
Abstract
Recent evidence suggests the Akt-mTOR pathway may play a role in development of low-grade gliomas (LGG). We sought to evaluate whether activation of this pathway correlates with survival in LGG by examining expression patterns of proteins within this pathway. Forty-five LGG tumor specimens from newly diagnosed patients were analyzed for methylation of the putative 5′-promoter region of PTEN using methylation-specific PCR as well as phosphorylation of S6 and PRAS40 and expression of PTEN protein using immunohistochemistry. Relationships between molecular markers and overall survival (OS) were assessed using Kaplan-Meier methods and exact log-rank test. Correlation between molecular markers was determined using the Mann-Whitney U and Spearman Rank Correlation tests. Eight of the 26 patients with methylated PTEN died, as compared to 1 of 19 without methylation. There was a trend towards statistical significance, with PTEN methylated patients having decreased survival (P = 0.128). Eight of 29 patients that expressed phospho-S6 died, whereas all 9 patients lacking p-S6 expression were alive at last follow-up. There was an inverse relationship between expression of phospho-S6 and survival (P = 0.029). There was a trend towards decreased survival in patients expressing phospho-PRAS40 (P = 0.077). Analyses of relationships between molecular markers demonstrated a statistically significant positive correlation between expression of p-S6(235) and p-PRAS40 (P = 0.04); expression of p-S6(240) correlated positively with PTEN methylation (P = 0.04) and negatively with PTEN expression (P = 0.03). Survival of LGG patients correlates with phosphorylation of S6 protein. This relationship supports the use of selective mTOR inhibitors in the treatment of low grade glioma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Low-grade gliomas (LGGs) are relatively slow growing neoplasms of the central nervous system (CNS) that comprise approximately 20% of all glial tumors. They can be divided into two broad categories based on their histological characteristics—Grade I and Grade II lesions. This paper will primarily focus on Grade II lesions, with astrocytomas being the predominant histological sub-type, constituting 35% of low-grade lesions and generally having a worse prognosis than the oligodendroglial or mixed variants.
The mainstay treatment for adult Grade II lesions, regardless of histology, has remained largely unchanged for decades, with surgical resection at presentation and radiation, often at progression. Recently, chemotherapy, namely temozolomide, has proven of some benefit in recurrent disease [1]. However, further therapeutic gain requires increased knowledge of the basic biology driving the tumor phenotype in these lesions.
Over the past few years, several key discoveries related to LGG biology have been made. Notably, in approximately 60% of astrocytomas, there is inactivation of the TP53 locus, while in over 80% of oligodendroglial lesions there is allelic loss of chromosome 1p and 19q [2, 3]. Both variants can also exhibit overexpression of the platelet derived growth factor (PDGF) and its receptor [4, 5]. 1p/19q status in oligodendrogliomas also appears to correlate with promoter methylation of the DNA repair enzyme O6-Methylguanine-DNA methyltransferase (MGMT); in patients with 1p/19q deletions, upwards of 85% have concomitant MGMT methylation. This is important from a prognostic standpoint as MGMT hypermethylated tumors tend to be more chemosensitive [6].
More recently, we found PTEN hypermethylation in upwards of 50% of Grade II astrocytomas, oligodendrogliomas, and mixed histologies. PTEN is a negative regulator of the phosphoinositide 3-kinase (PI3K) signal transduction pathway. Sitting downstream of PI3K is the mammalian target of rapamycin (mTOR), a 289-kDa serine/threonine kinase that phosphorylates a series of downstream effectors involved in protein biosynthesis, ribosome biogenesis, and the transcription of genes crucial to cell growth [7, 8]. Because of this pleiotropy, the mTOR pathway is frequently up-regulated in a variety of neoplasms and has been targeted for therapeutic intervention [9–11]. In fact, rapamycin has already been shown to have some antitumor activity in PTEN-deficient glioblastoma [12, 13]. Rapamycin, and its derivatives temsirolimus and everolimus inhibit the kinase activity of mTOR by binding to FK506 binding protein 12 (FKBP12), which then binds to the mTORC1 complex.
