
Abstract
In this paper, the preparation of calcium oxide (CaO) nanoparticles (NPs) is reported by a precipitation method, using CaCl2 and NaOH as starting raw materials. The produced NPs were characterized for chemical composition, phase composition, particle size distribution, morphological features, specific surface area, and crystallite sizes. It is shown that calcination of Ca(OH)2 in vacuum takes place faster/at a lower temperature compared to the calcination in air due to the higher entropy of the gaseous product of calcination. It is also shown that when these CaO nanoparticles are kept at room temperature in air, they fully and spontaneously transform into CaCO3 within 3 weeks. Therefore, if this material is disposed in open fields (not necessarily in industrial conditions), it is able to capture carbon dioxide from normal air slowly, but surely. However, when the CaO nanoparticles are kept in the air at 100–200 °C, they mostly capture water vapor from the air instead of carbon dioxide, and the resulting calcium hydroxide blocks the carbon dioxide capture by CaO nanoparticles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Calcium oxide (CaO) is an important inorganic compound. It can be used as a catalyst [1,2,3], pellet for CO2 capture and kinetic analysis [4, 5], toxic-waste remediation agent, or as an additive in refractory and paint industries [6], antimicrobial agent, a drug delivery agent, as well as in various other biomedical applications [7]. Calcium oxide has been regarded as one of the most promising candidates for carbon capture [8,9,10,11,12,13] due to its good kinetics and high capture capacity, low running cost [14], and even under low CO2 partial pressures. It is demonstrated even by the fact the CaO is unstable in air and will gradually convert back to CaCO3 when cooled back to room temperature [15]. Various methods such as sol–gel, thermal decomposition, hydrothermal technique, combustion method, co-precipitation technique, biogenic method, precipitation method, two-step thermal decomposition technique, one-step multi-component synthesis, and microwave synthesis [16,17,18,19,20,21,22,23,24,25,26,27,28,29] can be used for the preparation of CaO nanoparticles [30]. Varying these methods, all physical and chemical properties of nano-CaO can be changed; morphology, specific surface area, and capturing efficiency can be carefully controlled under specific synthesis conditions [31]. The calcination temperatures of CaO from Ca(OH)2 precursor are found in the range of 450–650 °C [32,33,34,35,36,37]. CaO can also be prepared from CaCO3 by decomposition above 900 °C [38, 39]. The adsorption capacity of CaO-based adsorbents decreases over several cycles of CaO carbonation/calcination [40,41,42,43]. To improve the sintering-resistant properties of CaO-based adsorbents, many factors should be considered, such as decreasing the particle size and increasing the surface area, dispersing CaO on an inert support, as well as surface modification [44, 45]. The CaO nanoparticles have comparatively high adsorption efficiency, but the capabilities of multiple carbonations and decarbonation cycles of raw CaO rapidly degrade. The CO2 capturing of CaO sorbent was done at high temperature between 550 and 800 °C [46, 47]. The grain size of CaO grows faster as the calcination temperature and heating time are increased [48]. The aim of this paper is to report on some novel results to produce CaO nanoparticles through precipitation method. Various analytical techniques are applied to characterize the product, and additionally, the results of carbon dioxide absorption experiments at low temperatures are presented.
Experimental
Materials and the synthesis method
Twenty milliliters of 1 M CaCl2 × H2O (≥ 95%, Sigma-Aldrich) and 20 ml of 2 M NaOH (≥ 98%, VWR Ltd.) were used as initial reagents for the synthesis of Ca(OH)2 powder. During synthesis, the reagents are used in stoichiometric Ca+2:OH− = 1:2 mol ratio. First, both aqueous solutions (CaCl2 and NaOH) were heated up to 80 °C. At this fixed temperature, a given NaOH solution was added dropwise to a given CaCl2 solution under stirring (1300 rpm) by magnetic stirrer for 30 min. The process takes place according to the following reaction:
The suspension obtained was washed five times with 120 ml of distilled water per occasion to remove NaCl from the suspension. The duration of each filtering process was 2.5 h. As a result, a white precipitate Ca(OH)2 occurred according to the process (left from/the solution passes the filter, while right from/the precipitate is captured by the filter):
The wet precipitate Ca(OH)2 was dried in air at room temperature over one night to collect a semi-dry precipitate Ca(OH)2. Of collected semi-dry precipitate of Ca(OH)2, 1.24 g was calcined in air at 650 °C for 1 h in the furnace with a heating rate of 15 °C/min. As a result, 0.92 g of calcium oxide (CaO) powder was produced by calcination while the dissociation product H2O was evaporated as shown in the following equation:
According to molar masses, from 1.24 g of dry Ca(OH)2, one expects to obtain about 0.939 g CaO. This is larger by 2.0% compared to the 0.92 g obtained in reality, meaning that our semi-dry Ca(OH)2 contained about 2 w% of water. In Table 1, all our samples used in this paper are collected.
