
Abstract
Mechanisms for core formation in differentiated bodies in the early solar system are poorly constrained. At temperatures below those required to extensively melt planetesimals, core formation could have proceeded via percolation of metallic liquids. Although there is some geochemical data to support such ‘low-temperature’ segregation, experimental studies and simulations suggest that percolation-driven segregation might have only contributed to core formation in a proportion of fully-differentiated bodies. Here, the effects low-temperature core-formation on elemental compositions of planetesimal cores and mantles are explored. Immiscibility of Fe-rich and FeS-rich liquids will occur in all core-formation models, including those involving large fraction silicate melting. Light element content of cores (Si, O, C, P, S) depends on conditions under which Fe-rich and FeS-rich liquids segregated, especially pressure and oxygen fugacity. The S contents of FeS-rich liquids significantly exceed eutectic compositions in Fe–Ni–S systems and cannot be reconciled with S-contents of parent bodies to magmatic iron meteorites. Furthermore, there is limited data on trace element partitioning between FeS-rich and Fe-rich phases, and solid/melt partitioning models cannot be readily applied to FeS-rich liquids. Interaction of metallic liquids with minor phases stable up to low fraction silicate melting could provide a means for determining the extent of silicate melting prior to initiation of core-formation. However, element partitioning in most core-formation models remains poorly constrained, and it is likely that conditions under which segregation of metallic liquid occurred, especially oxygen fugacity and pressure, had as significant a control on planetesimal composition as segregation mechanisms and extent of silicate melting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Differentiation within asteroids was a fundamental process in the early solar system. The meteoritic record provides evidence for a diverse range of differentiation processes within parent bodies, arising from differences in bulk composition of bodies, concentrations of heat-producing radioisotopes, timing of the onset of accretion, growth rates, size and oxygen fugacity (McCoy et al. 2006b; Kruijer et al. 2017). Core formation represents a group of large-scale differentiation processes involving efficient segregation of metal and formation Fe-rich cores and silicate mantles and crusts. Metal-silicate segregation in small bodies likely occurred very early in solar system history, and was protracted and episodic in rocky planets such as Earth (Jones and Drake 1986; Righter and Drake 1996; Kleine et al. 2002; Righter 2003; Wood et al. 2006; Kleine and Wadhwa 2017; Rushmer et al. 2000). The dominant process of core formation in planets was likely segregation of liquid metal from substantive silicate magma oceans (e.g. Righter and Drake 1996; Wood et al. 2006 ; Wood 2008; Brennan et al. 2020), although accretion of pre-differentiated bodies may have had a lasting legacy on the composition of planet mantles and cores (Rudge et al. 2010). However, although thermal models of planetesimal evolution provide insight into why only a portion retained sufficient heat to form Fe-rich cores, a complete understanding of the mechanisms of core formation in small bodies remains elusive.
Segregation of metallic, ‘core-forming’ liquids from a silicate magma ocean requires high temperatures to extensively melt interiors of rocky bodies. Although there is some geochemical evidence for widespread silicate melting on asteroids (e.g. Greenwood et al. 2005), lower-temperature core-formation models, or at least localised segregation of Fe-rich liquids from largely solid silicate via percolation, have also been proposed or invoked by various authors (e.g. Yoshino et al. 2003; McCoy et al. 2006b; Terasaki et al. 2008; Scheinberg et al. 2015; Berg et al. 2018; Dhaliwal et al. 2019; Lichtenberg et al. 2019; Neri et al. 2019). Under conditions of the interiors of planetesimals and large asteroids, FeS-rich liquids would be produced at temperatures significantly below the silicate solidus (Rushmer et al. 2000). This implies that during the thermal evolution of small bodies in the early solar system, there would be a continuum from the onset of metallic melting and progressive melting of metallic components, to onset of silicate melting, and at much higher temperatures, large-degree silicate melting. As such, if metallic liquids readily form interconnected melt networks within a solid silicate matrix, either localised segregation or large-scale core-formation can initiate at substantially lower temperatures than those required for segregation in magma ocean models. This would imply a higher fraction of rocky bodies in the early solar system retained sufficient heat during accretion to initiate core-formation, and might also imply a two-phase process of first low, and then high temperature core formation in bodies which later underwent large-fraction silicate melting.