Of interest, PTEN hypermethylation was found in approximately 80% of the examined secondary glioblastomas, lesions that themselves result from the malignant transformation of lower-grade lesions. This enrichment for hypermethylation in the high grade recurrences suggested that this epigenetic modification and the resultant up-regulation of the PI3K-mTOR pathway might play a crucial role in the biology of LGGs and thus have a significant prognostic impact.
Towards that end, we decided to examine Grade II adult lesions in order to determine the relationship between overall survival (OS) and the methylation status and expression of PTEN and the phosphorylation of proteins in the PI3K-mTOR pathway, specifically S6 and PRAS40 [14, 15]. We hypothesized that those patients with PTEN promoter hypermethylation, phosphorylation of S6, or phosphorylation of PRAS40 (because any of these changes might result in increased flux through the PI3K-mTOR pathway) would have decreased overall survival. Results we present here show that, in univariate analyses, there is a statistically significant inverse relationship between OS and phosphorylation of S6. Additionally, PTEN promoter methylation, PTEN protein expression, and phosphorylation of PRAS40 show a weaker statistical correlation with OS. The up-regulation of PI3K-mTOR pathway components and their apparent relationship to survival suggest that agents that selectively inhibit this pathway may have a role in the treatment of Grade II gliomas.
Materials and methods
Patients and tissue samples
Frozen tissue and formalin-fixed, paraffin-embedded sections from forty-five LGG (grade II) tumor specimens from newly diagnosed patients were obtained from the University of California, San Francisco Brain Tumor Research Center tissue bank, under appropriate institutional review board approval. Female to male ratio was 21:24. Twenty-two patients had astrocytomas (49%), 16 oligodendrogliomas (35%), and seven mixed histology (16%). Median age at diagnosis was 37 years (range: 20–64). All ages are given at the time of surgery, which occurred at the University of California, San Francisco between 1990 and 2003.
DNA preparation and bisulfite treatment
Genomic DNA was isolated from approximately 25 mg wet weight of frozen tissue sample using QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s instructions and eluted twice in a total of 100 μl of elution buffer. This procedure yielded 5–40 μg of DNA. Bisulfite modification of genomic DNA was performed as described previously. Briefly, 1 μg purified DNA was diluted in 36 μl H2O, 4 μl 3.0 M NaOH was added, and DNA was denatured at 37°C for 15 min. The samples were then treated with 416 μl 3.6 M sodium bisulfite solution (pH 5.0) and 24 μl of 10 mM hydroquinone. All solutions were prepared fresh for each analysis. Samples were incubated at 55°C for 16 h. Two drops of mineral oil were layered on top of the solution to prevent evaporation. Bisulfite-modified DNA was purified with the Wizard DNA Clean-up System and vacuum manifold (Promega, Madison, WI, USA) according to the manufacturer’s manual. Freshly prepared NaOH solution was added to a final concentration of 0.3 M, and samples were incubated at 37°C for 15 min, followed by neutralization with ammonium acetate (pH 7.0; final concentration, 3.0 M) and ethanol precipitation. Normal human peripheral blood lymphocyte DNA samples, treated and untreated with DNA methylase (M. Sss I; New England BioLabs, Beverly, MA, USA), were also modified as positive and negative controls, respectively.
Methylation-specific PCR
To examine whether the PTEN promoter is methylated in glioma specimens, we used methylation-specific primers that had previously been used to demonstrate methylation of the PTEN promoter in a subset of non-small-cell lung cancer samples. These primers amplify a 181-base pair region of the PTEN promoter that starts 2,477 nucleotides upstream from the translation start site—methylated primers: forward, 5′-GTTTGGGGATTTTTTTTTCGC-3′; reverse, 5′-AACCCTTCCTACGCCGCG-3′; unmethylated primers: forward, 5′-TATTAGTTTGGGGATTTTTTTTTTGT-3′; reverse, 5′-CCCAACCCTTCCTACACCACA-3′. It should be noted that these primers do not amplify the highly homologous PTEN pseudogene located on chromosome 9p21, as these sequences lie outside the region of similarity.