Characterization techniques
XRD investigations were performed using two instruments: both Cu K-alpha source, 40 kV and 40 mA generator settings, and parallel beam with Göbel mirror. The obtained CaO powder calcined in air was measured on Bruker D8 (Vantec-PSD with 1° opening, scanning rate 0.007°(2Th)/24 s). The wet Ca(OH)2 precipitate was in situ calcined between 25 and 650 °C, recording at each 50 °C in a Anton Paar HTK 1200 N heating chamber with 60 °C/min heating rate, 0.01 mbar when vacuum applied mounted on Bruker D8 Discover Lynx Eye XE-T solid-state ED detector in 0 D mode, and scanning rate 0.014°(2Th)/24 s. The SEM and EDS investigations are performed with a Thermo Fisher Helios G4 PFIB SEM equipped with an EDAX Octane ElectPlus detector. The average particle sizes of the samples are measured from the SEM images by “ImageJ” software using the mode of threshold adjustment. The TEM measurements were done by Fei Tecnai G2 20 XT win equipment with tungsten cathode and an acceleration voltage of 200 kV. The specific surface area of the CaO was examined by Brunauer–Emmett–Teller method (BET, Micrometrics TriStar 3000). The CO2 adsorption capacity was determined through the measurement of the mass increase at different temperatures (25, 100, 200 °C) in air as a function of time.
Results and discussion
XRD results
Figure 1 shows a series of XRD diffractograms of sample 1. It follows that the initial wet precipitate at 25 °C contains mostly Ca(OH)2 with very few amounts of CaCO3. The peaks for Ca(OH)2 disappeared in the temperature range between 250 and 300 °C; instead, new peaks of CaO appeared and remained stable till the maximum measured temperature of 650 °C. The small peak of CaCO3 disappeared in the temperature range between 500 and 550 °C. Similar results of sample 2 are shown in Fig. 2 for the case when the in situ calcination in the XRD equipment was performed in air. Compared to Fig. 1, calcination in air (instead of vacuum) leads to prolonged stability by about 150 °C for both initial compounds: from 300 to 450 °C for Ca(OH)2 and from 500 to 650 °C for CaCO3 (for the summary of data of Figs. 1 and 2, see Table 2). The difference between samples 1–2 is due to the high entropy of the gaseous reaction products (H2O and CO2) that drive the dissociation reaction in vacuum further and faster compared to the case when calcination is performed in air.

The XRD diffractograms of sample 1 during its calcination steps under vacuum in the temperature interval of 25–650 °C

The XRD diffractograms of sample 2 during its calcination steps under air in the temperature interval of 25–650 °C
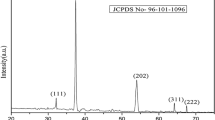
Figure 3 shows the temperature dependence of the crystallite sizes of CaO nanoparticles as function of calcination temperature in vacuum (sample 1) and in air (sample 2) using XRD technique. The crystal sizes of CaO calcined under vacuum goes through a maximum at 550 °C. Around this temperature, the CaO particles undergo recrystallization by eliminating lattice defects and dislocations. The same maximum is not present when CaO was obtained by calcination in air. Also, the final particle size obtained after calcination at 650 °C is lower when calcination is performed in air. That is why the samples obtained in this way are used here for further analysis. The XRD diffractogram of the corresponding sample 3 is shown after its calcination in Fig. 4. This figure confirms the formation of only one phase, CaO, being in agreement with Figs. 1 and 2. As follows from Fig. 4, the size of these crystals is below 100 nm, so they are truly nanoparticles [49].