Efficient low-temperature segregation of metallic liquids has implications for the thermal and geochemical evolution of rocky bodies in the early solar system, for both planetesimals and the larger rocky planets to which they may have accreted. However, evidence for segregation via percolation remains contentious. Here, the effects of low-temperature segregation on compositions of metallic and silicic/rocky components of planetesimals are investigated, using data from a wide range of experimental studies, to determine potential signatures for, and explore the implications of, percolation-driven core formation. Firstly, evidence from the meteoritic record for low temperature segregation is briefly summarised, along with constraints on the composition of core-forming liquids and timing of core-formation. Further constraints on timing of core formation based on models of thermal evolution are then outlined, and evidence from experimental studies reviewed, both experimental data on the viability and efficiency of percolation of metallic liquids, and geochemical and textural observations from partial melting experiments. From this information, different scenarios for core-formation via percolation, and their implications for the extent of chemical equilibration between solid and liquid components in differentiating planetesimals, are summarised. On this basis, data from a broad range of partial melting, element partitioning and other experimental studies are then used to explore how elemental compositions of metallic and silicic/rocky parts of planetesimals would be modified during low temperature segregation.
2 Geochemical Constraints on Low-Temperature Core-Formation from the Meteoritic Record
Magmatic iron meteorites and pallasites demonstrate that segregation of metallic, Fe-rich liquids occurred within a proportion of planetesimals/asteroids. The bulk composition of differentiated bodies can be assessed with reference to primitive meteorites. Broadly speaking, chondrites are considered representative of the precursor materials for bodies which were variably processed by large scale differentiation (Righter et al. 2006), including parent bodies for magmatic iron meteorites (Hilton et al. 2022 and references therein). Model radioisotope ages for magmatic irons imply remarkably short time periods for segregation in parent bodies, of the order of 1 Myr after the formation of the first solid material in the solar system, Ca-Al-rich inclusions (CAIs) (Kruijer et al. 2014). This is earlier than the age of many chondrites, implying that segregation was rapid and concurrent with accretion.
Unfortunately, direct evidence from magmatic irons on the nature of core-forming liquids is obscured, as their parent bodies were modified by fractional crystallisation (Chabot 2004). On the basis of highly siderophile element (HSE) modelling, Hilton et al. (2022) proposed that magmatic irons originated from differentiated planetesimals with core compositions ranging from 73 to 86 wt% Fe, 5–19 wt% Ni, 1–19 wt% S and 0.1-3 wt% P. Modelling also demonstrates a key effect of oxidation state on differentiation, with more reduced bodies forming Fe-rich, volumetrically larger cores, and more oxidising conditions resulting in smaller, S-rich cores with lower Fe:Ni ratios (Hilton et al. 2022). Although modelled S contents in parent bodies are variable, they are always substantially lower than those of eutectic liquids in the Fe,Ni-FeS system. On this basis it has been suggested that core segregation must have occurred at temperatures exceeding those of the eutectic, possibly following substantial silicate melting (Rushmer et al. 2000). Similarly, Fe isotope compositions of magmatic irons imply crystallisation from ≈ 1300 °C (Ni et al. 2020), consistent with higher segregation temperatures.
The S-poor nature of magmatic irons implies that complimentary S-rich components from parent bodies are unsampled in the meteoritic record (e.g. Ni et al. 2020). Furthermore, the low S-content of most modelled parent bodies similarly suggests that S-rich liquids formed during planetesimal differentiation are ‘missing’. This might indicate that (1) S-rich metallic liquids did not effectively segregate in parent bodies, or (2) accumulated S-rich liquids were rehomogenised with Fe-rich (S-poor) liquids, for example by mixing during latter stages of differentiation. Alternatively, (3) mechanically weaker FeS-rich bodies might simply be very poorly preserved in the meteoritic record compared to S-poor bodies (McKibbin et al. 2019; Ni et al. 2020), and/or (4) in contrast to Fe,Ni-rich bodies, deep-seated accumulations of S-rich material within parent bodies might simply be more inaccessible, and unsampled by impact events (Boesenberg et al. 2012). Finally, (5) Hirschmann et al. (2021) argued for substantial degassing and loss of S from early planetesimals.
