fluorometer, (n.d.)
Lonza, FlashGel System, (2012)
NCBI/NCI/The Cancer Imaging Archive (TCIA) (2015), VASARI (Visually AcceSAble Rembrandt Images) MRI feature set, NCBI/NCI, Bethesda MD. Available via https://wiki.cancerimagingarchive.net/display/Public/VASARI+Research+Project. Accessed 8 Mar 2017
Rupji M, Zhang X, Kowalski J (2017) CASAS: Cancer Survival Analysis Suite, a web based application. F1000Research 6:919. https://doi.org/10.12688/f1000research.11830.2
Zhang G, Huang S, Zhang J, Wu Z, Lin S, Wang Y (2016) Clinical outcome of gliosarcoma compared with glioblastoma multiforme: a clinical study in Chinese patients. J Neurooncol 127:355–362. https://doi.org/10.1007/s11060-015-2046-0
MetaCore, Regulation of IGF family activity in colorectal cancer, (n.d.). https://support.clarivate.com/LifeSciences/s/article/Metacore-How-to-cite-Metacore-in-publications?language=en_US
MetaCore, Inhibition of apoptosis in pancreatic cancer, (n.d.). https://support.clarivate.com/LifeSciences/s/article/Metacore-How-to-cite-Metacore-in-publications?language=en_US
Kleinschmidt-DeMasters BK, Meltesen L, McGavran L, Lillehei KO (2006) Characterization of glioblastomas in young adults. Brain Pathol 16:273–286. https://doi.org/10.1111/j.1750-3639.2006.00029.x
Yao K, Qi XL, Mei X, Jiang T (2015) Gliosarcoma with primitive neuroectodermal, osseous, cartilage and adipocyte differentiation: a case report. Int J Clin Exp Pathol 8:2079–2084
Shelly D, Epari S, Arora I, Pai T, Ahmed S, Moiyadi A, Chinnaswamy G, Gupta T, Goda J, Shetty P, Kane S, Desai S, Jalali R (2016) Epidermal growth factor receptor (EGFR) gene amplification in high-grade gliomas: Western Indian tertiary cancer center experience. Neurol India. https://doi.org/10.4103/0028-3886.173668
Pain M, Wang H, Lee E, Strahl M, Hamou W, Sebra R, Zhu J, Yong RL (2018) Treatment-associatedTP53DNA-binding domain missense mutations in the pathogenesis of secondary gliosarcoma. Oncotarget 9:2603–2621. https://doi.org/10.18632/oncotarget.23517
Lin JW, Wu YT, Chang IW (2011) The prognostic impact of O6-methylguanine DNA methyltransferase and epidermal growth factor receptor expressions on primary gliosarcoma: a clinicopathologic and immunohistochemical study of seven cases at a single institution. Indian J Pathol Microbiol 54:683–687. https://doi.org/10.4103/0377-4929.91491
OMIM, CYCLIN-DEPENDENT KINASE INHIBITOR 2A; CDKN2A, (n.d.). https://www.omim.org/entry/600160#mapping
OMIM, CYCLIN-DEPENDENT KINASE INHIBITOR 2B; CDKN2B, (n.d.). https://www.omim.org/entry/600431?search=cdkn2b&highlight=cdkn2b. Accessed 19 December 2018
Nishikawa R, Furnari FB, Lin H, Arap W, Cavenee WK, Huang HJS, Cavenee WK, Huang HJS, Cavenee WK, Arap W, Berger MS (1995) Loss of P16INK4 expression is frequent in high grade gliomas. Cancer Res 55:1941–1945
Shibahara I, Saito R, Zhang R, Chonan M, Shoji T, Kanamori M, Sonoda Y, Kumabe T, Kanehira M, Kikuchi T, So T, Watanabe T, Takahashi H, Iwabuchi E, Tanaka Y, Shibahara Y, Sasano H, Ishii N, Tominaga T (2015) OX40 ligand expressed in glioblastoma modulates adaptive immunity depending on the microenvironment: a clue for successful immunotherapy. Mol Cancer 14:1–13. https://doi.org/10.1186/s12943-015-0307-3
MetaCore, Role of activation of WNT signaling in the progression of lung cancer, (n.d.). https://support.clarivate.com/LifeSciences/s/article/Metacore-How-to-cite-Metacore-in-publications?language=en_US
Vibhakar R, Foltz G, Yoon J, Field L, Lee H, Ryu G, Pierson J, Davidson B, Madan A (2007) Dickkopf-1 is an epigenetically silenced candidate tumor suppressor gene in medulloblastoma1. Neuro-Oncology 9:135–144. https://doi.org/10.1215/15228517-2006-038
Shou J, Ali-Osman F, Multani AS, Pathak S, Fedi P, Srivenugopal KS (2002) Human Dkk-1, a gene encoding a Wnt antagonist, responds to DNA damage and its overexpression sensitizes brain tumor cells to apoptosis following alkylation damage of DNA. Oncogene 21:878–889. https://doi.org/10.1038/sj.onc.1205138
Soubannier V, Stifani S (2017) NF-κB signalling in glioblastoma. Biomedicines 5:29. https://doi.org/10.3390/biomedicines5020029
Friedmann-Morvinski D, Narasimamurthy R, Xia Y, Myskiw C, Soda Y, Verma IM (2016) Targeting NF-kB in glioblastoma: a therapeutic approach. Sci. Adv. https://doi.org/10.1126/sciadv.1501292
MetaCore, Lysophosphatidic acid signaling via GPCRs, (n.d.). https://support.clarivate.com/LifeSciences/s/article/Metacore-How-to-cite-Metacore-in-publications?language=en_US
Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL (2013) Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front. Oncol 3:1–11. https://doi.org/10.3389/fonc.2013.00241
Bhatlekar S, Fields JZ, Boman BM (2014) HOX genes and their role in the development of human cancers. J Mol Med 92:811–823. https://doi.org/10.1007/s00109-014-1181-y
