
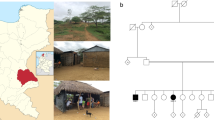
Ataxia with oculomotor apraxia (AOA) is a subgroup of the autosomal recessive ataxias with a characteristic oculomotor apraxia: difficulty in transferring and fixing gaze, inability to direct the eyes to a specified side, not due to muscle weakness. Types AOA1 (the APTX gene) and the relatively frequent AOA2 (the SETX gene) are known, along with the recently (2012) described AOA3 (the PIK3R5 gene). Oculomotor apraxia is also characteristic of Louis–Bar ataxia telangiectasia and its variants. DNA diagnosis of AOA1 and AOA2 has been initiated in the Laboratory of Inherited Metabolic Diseases, Medical Genetics Scientific Center. Clinically typical AOA2 in a patient aged 25 years is described – this is the first Russian case confirmed by DNA analysis. The SETX gene was found to contain a previously undescribed mutation, c.2623-2626 del 4, leading to a frameshift mutation, in the heterozygous state; detection of a single mutation with a corresponding clinical picture is sufficient to confirm the diagnosis; the search for the allelic mutation is under way. The diagnosis was made after seven years, though the inherited nature of the disease was proposed from the very beginning. AOA1 was also confirmed in a 15-year-old patient (not investigated clinically by ourselves): two new mutations were found in the compound heterozygotic state; diagnosis of this early form was also delayed. AOA and oculomotor apraxia, which requires specific detection, are probably underestimated in clinical practice. Timely detection and molecular genetic verification would permit effective medical genetic intervention.
Similar content being viewed by others


References
S. N. Illarioshkin, G. E. Rudenskaya, I. A. Ivanova-Smolenskaya, et al., Inherited Ataxias and Paraplegias [in Russian], MEDpress- Inform, Moscow (2006).
S. A. Klyushnikov, S. N. Illarioshkin, E. D. Markova, et al., “A family case of ataxia with oculomotor apraxia: first observation in the Russian population,” Ann. Klinich. Eksperim. Nevrol., No. 2, 34–39 (2007).
N. Al Tassan, D. Khalil, J. Shinwari, et al., “A missense mutation in PIK3R5 gene in a family with ataxia and oculomotor apraxia,” Hum. Mutat., 33, 351–354 (2012).
M. Anheim, “Autosomal recessive cerebellar ataxias,” Rev. Neurol., 167, 372–384 (2011).
M. Anheim, M. C. Fleury, J. Franques, et al., “Clinical and molecular findings of ataxia with oculomotor apraxia type 2 in 4 families,” Arch. Neurol., 65, 958–962 (2008).
M. Anheim, M. Fleury, B. Monga, et al., “Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management,” Neurogenetics, No. 11, 1–12 (2010).
M. Anheim, B. Monga, M. Fleury, et al., “Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients,” Brain, 132, 2688–2698 (2009).
L. Arning, L. Schöls, H. Cin, et al., “Identification and characterization of a large senataxin (SETX) gene duplication in ataxia with ocular apraxia type 2 (AOA2),” Neurogenetics, 9, 295–299 (2008).
A. Bassuk, Y. Chen, S. Batish, et al., “In cis autosomal dominant mutation of senataxin associated with tremor/ataxia syndrome,” Neurogenetics, 8, 45–49 (2007).
V. Bernard, M. Minnerop, K. Bürk, et al., “Exon deletions and intragenic insertions are not rare in ataxia with oculomotor apraxia 2,” BMC Med. Genet., 10, 87–91 (2009).
V. Bernard, S. Stricker, F. Kreuz, et al., “Ataxia with oculomotor apraxia type 2: novel mutations in six patients with juvenile age of onset and elevated serum alpha-fetoprotein,” Neuropediatrics, 39, 347–350 (2008).
S. Bohlega, J. Shinwari, L. Al Sharif, et al., “Clinical and molecular characterization of ataxia with oculomotor apraxia patients in Saudi Arabia,” BMC Med. Genet., 12, 27 (2011).
B. Castellotti, C. Mariotti, M. Rimoldi, et al., “Ataxia with oculomotor apraxia type 1 (OAO1): novel and recurrent aprataxin mutations, coenzyme Q10 analyses, and clinical findings in Italian patients,” Neurogenetics, 12, 193–201 (2011).