The meteoritic record also provides some evidence for conditions under which mobilisation of Fe-rich, potentially ‘core-forming’ liquids could occur. Although chondrites likely escaped modification by differentiation processes, they are variably metamorphosed. 2-pyroxene thermometry for some chondrites implies that temperatures exceeded the Fe,Ni-FeS eutectic, and whilst there is a lack of evidence for FeS veining and migration, there is textural evidence for sulfide melting and aggregation (Mare et al. 2014). Primitive achondrite meteorites provide evidence for migration of both silicate and metallic melts. Acapulcoites and lodranites may record processes of early metal and low-fraction (< 2–7%) silicate melt loss (Mittlefehldt et al. 1996; Dhaliwal et al. 2019). As noted by Hopp and Kleine (2021), complex patterns of S-poor and S-rich metallic veins, heterogeneous distribution of FeNi metal, and HSE systematics in several primitive achondrite groups indicate successive segregation and redistribution of both S-rich and S-free metallic liquids prior to silicate melting, and a complex series of differentiation processes. Wang et al. (2014) suggested that ungrouped feldspar-rich achondrites represent products of low-degree silicate melting, with Fe isotope systematics inferred to represent an earlier period of at least localised FeS-rich liquid accumulation. Day et al. (2019) argued that FeO-rich achondrite whole rock data, HSE concentrations, and O and Os isotopic data imply parent bodies formed by melt-rock interaction and as cumulates, requiring extensive Fe-Ni-S melting and 1–20% silicate melting. McCoy et al. (2006a) noted the presence of metallic veins in some lodranites, with a subset inferred to represent core segregation in parent bodies which experienced only minor silicate melting. However, textural evidence is challenging to interpret, and whether this represents a true core-formation process is uncertain (McCoy et al. 2006a). Tomkins et al. (2020) and Tomkins (2009) also caution that comparative studies of meteoritic samples and modelling small fraction melts remains challenging due to large inherent variations in meteorite chemistry. They instead suggest that textural evidence in chondrites and achondrites implies only localised S-poor and S-rich liquid mobilisation, largely due to shock impacts in parent bodies.
Evidence for large-scale mobilisation of core-forming liquids remains contentious. It is also possible that (1) the limited number of inferred parent bodies of differentiated meteorites are not strictly representative of larger-scale core-formation processes in the early solar system and (2) evidence of lower temperature core-formation processes has been obscured by later differentiation processes. As an added complication, there is growing evidence to support models where inferred meteorite parents instead represent different parts of large bodies. Chondrites and achondrites may have coexisited in the same parent, suggesting the accretion and differentiation were prolonged, possibly involving bodies with undifferentiated outer layers overlying differentiated and extensively molten interiors lids (e.g. Elkins-Tanton et al. 2011; Lichtenberg et al. 2019; Maurel et al. 2020; Dodds et al. 2021). Furthermore, although it is often assumed that dense core-forming liquids will have sunk to form metallic cores in broadly concentric parent bodies, it is possible that early stages of core formation resulted in only localised segregation (Day et al. 2019). Groups of meteorites could represent different regions a few large, only partly differentiated, multiply-processed and possible highly heterogeneous parent bodies.