The 25 μl PCR reaction contained 25 ng bisulfite DNA, 2% dimethylsulfoxide, 1.5 mM MgCl2, and 1 U Ampli Taq Gold DNA polymerase (Applied Biosystems, Foster City, CA, USA). PCR reactions were cycled in a Gene Amp 2700 thermocycler (Applied Biosystems) under the following conditions: preheat at 95°C for 10 min, 95°C for 30 s, 62°C for 30 s, 72°C for 30 s, 38 cycles, and a final extension at 72°C for 7 min. Aliquots (12 μl) of methylation-specific PCR (MSP) products were analyzed on 3% agarose gels, stained with ethidium bromide, and visualized under UV illumination. Results were recorded with a digital imaging system. For each PCR experiment, DNA from peripheral blood of normal blood donors treated with and without CpG methylase and bisulfite was included as positive and negative controls, respectively. We repeated MSP assays on all samples and found no discordant results among replicates. The MSP assay is sensitive to approximately 5% methylated product. To confirm the efficiency of the bisulfite modification and the specificity of MSP, bisulfite sequencing of the PCR products was carried out using the procedure reported previously.
Immunohistochemistry
All tissue was routinely fixed in either phosphate buffered 4% formalin or Zn-4% Formalin, dehydrated by graded ethanols and embedded in wax (Paraplast Plus, McCormick Scientific) using routine techniques. All sections were 5 μ on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA, USA). The following antibodies were obtained from commercial sources and used at following dilutions and incubation times/temperatures: 1) rabbit polyclonal phospho-S6 ribosomal protein (Ser235/236; Cell Signaling #2211) 1:200, 32 min/37°; 2) rabbit polyclonal phospho-S6 ribosomal protein (Ser240/244; Cell Signaling #2215) 1:200, 32 min/37°; 3) rabbit monoclonal PTEN (138G6; Cell Signaling #9559) at 1:50-1:100, 32 min/37°; 4) rabbit polyclonal phospho-PRAS40 (Invitrogen#441100G) 1:100, 32 min/37°; and 5) rabbit polyclonal MIB-1; [Ventana Medical Systems Anti-Ki-67(30-9)], 2μg/ml/32 min/37°. Antigen retrieval for phospho-S6 (both epitopes) and phospho-PRAS40 was performed for 30 min in Tris buffer pH 8 at 90°; for PTEN and MIB-1 retrieval time was 16 min. Following antigen-retrieval, sections were treated with 3% methanol-hydrogen peroxide at 22C° for 16 min for PTEN and MIB-1 and for 32 min for the phospho-PRAS40 and phospho-S6 epitopes. All immunohistochemistry assays were performed on the Ventana Medical Systems Benchmark XT using the Ultraview (multimer) detection system.
All slides were then scored. One score was assigned to each slide based upon the percentage of visible cells that stained positive. For the percentage staining positive, a score of 0 denoted no positive cells; a score of 1 meant that 1–33% of cells stained positive; 2 for 33–66% of cells; and 3 for >66% of cells. Immunohistochemistry scoring was performed in an entirely blinded fashion.
Statistics
All statistical analyses were performed using Statistica 6.0 (StatSoft, Tulsa, OK) and StatXact-8 (Cytel, Cambridge, MA). Overall survival was defined as the period between resection and death or date last known alive. Kaplan-Meier methods were used to estimate overall survival. All P values in the survival analysis were determined by the exact log–rank test and are two-tailed. Because of the small sample size and few events, only univariate analysis was possible. Correlative analysis between the biological variables investigated was done using the same software. P values were derived using the Mann-Whitney U test or Spearman’s rank test for correlation. Due to the exploratory nature of these analyses, no adjustment for multiple comparisons was made; statistical significance was defined as a P value <0.05 in all cases.
In analyzing IHC scores in relationship with survival, we considered the following approaches to divide the samples into two groups: (1) no (0) vs. positive (≥1) staining; or (2) scores of 0/1 vs. scores of 2/3 staining. We found that while the latter approach did not yield statistically significant relationships to OS (P = 0.104 for PTEN; P = 0.38 for p-S6 235/236; P = 0.39 for p-S6 240; P = 0.37 for p-PRAS40), we were able to establish a more robust relationship to OS with the former method of grouping. This discrepancy may be explained by the impact of small sample size on the sampling distributions. Nevertheless, there was generally a consistent survival trend using either approach.
Results
Table 1 summarizes patient baseline characteristics and length of follow-up among surviving patients, according to status of each molecular marker. At the time of analysis, nine patients had died. Median follow up for those still alive was 5.8 years (range: 0.01–11.7). Survival data was acquired for all 45 patients; however in a small number of patients there was not sufficient material to complete each and every molecular analysis.