P. Coutinho and C. Barbot, “Ataxia with oculomotor apraxia type 1,” Gene Reviews, www.ncbi.nlm.nih.gov/books/NBK1456/.
Y. Chen, S. Hashemi, S. Anderson, et al., “Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease,” Neurobiol. Dis., 23, 97–108 (2006).
C. Criscuolo, L. Chessa, S. Di Giandomenico, et al., “Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study,” Neurology, 66, 1207–1210 (2006).
A. De Amicis, M. Piane, F. Ferrari, et al., “Role of senataxin in DNA damage and telomeric stability,” DNA Repair (Amst.), 10, 199–209 (2011).
A. Duquette, K. Roddier, J. McNabb-Baltar, et al., “Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy,” Ann. Neurol., 57, 408–414 (2005).
M. Fernet, M. Gribaa, and M. Salih, et al., “Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder,” Hum. Molec. Genet., 14, 307–318 (2005).
J. Gazulla, I. Benavente, I. López-Fraile, et al., “Sensorimotor neuronopathy in ataxia with oculomotor apraxia type 2,” Muscle Nerv., 40, 481–485 (2009).
B. H’mida, A. M’zahem, M. Assoum, et al., “Molecular diagnosis of known recessive ataxias by homozygosity mapping with SNP arrays,” J. Neurol., 258, 56–67 (2011).
Human Gene Mutation Database, Cardiff (NGMD), www.hgmd.cf.ac.uk.
I. Le Ber, N. N. Bouslam, S. Rivaud-Pechoux, et al., “Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients,” Brain, 127, 759–767 (2004).
D. Lynch, C. Braastad, and N. Nagan, “Ovarian failure in ataxia with oculomotor apraxia type 2,” Am. J. Med. Genet. A, 143, 1775–1777 (2007).
M. Moreira and M. Koenig, “Ataxia with oculomotor apraxia type 2,” Gene Reviews, www.ncbi.nlm.nih.gov/books/NBK1154/.
M. Moreira, S. Klur, M. Watanabe, et al., “Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2,” Nat. Genet., 36, 225–227 (2004).
A. H. Nemeth, E. Bochukova, E. Dunne, et al., “Autosomal recessive cerebellar ataxia with oculomotor apraxia (ataxia-telangiectasia-like syndrome) is linked to chromosome 9q34,” Am. J. Hum. Genet., 67, 1320–1326 (2000).
OMIM, www.ncbi.nlm.nih.gov.
L. Schöls, and R. Schüle, et al., “‘Pseudodominant inheritance’ of ataxia with ocular apraxia type 2 (OAO2),” J. Neurol., 255, 495–501 (2008).
A. Suraweera, Y. Lim, R. Woods, et al., “Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation,” Hum. Mol. Genet., 18, 3384–3396 (2009).
B. Swartz, M. Murmeister, J. Somers, et al., “A form of inherited cerebellar ataxia with saccadic intrusions, increased saccadic speed, sensory neuropathy, and myoclonus,” Ann. N.Y. Acad. Sci., 956, 441–444 (2002).
M. Tazir, L. Ali-Pacha, A. M’Zahem, et al., “Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients,” J. Neurol. Sci., 278, 77–81 (2009).
C. Vantaggiato, S. Bondioni, G. Airoldi, et al., “Senataxin modulates neurite growth through fibroblast growth factor 8 signalling,” Brain, 134, 1808–1828 (2011).
Washington University Neuromuscular Disease Database, http://neuromuscular.wustl.edu.
C. Zühlke, V. Bernard, and G. Gillessen-Kaesbach, “Investigation of recessive ataxia loci in patients with young age of onset,” Neuropediatrics, 38, 207–209 (2007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Zhurnal Nevrologii i Psikhiatrii imeni S. S. Korsakova, Vol. 112, No. 10, Iss. I, p. 58–63, October, 2012.
Rights and permissions
About this article
Cite this article
Rudenskaya, G.E., Kurkina, M.V. & Zakharova, E.Y. Ataxia with Oculomotor Apraxia: Clinical Genetic Characteristics and DNA Diagnosis. Neurosci Behav Physi 43, 1143–1149 (2013). https://doi.org/10.1007/s11055-013-9863-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11055-013-9863-4