3 Timescales for core-formation Inferred from Thermal Models of Planetesimal Evolution
Thermal models of planetesimals provide additional insight into timescales for differentiation. Models typically invoke rapid internal radiogenic heating by 26Al, and to a lesser degree 60Fe, as the only mechanisms capable of driving differentiation (e.g. Elkins-Tanton et al. 2011; Lichtenberg et al. 2019; Dodds et al. 2021 and references therein). Rapid accretion and fast growth allow retention of sufficient heat from radioactive decay to allow melting and segregation. Larger/earlier-formed/more rapidly accreting planetesimals experience higher maximum temperatures and a greater degree of internal melting and processing, with a gradation to bodies which experience limited heating and do not segregate (Lichtenberg et al. 2021), and/or outer (late accreted) undifferentiated surface regions of differentiated bodies (Elkins-Tanton et al. 2011). Within bodies which subsequently undergo substantial silicate melting, thermal models predict a significant time lag, e.g. 0.4–0.6 Myr (Lichtenberg et al. 2021), from the onset of melting of metallic components to formation of silicate ‘magma mush’.
The cut-off size and accretion rate required to allow silicate melting is model dependent. Elkins-Tanton et al. (2011) suggest that bodies that exceed ≈ 200 km by 1.5 Myr after CAI can melt from the interior outwards, resulting in an interior magma ocean underlying a solid, convective, undifferentiated shell. In such models, melting is likely to be both protracted and highly variable, between and within bodies. Some thermal models assume that substantial silicate melting is required to effectively initiate core formation, e.g. >20% silicate melting and the onset of convection (Dodds et al. 2021), some assume that onset of Fe,Ni-FeS melting is sufficient to initiate percolation-driven segregation (Lichtenberg et al. 2021), whilst others invoke a potential role of early percolation and later metal accumulation due to localised silicate melting (Lichtenberg et al. 2019).
4 Insight from Experimental Studies
Aside from experiments in fully molten systems which aim to simulate core segregation in silicate magma oceans, experimental studies which provide insight into core segregation at lower temperatures can largely be divided into: (1) those which constrain the efficiency and mechanisms of percolation, and (2) studies of melt relations in primitive (e.g. chondritic) or other model compositions.
4.1 Experimental Studies of Percolation of Core-Forming Melts
For fully interconnected melt networks, Darcy flow calculations indicate that dense core-forming liquids can rapidly segregate from the solid silicate portion of a planetesimal (Rushmer et al. 2000). Therefore, the aim of many studies has been to constrain conditions under which interconnected metallic melt networks form. Studies typically demonstrate high dihedral angles ( > > 60°) for metallic liquids in olivine-dominant systems, with interconnected melt networks only forming above a critical melt threshold, estimated to range from 5 to 17 vol% for Fe-S liquids (e.g. Yoshino et al. 2003; Terasaki et al. 2005; Roberts et al. 2007; Walte et al. 2007; Bagdassarov et al. 2009a; Ghanbarzadeh et al. 2017). Solferino et al. (2020) argued that sluggish kinetics in olivine-metallic liquid systems explain much of this discrepancy, with long run duration experiments implying critical melt thresholds of around 14 vol%, consistent with the results of numerical simulations which suggest values of 10–17 vol% (Ghanbarzadeh et al. 2017). Experimentally determined interfacial energies support the assertion that segregation is more feasible for S-rich melts used in most experimental studies, and that dihedral angles and critical thresholds are considerably higher for low S metallic melts (Neri et al. 2019). Terasaki et al. (2008) demonstrated that solubility of O in FeS-rich liquids results in a marked decrease in dihedral angles, implying that core-forming liquids can form interconnected melt networks in more oxidised planetesimals at very low melt fractions. It has also been suggested that dihedral angles for Fe-liquids/silicate systems are reduced at pressures corresponding to the Earth’s lower mantle (Shannon and Agee 1998; Shi et al. 2013), although the extreme conditions required to promote melt network development via this mechanism limit the importance of this process to the later stages of accretion of Earth-sized planets.