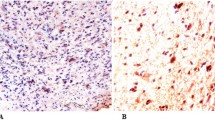
Table 2 presents the distribution of the IHC based assay results. Only a small fraction of samples scored three for all biological variables while the majority of the samples were scored 0 or 1. Figure 1 illustrates positive staining for each of the four immunohistochemical assays performed; each of the four panels in the figure displays a separate tumor that received a score of 3 for PTEN, phospho-PRAS40, phospho-S6 (Ser235/236), or phospho-S6 (240/244), respectively.

Examples of scores of 3 for each of the four immunohistochemical assays performed; each of the four panels in the figure displays a separate tumor that received a score of 3 for PTEN, phospho-PRAS40, phospho-S6 (Ser235/236), or phospho-S6 (240/244), respectively
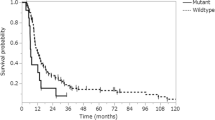
Figure 2 presents the Kaplan-Meier plots of survival with respect to each biological variable. For PTEN methylation, there was no statistically significant relationship between methylation status and overall-survival (OS) (P = 0.128). At the time of the analysis, 4 of the 10 patients with PTEN expression by IHC died, as compared to 3 of 23 patients who did not express PTEN. There was a trend towards a statistically significant difference in survival (P = 0.098) that favored those patients whose tumors did not express PTEN (Fig. 2). The estimated 5-year survival for patients with PTEN expression was 68%, as compared to 90% for those patients not expressing PTEN. Median follow-up for surviving patients who did not express PTEN was 5.76 years (0.01–11.69 years); for those surviving patients who were PTEN positive, median follow-up was 6.06 years (3.54–9.98 years).

Kaplan-Meier plots by molecular marker status. The vertical tick marks represent patients censored for survival at the time of analysis. P-values are based on the exact log-rank test
We assessed the phosphorylation status of two S6 epitopes: Ser-235/236 and Ser-240. Data on phosphorylation of Ser-235/36 was available on 38 patients (84%) and of those, 29 (76%) were phosphorylated. There was a statistically significant inverse relationship between OS and phosphorylation of the 235/236 epitope (P = 0.045) (Fig. 2); eight patients with phosphorylation at the 235/236 site died, whereas all patients without phosphorylation were alive at last follow-up. Median follow-up for surviving patients with phosphorylation was 5.3 years (0.10–10.98) versus 8.59 years (0.57–11.69) for those without. The estimated 5-year survival for patients with phosphorylation of S6 Ser-235/236 was 76%, as compared to 100% for patients without.
With respect to phosphorylation of the S6 Ser-240 epitope, again 84% of samples were included in the analysis, with 76% phosphorylated. There was an even more robust, statistically significant inverse relationship between phosphorylation of Ser-240 and OS (P = 0.029) (Fig. 2). Similar to the 235/236 epitope, there were eight deaths in the phosphorylated group and none in the negative group. Median follow-up for surviving patients with phosphorylation was 5.58 years (0.01–11.69) versus 7.19 years (0.57–11.5 years) for the negative group. The estimated 5-year survivals were the same as they were for the Ser-235/36 groupings. Since tumor histology may play an important role in evaluating the relationship between molecular variables and survival, we further examined the association between survival and phosphorylation of Ser-240 (the marker that was found to be most predictive of survival in our study) separately for astrocytomas (n = 22) and oligodendrogliomas (n = 16). Among these patients, 6/22 astrocytomas and 2/16 oligodendragliomas had died. All eight deaths demonstrated Ser-240 phosphorylation. Within each histologic subtype, we again found a consistent pattern of decreased survival among patients with Ser-240 phosphorylation even though the log-rank test did not reach statistical significance for either subgroup due to small event number (P = 0.137 and P = 0.301 for astrocytoma and oligodendraglioma, respectively).
We also assessed the relationship between phosphorylation of PRAS40 and OS in 35 patients. Eighteen of these patients (51%) had positive p-PRAS40 expression. There were six deaths in the positive group, and one in the negative group. There was a trend towards decreased survival for those patients whose tumors expressed any p-PRAS40 (P-value = 0.077). The 5-year survival for patients who expressed p-PRAS40 was 75 versus 94% for patients without expression. Median follow-up for survivors with and without p-PRAS40 was 5.8 years (0.01–10.98) and 5.8 years (0.57–11.69), respectively.