The crystallite size of CaO as function of calcination temperature of samples 1 and 2. According to Table 2, the dotted line obtained in vacuum starts from 200 °C, while the bold line obtained in air starts from 450 °C. At 450 °C and at 600 °C, two values coincide

The XRD diffractogram of sample 3: dry Ca(OH)2 precipitate calcined in air at 650 °C with heating rate of 15 °C/min. Only nano-CaO is found
SEM, EDS, and TEM investigations of the CaO particles
Figure 5 shows SEM and EDS examination of samples 3–4. The SEM images prove that the CaO particles have flake shape morphology. The particle sizes found from the SEM images are too large (115–255 nm), proving that the particles are agglomerated. The EDS investigation confirmed the presence of Ca and O (a small amount of C comes from the sample holder). Figure 5b shows the SEM image of CaO (sample 4) after the sample was kept in normal air at room temperature for 3 weeks. As follows from the comparison of Fig. 5a, b, the C and O contents of CaO increased significantly proving that it adsorbed CO2 with the formation of CaCO3 (for details see below).

SEM image (left) and EDS spectrum (right) of fresh CaO (sample 3; a) and for the sample kept in air at room temperature for 3 weeks (sample 4; b). The peaks for gold in EDS spectra correspond to the gold coating added during sample preparation
TEM images of different CaO particles (sample 3) are shown in Fig. 6. Both single particles with their sizes below 100 nm and agglomerated particles with their sizes around 500 nm are present. The comparison of Figs. 5 and 6 proves that the particles of Fig. 5 are agglomerated particles. The comparison of Fig. 3 with Fig. 6 proves that the sizes for particles equaling crystallites obtained by XRD and TEM are similar; thus, the CaO nanoparticles were successfully produced. According to SEM and TEM images, this sorbent did not exhibit a “fluffy” structure, which probably contributes to its high surface area and large pore volume enhancing the uptake capacity and reversibility of CaO sorbent.

TEM images of fresh CaO (sample 3) after the calcination of dry Ca(OH)2 precipitate in air at 650 °C for 1 h: single particles (left) and agglomerated particles (right)
Absorption measurements
The CaO nanoparticles described above (samples 4–6) were used for absorption measurements. Samples 4–6 were exposed to air at different temperatures (25, 100, and 200 °C) for three weeks (= 504 h). The time dependences of the mass increase of the three CaO samples are shown in Fig. 7. Table 3 shows the initial and final masses of the samples kept at different temperatures. As follows from Fig. 7, the initial mass increase is much faster compared to the same at longer times. Moreover, after about 450 h, all samples reach saturation, i.e., their masses do not increase any more with further holding time. The half-time of curves in Fig. 7 decreases with increasing temperature, i.e., the relative rate of the process increases with temperature. More importantly, the relative mass increase of the samples decreases with increasing temperature.

The time dependence of the relative mass change of CaO samples 4–6 exposed to normal air at different temperatures
The expected primary reaction of the air components with CaO nanoparticles is the capture of carbon dioxide from air: \({\mathrm{CaO}}_{(\mathrm s)}+{\mathrm{CO}}_{2(\mathrm g)}={\mathrm{CaCO}}_{3(\mathrm s)}\). This reaction is accompanied by the following standard Gibbs energy changes in kilojoules per mole (at different temperatures given in °C): − 129.9 (25), − 117.8 (100), and − 101.6 (200) [50]. Indeed, when CaO was kept for 3 weeks at 25 °C, it was fully transformed into the calcite form of CaCO3 (see also Fig. 8). The theoretical ratio of the molar masses of CaCO3 to CaO equals 1.78. This ratio is practically the same as found at the end of the experiment performed at 25 °C (see Fig. 7 and Table 3), further confirming that at room temperature, CaO nanoparticles are able to capture carbon dioxide from air and the full chemical capacity of CaO is used for this process within 3 weeks. Note that although the 3 weeks seems a long time, this process does not need industrial conditions: The CaO nano-particles deposited on field will slowly but surely absorb CO2 from surrounding air.

The XRD patterns of the CaO nanoparticles (samples 4–6) after being kept in air at 25, 100, and 200 °C for 3 weeks
In contrary to the results found at room temperature, the same CaO sample also captured some moisture from air when it was kept in air at 100–200 °C during 3 weeks (see Fig. 8). These samples transformed into the mixture of two different phases: Ca(OH)2 and CaCO3. The calcite form of CaCO3 was formed at both temperatures, but at 100 °C, CaCO3 was found partly as aragonite. The sample kept for 3 weeks at 100 °C is composed of 74 w% of Ca(OH)2 and 26 w% of CaCO3; the latter divided into 17 w% of calcite and 9 w% of aragonite. The sample kept for 3 weeks at 200 °C is composed of 60 w% of Ca(OH)2 and 40 w% of CaCO3 in the form of calcite. This is in agreement with Fig. 2 and Table 2, where the mixture of (Ca(OH)2 + CaCO3) is reported at temperatures below 400 °C when Ca(OH)2 precipitate is calcined in air.