Additional complexities are the role of non-hydrostatic conditions, concurrent deformation of silicate matrices and the effect of silicate melting. Non-hydrostatic conditions significantly alter melt geometry, resulting in alignment of melt pockets, incipient melt network formation, and even the formation of fully interconnected metallic melt networks in olivine-dominated matrices (Bruhn et al. 2000; Rushmer et al. 2000, 2005; Groebner and Kohstedt 2006; Hustoft and Kohlstedt 2006; Rushmer and Petford 2011; Walte et al. 2011; Berg et al. 2017). This can mobilise metallic liquids below critical threshold values. However, high strain rates and high strains from experiments are not easily extrapolated to natural systems, and it is unclear whether ‘deformation-aided percolation’ of metallic melts is inhibited at low strain rates or not (Walte et al. 2011; Berg et al. 2017). Onset of silicate melting will have an additional control on segregation processes, with experimental studies indicating that the presence of low fraction silicate melt inhibits segregation of metallic liquids (Holzheid et al. 2000; Rushmer and Petford 2011; Cerantola et al. 2015). In contrast, higher fraction partial melting experiments at ambient pressure appear to suggest that the onset of silicate melting promotes, rather than inhibits, formation of FeS-rich melt channels (McCoy et al. 1999). On the basis of experimentally determined interfacial energies, Neri et al. (2019) also noted that rapid segregation of low fraction silicate melts results in an effective increase in FeS-rich liquid fractions above critical melt thresholds. As such, in larger systems, silicate melting and migration can promote segregation of core-forming liquids. At higher temperatures, increasing silicate melt fraction and the transition to a ‘crystal mush’ will eventually allow segregation of metallic melts through a rapid process of melt immiscibility and gravitational settling.
The efficacy of core-formation by percolation also depends on timescales for segregation of core-forming liquids. Percolation is an inherently slower process and only a viable mechanism for planetesimal differentiation if it is able to account for core segregation within, at most, a few Myr. For a fully interconnected melt network based on models of Ghanbarzadeh et al. (2017) and using a simple Darcy flow type calculation, Solferino et al. (2020) estimated core segregation in a 100 km radius body in < 1 Myr. By contrast, high-pressure centrifuge experiments by Bagdassarov et al. (2009b) imply that segregation of Fe-S liquid is an order of magnitude too slow to account for core-formation in planetesimals. Similarly, Todd et al. (2016) estimated core formation in planetesimals exceeding 1–5 Myr timescales based on Fe-S melt permeabilities obtained from high pressure/temperature deformation experiments. In contrast, Berg et al. (2018) directly measured metallic melt segregation velocities in-situ under core-forming conditions in an analogue system. They noted rapid melt migration due to hydraulic fracturing, and prolonged melt migration during matrix compaction, as metallic melts migrated through a solid matrix. Data from that study are consistent with rapid core formation via percolation in planetesimals following development of melt networks. However, aside from conflicting results based on experimental methodology used, there are limitations in extrapolating results from experimental studies to natural systems. Kinetics in olivine-rich/metallic liquid systems are sluggish, and time- and length-scales must be extrapolated over many orders of magnitude. Melt geometry remains key. If it is assumed that pore alignment and micron-sized melt channels are only an initial texture in partially molten systems, with segregation resulting in channelization and formation of melt-rich bands as noted in some studies (e.g. Groebner and Kohstedt 2006; Hustoft and Kohlstedt 2006; Berg et al. 2018), percolation can make a significant contribution to core formation. In other scenarios, the efficacy of percolation may be limited, with only localised accumulation of FeS-rich liquids.
On balance, it is possible that percolation, or ‘non-magma ocean segregation’, could have contributed to planetesimal differentiation under some circumstances, either as a primary process, or as a secondary processes in bodies which then undergo ‘greater than low-fraction’ silicate melting (Watson and Roberts 2011). A key-step in any model of percolation-driven segregation is initial development of a melt network. In most systems metallic melts will be non-wetting below a critical melt volume threshold, likely to be ≈ 10–14 vol% for S-rich melts, and likely higher from S-poor melts. High O contents in FeS-rich liquids may significantly enhance network development even at low melt fractions, promoting segregation in more oxidised bodies. Deformation could also enhance development of metallic melt networks, either locally due to shock impacts and/or during the onset of melting and hydraulic fracturing (Berg et al. 2017), or in large bodies undergoing vigorous convection. The temperature interval over which metallic liquids can segregate may also be limited by the onset of silicate melting, which then inhibits segregation until higher melt fractions/temperatures are reached. Aside from this, simulations suggest that once melt networks form, they can effectively drain regions of melt down to melt fractions considerably lower than critical melt fractions needed to initiate segregation of core-forming liquids (Ghanbarzadeh et al. 2017). However, a secondary process required for larger-scale segregation is concurrent deformation of the silicate matrix. Flow of the solid matrix is required to accommodate substantial metallic liquid redistribution, and there may be a lower temperature threshold at which the viscosity of the solid portion of a rocky body prevents core formation over realistic timescales. The complexity of physical processes of melt network formation, of melt segregation via percolation, of limitations imposed by low degree silicate melting and deformation of silicate matrices, and timescales of melt segregation mean that we have, at best, limited constraints on the importance of percolation in planetesimal segregation in the early solar system. An alternative approach to assessing whether percolation contributed to core-formation is clearly required.