There was a positive correlation between p-S6 (235/36) staining and p-PRAS40 staining (P-value = 0.04); there was also a near perfect positive correlation between the staining for the two S6 epitopes (P ≤ 0.001). The expression of p-S6 (240) was correlated positively with PTEN methylation (P = 0.04) and negatively with PTEN-expression (P = 0.03).
In examining the baseline variables, there did not appear to be any severe imbalance in the distributions with respect to the biological variables (Table 1). However, in the variable found to be most predictive of survival based on univariate analysis (i.e. p-S6 Ser-240), a relatively higher percentage of phospho-negative patients had gross total resections. To confirm that the significant survival advantage we observed among patients without p-S6-240 phosphorylation was not a result of a larger fraction with gross total resection, we further assessed if p-S6-240 was predictive of survival in the subset of patients who only had subtotal resections. Among this subset of 23 patients, the trend towards decreased survival among those with p-S6-240 phosphorylation persisted (Fig. 2), even though the statistical test was no longer significant (P = 0.068). This may be due to the decease in sample size. It is also noteworthy that the eight patients with p-S6-240 phosphorylation who died all had had subtotal resections.
Discussion
We know that the mTOR pathway plays a crucial role in the tumorigenesis of a variety of solid malignancies. The pathway, when activated, amplifies protein transcription, intensifies cell growth, and halts apoptosis. More recent evidence implicates PTEN promoter methylation, and by extension mTOR activation, in the malignant progression of LGGs. We reasoned that if this association were clinically significant, mTOR activation in LGG patients would portend a worse prognosis. We therefore sought to demonstrate a relationship between activation of this pathway and survival in LGG by examining several key proteins in the pathway. Such an association would provide justification for the use mTOR inhibitors in the treatment of LGG.
PTEN is a tumor suppressor that inhibits flux through the mTOR pathway. We would expect patients with PTEN promoter hypermethylation to have increased mTOR activity and thus decreased OS. The relationship between promoter hypermethylation and OS demonstrated a statistical trend that did not reach significance. It is possible that a larger study will provide greater statistical power to demonstrate a true correlation between PTEN promoter hypermethylation and OS if one exists; however, such an endeavor would require a much greater multi-institutional effort that we hope to accomplish in the future. With regards to PTEN protein expression and contrary to what we know about PTEN’s tumor suppressive activities, we found those patients with expression had a trend towards decreased OS, although this association was not statistically significant. However, because of the challenges involved with the PTEN IHC assay, one must use caution in interpreting this particular result.
PRAS40 is a protein that, when phosphorylated, no longer acts to inhibit the mTOR pathway; S6 is a downstream target of the mTOR effector p70S6K that is activated by phosphorylation. In univariate analyses, we established a correlation between phosphorylation of these two proteins and OS in LGG; patients whose tumors had evidence of either PRAS40 or S6 phosphorylation had decreased OS. This effect is presumably mediated by the pro-tumorigenic consequences of mTOR signaling, of which these two proteins are participants. One would additionally assume that if the common mTOR pathway linking these proteins was activated, their phosphorylation would correlate. Indeed, we found significant positive correlations between S6 phosphorylation and both PRAS40 phosphorylation and PTEN methylation as well as a significant negative correlation between S6 phosphorylation and PTEN expression. Finally, the near-perfect correlation between phosphorylation at the two S6 epitopes provides good internal validation of our IHC protocol.
Because small patient numbers precluded multivariate analysis, possible confounding exists. The most likely confounders are differences in age, treatments used, and surgical outcomes between groups of patients whose tumors varied in their molecular markers. We found no statistically significant relationship between treatment with chemotherapy or radiation and OS. Therefore, it is unlikely that differences in the number of patients who had received either chemotherapy or radiation between groups with and without phosphorylation of S6 and PRAS40 and with and without expression of PTEN confounded our conclusions. Furthermore, there appeared to be no dramatic differences between median age and histological mix when comparing groups with different molecular markers.