Based on the above, at 100–200 °C, the following additional reaction also took place: \({\mathrm{CaO}}_{\mathrm{s}}+{\mathrm{H}}_{2}{\mathrm{O}}_{(\mathrm{g})}=\mathrm{Ca}{(\mathrm{OH})}_{2(\mathrm{s})}\). This side reaction is accompanied by the following standard Gibbs energy changes in kilojoules per mole (at different temperatures given in °C): − 66.3 (25), − 55.8 (100), and − 41.9 (200) [50]. Although these standard Gibbs energy changes are less negative compared to those accompanying the formation of CaCO3, nevertheless these values are also sufficiently negative to allow the formation of Ca(OH)2 in addition to CaCO3. The fact that Ca(OH)2 is formed at 100 and 200 °C but not at 25 °C cannot be explained solely by thermodynamic reasons. It is probably due to kinetic reasons, namely to the lower activation energy of Ca(OH)2 formation compared to that of CaCO3 formation. In summary, with increasing temperature, thermodynamics prefers the formation of CaCO3, while kinetics prefers the formation of Ca(OH)2. The interplay between these two opposite trends might be the reason for the observed values. Certainly, further experiments are needed to clarify the temperature dependence in more details.
The ratio of the molar masses of Ca(OH)2 to CaO equals 1.32, and this ratio is considerably lower than 1.78 valid for CaCO3. This, together with Fig. 8, explains in Fig. 7 why the final masses of CaO samples kept at 100–200 °C are considerably lower compared to that of the sample kept at 25 °C. The final masses of the CaO samples can be estimated by the following equation:
where \(\mathrm{C}_{\mathrm{CaCO}3}\) (w%) and \(\mathrm{C}_{\mathrm{Ca}(\mathrm{OH})2}\) (w%) are the CaCO3 and Ca(OH)2 contents (in mass percent) of the final sample. Substituting the above values and the values given in Table 3, the following results are obtained:
-
i.
At 25 °C: \({\mathrm C}_{\mathrm{CaCO}3}\) = 100% and \({\mathrm C}_{\mathrm{Ca}(\mathrm{OH})2}\) = 0%: \({\mathrm m}_{\mathrm{final}}\) = 1.55 g, deviating less than 1% from the measured value of 1.54 g (see Table 3),
-
ii.
At 100 °C: \({\mathrm C}_{\mathrm{CaCO}3}\) = 26% and \({\mathrm C}_{\mathrm{Ca}(\mathrm{OH})2}\) = 74%: \({\mathrm m}_{\mathrm{final}}\) = 1.08 g, being larger by 4.8% compared to the measured value of 1.03 g (see Table 3),
-
iii.
At 200 °C: \({\mathrm C}_{\mathrm{CaCO}3}\) = 40% and \({\mathrm C}_{\mathrm{Ca}(\mathrm{OH})2}\) = 60%: \({\mathrm m}_{\mathrm{final}}\) = 1.32 g, being larger by 21% compared to the measured value of 1.09 g (see Table 3).
These results indicate that the formation of Ca(OH)2 kinetically blocks the formation of CaCO3, and this mechanism is enhanced with increasing temperature at least up to 200 °C. This can be explained by the large difference between molar volumes of CaO (16.8 cm3/mol) and Ca(OH)2 (33.5 cm3/mol). The fact that Eq. (1) predicts higher mass increase than experimentally observed indicates that some residual, unreacted CaO should be present in the samples, kinetically blocked by Ca(OH)2. However, no peak of CaO is visible in Fig. 8. This might be explained by the nano-size of the blocked, un-reacted islands of CaO, which might be not visible by XRD. From the measured values, the remaining concentration of CaO can be found extending Eq. (1) as follows:
From Eq. (2):
Substituting the above values into Eq. (3):
-
i.