4.2 Experimental Studies of Partial Melting in Chondritic Systems
There have been various experimental studies of melting in primitive, broadly chondritic compositions (Agee 1993; Agee et al. 1995; Jurewicz et al. 1991, 1993, 1995; Ford et al. 2008; Berthet et al. 2009; Usui et al. 2015; Collinet and Grove 2020a, b). However, insight provided on mechanisms and consequences of core formation is limited because: (1) most studies focussed on silicate melting, with experiments conducted at higher temperatures only. The onset and composition of lower temperature metallic liquids, and phase relations between solid and liquid metallic phases, are less well contained. (2) Some experiments were conducted in S-free systems and provide limited insight into core formation in planetesimals in which S is an important component. (3) Most experimental work has been performed at ambient pressure using gas-mixing apparatus. In some studies, this likely resulted in variable alkali loss and a change in melt relations and compositions, as discussed in Collinet and Grove (2020a, b). Variable S loss may also have occurred (McCoy et al. 1999). The effects of even moderate pressure remain less well constrained. (4) Many studies report limited data on phase composition, sometimes with important omissions such as O and C in metallic liquids.
From available experimental work the following can be concluded. As expected, metallic components always melt at substantially lower temperatures than silicate components. In Fe,Ni-FeS systems a eutectic governs composition of first formed melts, and the S content of the eutectic linearly decreases with increasing pressure, from 31% S at 1 bar to 20.7% at 7 GPa (Fei et al. 1997 and references therein). The eutectic temperature linearly decreases with increasing pressure from 988 °C to 1 bar to 860 °C at 14 GPa. For pressures below 5 GPa, which encompasses conditions within planetesimals and planetary embryos, this shift in solidus is minor. However, silicate solidii increase significantly with pressure and there is a substantial region of temperature-time space in progressively heating planetesimals over which metallic liquids are in equilibrium with solid silicate, and then low fraction silicate melt, especially in larger bodies.
Two immiscible liquid metal alloys coexist from ambient pressure to about 5.5 GPa in chondritic compositions, one S-rich and one S-poor, termed FeS-rich and Fe-rich here for clarity. This is not simply a textural effect due to metallic melt migration or unmixing during quenching, but represents an important immiscibility gap in systems with a free Fe-rich phase which closes at pressures above ≈ 5–6 GPa (Corgne et al. 2008). In some studies, additional multiple sulfide phases are noted in run products, especially at lower temperatures (e.g. McCoy et al. 1997). However, these may partly represent local reaction between specific crystalline phases and percolating FeS-rich liquid. Corgne et al. (2008) noted that in fully molten systems, immiscible Fe-rich liquids are C-rich, and FeS liquids C-poor. Berthet et al. (2009) similarly noted that extent of immiscibility is dependent on C content in complex systems over a range of temperatures. In many experimental studies FeS-rich liquids are described as having ‘near-eutectic compositions’, although S contents are recorded as decreasing in higher temperature experiments, coupled to increasing dissolution of S in silicate melts (e.g. McCoy et al. 1999; Berthet et al. 2009). Composition of FeS-rich and Fe-rich liquids is also dependent on oxygen fugacity, fO2, which can be described in terms of log units relative to the iron-wüstite (Fe-FeO) solid buffer (IW). At fO2 several log units below IW, reduction of FeO in silicates drives changes in Fe:Ni ratios in metallic components, and proportions of Fe-rich and FeS-rich liquids (e.g. Berthet et al. 2009; Usui et al. 2015; Collinet and Grove 2020a, b). Very low fO2 may also result in changes in Si and S partitioning between FeS-rich and Fe-rich liquids due to SiO2 reduction and increase in Si content of Fe-rich liquid (Berthet et al. 2009). Other changes in sulfide melt chemistry at low fO2 (log fO2 ≈ IW−4) can be attributed to reduction of silicate by FeS, resulting in the formation of Ca–, Mg–, Cr–, Mn–, Fe–S bearing liquids. Highly reducing conditions also change the chemical nature and silicate-metal partitioning of Cr and Mn (Berthet et al. 2009). In contrast, more oxidising conditions (log fO2 = IW > + 2) eventually result in extensive Fe oxidation and preclude the presence of a metallic component entirely (Jurewicz et al. 1993).