Duan R, Han L, Wang Q, Wei J, Chen L, Zhang J, Kang C, Wang L (2015) HOXA13 is a potential GBM diagnostic marker and promotes glioma invasion by activating the Wnt and TGF-β pathways. Oncotarget 6:6. https://doi.org/10.1136/bmj.2.4933.263-b
Schmid RS, Simon JM, Vitucci M, McNeill RS, Bash RE, Werneke AM, Huey L, White KK, Ewend MG, Wu J, Miller CR (2016) Core pathway mutations induce de-differentiation of murine astrocytes into glioblastoma stem cells that are sensitive to radiation but resistant to temozolomide. Neuro-Oncology 18:962–973. https://doi.org/10.1093/neuonc/nov321
Svoboda LK, Bailey N, Van Noord RA, Krook MA, Harris A, Cramer C, Jasman B, Patel RM, Thomas D, Borkin D, Cierpicki T (2017) Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget. 8(1):458
Svoboda LK, Harris A, Bailey NJ, Schwentner R, Tomazou E, von Levetzow C, Magnuson B, Ljungman M, Kovar H, Lawlor ER (2014) Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 9(12):1613–1625
Small U, Round B, Sarcomas C (2016) Novel BCOR-MAML3 and ZC3H7B-BCOR gene fusions in undifferentiated small blue round cell sarcomas. Katja 40:433–442. https://doi.org/10.1097/pas.0000000000000591.novel
Gladson CL, Prayson RA, Liu WM (2010) The pathobiology of glioma tumors. Annu Rev Pathol Mech Dis 5:33–50. https://doi.org/10.1146/annurev-pathol-121808-102109
Tabuchi S (2015) The autotaxin-lysophosphatidic acid–lysophosphatidic acid receptor cascade : proposal of a novel potential therapeutic target for treating glioblastoma multiforme. Lipids Health Dis. https://doi.org/10.1186/s12944-015-0059-5
Manning TJ, Parker JC, Sontheimer H (2000) Role of lysophosphatidic acid and rho in glioma. Cell Motility 199:185–199
Ferluga S, Debinski W (2015) HHS Public Access 32:190–201. https://doi.org/10.3109/08977194.2014.985787.Ephs
Sigismund S, Avanzato D, Lanzetti L (2018) Emerging functions of the EGFR in cancer. Mol. Oncol. 12:3–20. https://doi.org/10.1002/1878-0261.12155
Ålgars A, Lintunen M, Carpén O, Ristamäki R, Sundström J (2012) Reply: eGFR alterations and response to anti-EGFR therapy: is it a matter of gene amplification or gene copy number gain? Br J Cancer 106:428. https://doi.org/10.1038/bjc.2011.570
Newman S (2015) Interactive analysis of large cancer copy number studies with Copy Number Explorer. Bioinformatics 31:2874–2876. https://doi.org/10.1093/bioinformatics/btv298
Smith DR, Wu CC, Saadatmand HJ, Isaacson SR, Cheng SK, Sisti MB, Bruce JN, Sheth SA, Lassman AB, Iwamoto FM, Wang SH, Canoll P, McKhann GM, Wang TJC (2018) Clinical and molecular characteristics of gliosarcoma and modern prognostic significance relative to conventional glioblastoma. J Neurooncol 137:303–311. https://doi.org/10.1007/s11060-017-2718-z
Acknowledgements
Research reported in this publication was supported in part by the Biostatistics and Bioinformatics shared resource of Winship Cancer Institute of Emory University and NIH/NCI under Award Number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
Funding for data interpretation and analysis (Winship Cancer Institute Biostatistics and Bioinformatics Shared Resource) was provided by K08 award NS083626 (Principal investigator: Soma Sengupta, M.D., Ph.D.).
Author information
Authors and Affiliations
Contributions
LL, JH, DS, SN analyzed and interpreted the DNA microarray analysis. AW and LL provided the introduction. LL, SN, MS performed the histological examination of the brain samples. HC performed the pathway enrichment. LL, HC, JK, SS performed the pathway interpretation. MR and JK determined survival characteristics. BW, SS and JO provided the clinical data including surgical and imaging characteristics. LL was the main contributor in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Consent for publication was provided by the Emory University Institutional Review Board (IRB, 1599 Clifton Road, NE, 5th floor, Atlanta, GA 30322 United States; IRB@emory.edu) under the Emory University Brain Tumor Sample and Information Resource (BTSIR; AM16 IRB00045732).
Ethics approval and consent to participate
Ethics approval and consent to participate was provided by the Emory University Institutional Review Board (IRB, 1599 Clifton Road, NE, 5th floor, Atlanta, GA 30322 United States; IRB@emory.edu) under the Emory University Brain Tumor Sample and Information Resource (BTSIR; AM16 IRB00045732).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lowder, L., Hauenstein, J., Woods, A. et al. Gliosarcoma: distinct molecular pathways and genomic alterations identified by DNA copy number/SNP microarray analysis. J Neurooncol 143, 381–392 (2019). https://doi.org/10.1007/s11060-019-03184-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-019-03184-1