At 25 °C: \({\mathrm m}_{\mathrm{final}}/{\mathrm m}_{\mathrm{initial}}\) = 1.77, \({\mathrm C}_{\mathrm{CaCO}3}\) = 100% and \({\mathrm C}_{\mathrm{Ca}(\mathrm{OH})2}\) = 0%: \({\mathrm C}_{\mathrm{CaO}}\) = 1.3%,
-
ii.
At 100 °C: \({\mathrm m}_{\mathrm{final}}/{\mathrm m}_{\mathrm{initial}}\) = 1.37, \({\mathrm C}_{\mathrm{CaCO}3}\) = 26% and \({\mathrm C}_{\mathrm{Ca}(\mathrm{OH})2}\) = 74%: \({\mathrm C}_{\mathrm{CaO}}\) = 15.8%,
-
iii.
At 200 °C: \({\mathrm m}_{\mathrm{final}}/{\mathrm m}_{\mathrm{initial}}\) = 1.24, \({\mathrm C}_{\mathrm{CaCO}3}\) = 40% and \({\mathrm C}_{\mathrm{Ca}(\mathrm{OH})2}\) = 60%: \({\mathrm C}_{\mathrm{CaO}}\) = 52.4%.
As follows from these results, the higher is the temperature, the higher is the ratio of blocked and unreacted CaO, at least until 200 °C. Certainly, further experiments are needed to clarify this question.
Finally, it is suggested here to perform capturing of carbon dioxide by CaO nanoparticles produced here at room temperature, as at higher temperature (at least up to 200 °C), the efficiency of this process is considerably decreased due to the partial formation of Ca(OH)2. Even if the rate of CO2 capturing is lower at room temperature, it does not require any industrial equipment: When CaO nano-particles are disposed on a field, they will capture the stoichiometric amount of carbon dioxide slowly, but surely.
The specific surface area of the CaO samples
Freshly prepared pure CaO sample 3 (obtained by calcination in air at 650 °C for 1 h) was found to have the measured specific surface area of 4.34 m2/g (Table 4). After keeping the same sample at room temperature in air for 3 weeks (sample 4), its specific surface area is increased to 6.05 m2/g (Fig. 4). This difference looks quite large, about 39%. As follows from the above, this is due to the absorption of CO2 by CaO nanoparticles and by the transformation of the latter into CaCO3. Now, let us re-calculate these measured values to somewhat more scientific values expressed in m2/m3 = 1/m. Using the density of 3.34 g/cm3 of CaO [51], the initial specific surface area of 4.34 m2/g is recalculated to 1.45 E7 1/m. On the other hand, using the density of 2.71 g/cm3 of CaCO3 in the calcite form [50], the final specific surface area of 6.05 m2/g is recalculated to 1.64 E7 1/m. Thus, the difference between the initial and final values is reduced to 13%. Modeling the particles in the first approximation as cubes with their specific surface area expressed as 6/a (with a = the side length of a cube), the cube side is found to decrease during the carbonization process from about 414 nm to about 359 nm. It means that the specific surface area measurements correspond to the agglomerated particles, as explained above in relation with Fig. 6.
As follows from Table 4, the specific surface areas of pores (BJH) are increased from 3.17 m2/g for freshly prepared CaO sample 3 to 5.17 m2/g after 3 weeks explosion to air at room temperature (sample 4). The cumulative specific pore volumes are increased from 0.016 m3/g of the freshly prepared CaO sample to 0.031 m3/g after it is kept in air during 3 weeks at room temperature. The average pore diameters are increased from 11 nm for fresh CaO to 17 nm after it is kept for 3 weeks in fresh air at room temperature.
Conclusions
Ca(OH)2 precipitates were prepared from the reaction of aqueous solutions containing dissolved CaCl2 and NaOH. After the dissolved NaCl is washed out and the Ca(OH)2 precipitates are filtered and dried, they were calcined to form CaO nano-particles. It was found that calcination in vacuum leads to the formation of CaO nanoparticles at a somewhat lower temperature compared to the same calcination performed in air, due to high entropy of low-pressure gaseous products. However, applying vacuum during calcination also leads to a larger CaO grain size compared to calcination in air. That is why calcination in air is recommended here to obtain CaO nano-particles of smaller size and larger specific surface area.
It is shown that when these CaO nanoparticles are kept at room temperature in normal air, they fully and spontaneously transform into CaCO3 within 3 weeks, i.e., they are able to capture carbon dioxide from air. This process does not require any industrial condition; it rather happens spontaneously if the CaO nano-particles are disposed to open fields.