Although complete melting in Fe,Ni-FeS systems only occurs at very high temperature (Fei et al. 1997), experimental studies note complete melting of metallic components in chondritic systems at much lower temperatures (e.g. Jurewicz et al. 1995; McCoy et al. 1999). Onset of melting of Fe-rich components in run products can be difficult to determine, although textural observations appear to be consistent with complete melting of metallic components before substantial silicate melting occurs (Ford et al. 2008). Onset of silicate melting can also be difficult to ascertain, and can be overestimated if volatile loss occurs. At ambient pressure, Jurewicz et al. (1993) noted onset of silicate melting in a CV3 composition from 1130 to 1150 °C, with ≈ 35% silicate melting at 1325 °C. Jurewicz et al. (1995) noted, in LL and H chondritic compositions, progressively increasing silicate melt fractions from 1170 °C, and full silicate melting at 1500 °C. In an H6 composition, Ford et al. (2008) noted low fraction silicate melts from 1200 °C. At low confining pressures in a range of synthetic, S-free chondritic compositions Collinet and Grove (2020a,b) noted progressively increasing silicate melt fractions from approx. 1060-1163 °C. Bulk composition has a strong effect on silicate melting, although both silicate solidus and liquidus temperatures are also dependent on fO2, and can increase by around 30–50 °C under more reducing conditions (log fO2 < IW – 1) (Collinet and Grove 2020a). At extremely reducing conditions (log fO2 < IW – 4 and below) at 1 GPa, Berthet et al. (2009) noted that the onset of silicate melting can be delayed to 1300–1400 °C.
Most partial melting studies demonstrate the dominance of olivine and low-Ca pyroxene in chondritic systems. Phosphates are rapidly consumed during low degree silicate melting, followed, with increasing melt fraction, by plagioclase and high Ca-pyroxene, both major phases in subsolidus chondritic compositions (Jurewicz et al. 1995; Ford et al. 2008; Berthet et al. 2009; Usui et al. 2015; Lunning et al. 2017). Spinel-group minerals are a minor phase, and may react with FeS-rich liquid (McCoy et al. 1999), although are stable to moderate silicate melt fractions in some systems (Jurewicz et al. 1993). Under highly reducing conditions (log fO2 ≈ IW-4), olivine is destabilised, and silicate liquidus assemblages are pyroxene + quartz (Berthet et al. 2009). As noted above, fO2 has a direct effect on Fe oxidation and the FeO content of silicate phases, and as a result, also strongly influences element partitioning. For example, olivine in more reduced systems is more forsteritic (MgO rich), with lower CaO and NiO (Collinet and Grove 2020a, b).