However, again looking at the patient population as a whole, we did find a statistically significant relationship between extent of resection (EOR) and OS; those patients who had undergone gross-total resections of their tumors had longer survival. As mentioned, for the variable most predictive of survival (p-S6-Ser 240), we did find that the negative expression group did have a higher percentage of patients with gross-total resection. However, even by looking exclusively at the group of patients with subtotal resections, phospho-S6 expression still portended a worse overall survival, exhibiting a statistical trend nearing significance (P = 0.068).
Of note, inhibitors of mTOR have proven largely ineffectual in glioblastoma [16]. This may be due to greater genetic complexity and activation of redundant pathways in glioblastoma compared to lower grade gliomas. In addition, mTOR drives a feedback loop that normally keeps PI3K activity in check and mTOR inhibition can alleviate this negative feedback loop and result in activation of PI3K. Therefore, abrogation of this negative loop may impede anti-tumor efficacy of mTOR inhibitors [13].
Ultimately, larger cohorts that will likely require multi-institutional efforts, are needed to validate our findings. Our data support a novel role for activation of PI3K/mTOR signaling in LGGs, and provide a compelling rationale for the use of mTOR inhibitors in the treatment of LGG. Such studies are ongoing in adults and children and will greatly benefit from incorporation of molecular analyses that may predict clinical outcome and identify those patients most likely to benefit from targeted therapies.
References
Agulnik M, Mason WP (2006) The changing management of low-grade astrocytomas and oligodendrogliomas. Hematol Oncol Clin North Am 20:1249–1266
Rasheed BK, McLendon RE, Herndon JE et al (1994) Alterations of the TP53 gene in human gliomas. Cancer Res 54:1324–1330
Reifenberger J, Reifenberger G, Liu L et al (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175–1190
Varela M, Ranuncolo SM, Morand A et al (2004) EGF-R and PDGF-R, but not bcl-2, overexpression predict overall survival in patients with low-grade astrocytomas. J Surg Oncol 86:34–40
Maiuri F, Del Basso De Caro ML, Iaconetta G, Peca C, Esposito M, de Divitiis E (2006) Prognostic and survival-related factors in patients with well-differentiated oligodendrogliomas. Zentralbl Neurochir 67:204–209
Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G (2005) Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 113:379–385
Contreras AG, Dormond O, Edelbauer M et al (2008) mTOR-understanding the clinical effects. Transplant Proc 40:S9–S12
Geissler EK (2008) The impact of mTOR inhibitors on the development of malignancy. Transplant Proc 40:S32–S35
Bjornsti MA, Houghton PJ (2004) The TOR pathway: a target for cancer therapy. Nat Rev Cancer 4:335–348
Strimpakos AS, Karapanagiotou EM, Saif MW, Syrigos KN (2009) The role of mTOR in the management of solid tumors: an overview. Cancer Treat Rev 35:148–159
Xu G, Zhang W, Bertram P, Zheng XF, McLeod H (2004) Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR pathway in common human tumors. Int J Oncol 24:893–900
Neshat MS, Mellinghoff IK, Tran C et al (2001) Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA 98:10314–10319
Cloughesy TF, Yoshimoto K, Nghiemphu P et al (2008) Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med 5:e8
Volarevic S, Thomas G (2001) Role of S6 phosphorylation and S6 kinase in cell growth. Prog Nucleic Acid Res Mol Biol 65:101–127
Wang L, Harris TE, Lawrence JC (2008) Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J Biol Chem 283:15619–15627
Dancey JE (2006) MTOR and related pathways. Cancer Biol Ther 5(9):1065–1073
Acknowledgments
This research was supported in part by NIH-PO1 NS-42927-27A2 (D.A.H–K, K.R.L), NIH Brain Tumor SPORE grant P50 CA097257 (D.A.H–K, K.R.L), The Nancy and Stephen Grand Philanthropic Fund (D.A.H–K), CA 82103 (K.R.L., M.S.B), and the Doris Duke Charitable Foundation (S.M.M).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
McBride, S.M., Perez, D.A., Polley, MY. et al. Activation of PI3K/mTOR pathway occurs in most adult low-grade gliomas and predicts patient survival. J Neurooncol 97, 33–40 (2010). https://doi.org/10.1007/s11060-009-0004-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-009-0004-4