However, when the CaO nanoparticles are kept in air at 100–200 °C, then they mostly capture water vapor from air instead of carbon dioxide, and calcium hydroxide forming as a result blocks the carbon dioxide capture by CaO nanoparticles.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Bharti P, Singh B et al (2019) Process optimization of biodiesel production catalyzed by CaO nanocatalyst using response surface methodology. J Nanostruc Chem 9(4):269–280. https://doi.org/10.1007/s40097-019-00317-w
Banković-Ilić MMR et al (2017) Application of nano CaO–based catalysts in biodiesel synthesis. Renew Sustain Energy Rev 72:746–760. https://doi.org/10.1016/j.rser.2017.01.076
Colombo K, Ender L et al (2017) The study of biodiesel production using CaO as a heterogeneous catalytic reaction. Egypt J Pet 26(2):341–349. https://doi.org/10.1016/j.ejpe.2016.05.006
Zhang Y, Gong X et al (2018) Performance of synthetic CaO-based sorbent pellets for CO2 capture and kinetic analysis. Fuel 232:205–214. https://doi.org/10.1016/j.fuel.2018.05.143
Sun J, Liu W et al (2017) Stabilized CO2 capture performance of extruded–spheronized CaO-based pellets by microalgae templating. Proc Combust Inst 36(3):3977–3984. https://doi.org/10.1016/j.proci.2016.09.001
Arul E, Raja K et al (2018) Bio-directed synthesis of calcium oxide (CaO) nanoparticles extracted from limestone using honey. J Nanosci Nanotechnol 18(8):5790–5793. https://doi.org/10.1166/jnn.2018.15386
Abraham S, Sarathy VP (2018) Biomedical applications of calcium oxide nanoparticles - a spectroscopic study. Int J Pharm Sci Rev Res 49(1):121–125
Benedetti A, Llavsky J et al (2019) Analysis of textural properties of CaO-based CO2 sorbents by ex situ USAXS. Chem Eng J 355:760–776. https://doi.org/10.1016/j.cej.2018.07.164
Ma X, Li Y et al (2019) Preparation of a morph-genetic CaO-based sorbent using paper fiber as a biotemplate for enhanced CO2 capture. Chem Eng J 361:235–244. https://doi.org/10.1016/j.cej.2018.12.061
Zhao B, Ma L et al (2018) Calcium precursor from stirring processes at room temperature for controllable preparation of nano-structure CaO sorbents for high-temperature CO2 adsorption. J CO2 Util 25:315–322. https://doi.org/10.1016/j.jcou.2018.04.012
Granados-Pichardo A, Granados-Correa F et al (2020) New CaO-based adsorbents prepared by solution combustion and high-energy ball-milling processes for CO2 adsorption: textural and structural influences. Arab J Chem 13(1):171–183. https://doi.org/10.1016/j.arabjc.2017.03.005
Donat F, Müller CR (2018) A critical assessment of the testing conditions of CaO-based CO2 sorbents. Chem Eng J 336:544–549. https://doi.org/10.1016/j.cej.2017.12.050
Azimi B, Tahmasebpoor M et al (2019) Multicycle CO2 capture activity and finitizability of Al-based synthesized CaO sorbents. Chem Eng J 358:679–690. https://doi.org/10.1016/j.cej.2018.10.061
Chen H, Khalili N et al (2018) Development of stabilized Ca-based CO2 sorbents supported by fly ash. Chem Eng J 345:312–319. https://doi.org/10.1016/j.cej.2018.03.162
Liu W, An H et al (2012) Performance enhancement of calcium oxide sorbents for cyclic CO2 capture—a review. Energy Fuels 26(5):2751–2767. https://doi.org/10.1021/ef300220x
Gupta J, Agarwal, (2016) Preparation and characterization of CaO nanoparticle for biodiesel production. AIP Conf Proc 1724(1):020066. https://doi.org/10.1063/1.4945186
Sun H, Wu C et al (2018) Progress in the development and application of CaO-based adsorbents for CO2 capture—a review. Mater Today Sustain 1:1–27. https://doi.org/10.1016/j.mtsust.2018.08.001
Roy A, Bhattacharya J (2011) Microwave assisted synthesis of CaO nanoparticles and use in waste water treatment. Nano Technol 3:565–568. https://doi.org/10.1142/S0219581X11008150
Mohadi R, Anggraini K et al (2016) Preparation calcium oxide from chicken eggshells. Sriwijaya J Environ 1(2):32–35. https://doi.org/10.22135/sje.2016.1.2.32-35
El-Dafrawy SM, Youssef HM et al (2015) Synthesis of nano-CaO particles and its application for the removal of copper (II), lead (II), cadmium (II) and iron (III) from aqueous solutions. Egypt J Chem 58(6):579–589
Tang ZX, Yu Z et al (2013) Sonication-assisted preparation of CaO nanoparticles for antibacterial agents. Quim Nova 36(7):933–936. https://doi.org/10.1590/S0100-40422013000700002
Sadeghi M, Husseinin MH et al (2013) A novel method for the synthesis of CaO nanoparticle for the decomposition of sulfurous pollutant. J Appl Chem Res 7(4):39–49
Tang ZX, Claveau D et al (2008) Preparation of nano-CaO using thermal-decomposition method. Mater Lett 62(14):2096–2098. https://doi.org/10.1016/j.matlet.2007.11.053
Szalai AJ, Manivannan N et al (2019) Super-paramagnetic magnetite nanoparticles obtained by different synthesis and separation methods stabilized by biocompatible coatings. Colloids Surf, A 568:113–122. https://doi.org/10.1016/j.colsurfa.2019.02.006
Habte L, Shiferaw N et al (2019) Synthesis of nano-calcium oxide from waste eggshell by sol-gel method. Sustainability 11(11):3196. https://doi.org/10.3390/su11113196
Aseel MA, Itab FH et al (2018) Producing high purity of metal oxide nano structural using simple chemical method. J Phys: Conf Ser 1032(1):012036. https://doi.org/10.1088/1742-6596/1032/1/012036
Jamrunroj P, Wongsakulphasatch S et al (2019) Surfactant assisted CaO-based sorbent synthesis and their application to high-temperature CO2 capture. Powder Technol 344:208–221. https://doi.org/10.1016/j.powtec.2018.12.011
Saket J, Ravikant S et al (2018) Bacterial toxicological assay of calcium oxide nanoparticles against some plant growth promoting rhizobacteria. Int J Res Appl Sci Eng Technol 6(11):460–466
Salaudeen SA, Acharya B et al (2018) CaO-based CO2 sorbents: a review on screening, enhancement, cyclic stability, regeneration and kinetics modelling. J CO2 Util 23:179–199. https://doi.org/10.1016/j.jcou.2017.11.012
Ramli M, Rossani RB et al (2019) Nanoparticle fabrication of calcium oxide (CaO) mediated by the extract of red dragon fruit peels (Hylocereus polyrhizus) and its application as inorganic–anti-microorganism materials. IOP Conf Ser: Mater Sci Eng 509(1):012090. https://doi.org/10.1088/1757-899X/509/1/012090
Kulkarni D, Wachs IE (2002) Isopropanol oxidation by pure metal oxide catalysts: number of active surface sites and turnover frequencies. Appl Catal A 237(1–2):121–137. https://doi.org/10.1016/S0926-860X(02)00325-3
Mirghiasi Z, Bakhtiari F et al (2014) Preparation and characterization of CaO nanoparticles from Ca(OH)2 by direct thermal decomposition method. J Ind Eng Chem 20(1):113–117. https://doi.org/10.1016/j.jiec.2013.04.018
Karami D, Mahinpey N et al (2012) Highly active CaO-based sorbents for CO2 capture using the precipitation method: preparation and characterization of the sorbent powder. Ind Eng Chem Res 51(12):4567–4572. https://doi.org/10.1021/ie2024257
Yang J, Ma L et al (2019) Thermodynamics and kinetics analysis of Ca-looping for CO2 capture: application of carbide slag. Fuel 242:1–11. https://doi.org/10.1016/j.fuel.2019.01.018
Imtiaz A, Farrukh MA et al (2013) Micelle-assisted synthesis of Al2O3·CaO nanocatalyst: optical properties and their applications in photodegradation of 2,4,6-trinitrophenol. Sci World J. https://doi.org/10.1155/2013/641420
Butt AR, Ejaz S et al (2015) CaO Nanoparticles as a potential drug delivery agent for biomedical applications. Dig J Nanomater Biostruct (DJNB) 10(3):799–809
Khine EE, Baumli P et al (2020) Preparation of calcium oxide by a precipitation method. Mater Sci Eng 45(1):182–190. https://doi.org/10.32974/mse.2020.018
Nimmas T, Jamrunroj P et al (2019) Influence of CaO precursor on CO2 capture performance and sorption-enhanced steam ethanol reforming. Int J Hydrogen Energy 44(37):20649–20662. https://doi.org/10.1016/j.ijhydene.2018.07.095
Dieter H, Hawthorne C et al (2013) Progress in calcium looping post combustion CO2 capture: successful pilot scale demonstration. Energy Procedia 37:48–56. https://doi.org/10.1016/j.egypro.2013.05.084
López JM, Grasa G et al (2018) Evaluation of the effect of inert support on the carbonation reaction of synthetic CaO-based CO2 sorbents. Chem Eng J 350:559–572. https://doi.org/10.1016/j.cej.2018.05.014
Diego ME, Arias B et al (2017) Evolution of the CO2 carrying capacity of CaO particles in a large calcium looping pilot plant. Int J Greenhouse Gas Control 62:69–75. https://doi.org/10.1016/j.ijggc.2017.04.005
Bazaikin YV, Malkovich EG et al (2019) Evolution of sorptive and textural properties of CaO-based sorbents during repetitive sorption/regeneration cycles: part II. Modeling of sorbent sintering during initial cycles. Chem Eng Sci 199:156–163. https://doi.org/10.1016/j.ces.2018.12.065
Cai J, Wang S et al (2017) Modeling of carbonation reaction for CaO-based limestone with CO2 in multitudinous calcination-carbonation cycles. Int J Hydrogen Energy 42(31):19744–19754. https://doi.org/10.1016/j.ijhydene.2017.06.173
Wang J, Huang L et al (2014) Recent advances in solid sorbents for CO2 capture and new development trends. Energy Environ Sci 7(11):3478–34518. https://doi.org/10.1039/b000000x
Sun H, Wang J et al (2019) Fundamental studies of carbon capture using CaO-based materials. J Mater Chem A 7(16):9977–9987. https://doi.org/10.1039/C8TA10472G
Lu H, Ettireddy PR et al (2006) Calcium oxide-based sorbents for capture of carbon dioxide at high temperatures. Ind Eng Chem Res 45(11):3944–3949. https://doi.org/10.1021/ie051325x
Naeem MA, Armutlulu A et al (2018) Optimization of the structural characteristics of CaO and its effective stabilization yield high-capacity CO2 sorbents. Nat Commun 9(1):1–11
Zhu Y, Wu S et al (2011) Nano CaO grain characteristics and growth model under calcination. Chem Eng J 175:512–518. https://doi.org/10.1016/j.cej.2011.09.084
Kaptay G (2018) On the size dependence of molar and specific properties of independent nano-phases and those in contact with other phases. J Mater Eng Perform 27:5023–5029. https://doi.org/10.1007/s11665-018-3411-x
Barin I, Platzki G (1993) Thermochemical data of pure substances. ISBN 3–527–28531–8(VCH, Weinheim), ISBN 1–56081–717–8(VCH, New York). https://doi.org/10.1002/9783527619825
Lide DR, editor (2004) CRC handbook of chemistry and physics, June 29. CRC Press, pp 1993–1994
Funding
Open access funding provided by University of Miskolc. This study was carried out as part of the GINOP-2.3.2- 15–2016-00027 “Sustainable operation of the workshop of excellence for the research and development of crystalline and amorphous nanostructured materials” project implemented in the framework of the Szechenyi 2020 program. The realization of this project was supported by the European Union.
Author information
Authors and Affiliations
Contributions
EEK: experiments and writing original draft, DKH: SEM/EDS, FK: XRD and review/editing, TF: specific surface area, G. Karacs: TEM, PB: supervision, GK: conceptualization, review/editing, corresponding.
Corresponding author
Ethics declarations
Ethics approval
The authors declare that the current research was conducted in agreement with all existing ethical standards.
Consent to participate
All authors have their consent to participate in this research.
Consent for publication
All authors have their consent to participate in this publication as it is presented here.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khine, E.E., Koncz-Horvath, D., Kristaly, F. et al. Synthesis and characterization of calcium oxide nanoparticles for CO2 capture. J Nanopart Res 24, 139 (2022). https://doi.org/10.1007/s11051-022-05518-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-022-05518-z