To summarise, studies of partial melting in chondritic systems imply that: (1) substantial to full melting of metallic components occurs at low temperatures; (2) metallic liquids are immiscible resulting in coexisting FeS-rich and Fe-rich/S-poor liquids; (3) the onset of silicate melting is variable, but typically occurs at higher temperatures, and that there is a moderate to very large temperature window over which silicate melt fraction progressively increases; (4) generally, olivine and orthopyroxene (± spinel) are the dominate silicate minerals during partial melting. Minor phosphates are only stable subsolidus or during very low fraction melting, and plagioclase and clinopyroxene are consumed during low to moderate fraction melting.
5 Scenarios for Percolation-Driven Metallic Melt Segregation in Planetesimals
In comparison to planets like the Earth, asteroidal differentiation occurred at much lower gravity, from 0.03 terrestrial for a < 500 km body, over a wide range of log fO2: IW - 1 to IW + 2 for angrites to IW - 5 for aubrites, and under both volatile-rich and volatile-poor conditions (McCoy et al. 2006b). Core-formation models can be categorised based on inferred degrees of interaction between solid and liquid silicate and metallic components:
-
1.
Segregation of metallic liquid from a solid silicate matrix (Fig. 1, scenario 1). Before the onset of silicate melting, Fe,Ni-FeS liquids separate from a crystalline silicate matrix under certain conditions, e.g. in more oxidised or anion-rich bodies, in vigorously convecting or deforming bodies, and/or in bodies with higher metallic fractions. Processes range from localised redistribution of FeS-rich liquid to protocore formation, likely involving segregation of Fe-rich and FeS-rich liquids, and chemical equilibration between FeS-rich liquid, Fe,Ni-rich solid or liquid, and solid phases within the silicate portion of bodies. Once initiated, melt networks may be effective in draining core-forming liquids to below critical thresholds needed to form melt networks (Ghanbarzadeh et al. 2017). However, the silicate portion of differentiating bodies also needs to deform and displace to accommodate core-segregation, placing constraints on the effectiveness of percolation (Scheinberg et al. 2015).
-
2.
Fe,Ni-rich solid accumulation followed by later segregation during silicate melting (Figs. 1, scenarios 2a to 2c). In this scenario (Tomkins et al. 2013, 2020) shock processes drive FeS melting and localised melt migration, and the formation of enlarged, S-depleted (solid) Fe,Ni accumulations which do not fully equilibrate with surrounding silicate. As planetesimals grow large enough to initiate silicate melting, these large metal accumulations rapidly sink to form protocores, leaving sulfide and small metal particles behind. This model is consistent with the S-depleted nature of magmatic irons, and implies variable chemical disequilbrium. Variable degrees of chemical interaction between Fe,Ni-rich and FeS-rich portions are possible, although the model does rely on effective segregation of S-rich and S-poor components.
-
3.
Metal percolation following partial silicate melting (Fig. 1, scenarios 3a to 3b). Although small fraction silicate melting initially inhibits percolation of metallic melts, greater degrees of silicate melting may promote effective segregation of core-forming liquids by allowing rapid segregation of metallic liquids, and/or by aiding compaction and deformation of the remaining solid silicate portion (Taylor and Norman 1990; Scheinberg et al. 2015 and references therein). Extraction of silicate melt also results in an effective increase in the proportion of metallic phases above percolation thresholds, promoting segregation (Neri et al. 2019). This general set of scenarios involves various periods of chemical equilibration between solid silicate, liquid silicate, and core-forming liquids, and may also include equilibration between Fe-rich liquid/solid and FeS-rich liquid.
-
4.
Magma ocean/mush differentiation (Fig. 1, scenario 4). At higher temperatures, liquid silicate-liquid metal immiscibility dominates segregation, via gravitational settling of denser metallic melt droplets or larger accumulations of metal formed during inefficient core formation. The silicate melt fraction required to allow this is poorly constrained and may approach 50% (Taylor et al. 1993). Liquid metal/ liquid silicate partitioning governs composition of the two reservoirs, with equilibration over a broad range of conditions throughout planetesimal interiors (P–T– fO2). This contrasts magma ocean models for larger planet-sized bodies such as the Earth, in which full metal liquid-silicate liquid chemical equilibration occurs under conditions of the base of very deep, fully molten magma oceans.
Scenarios could represent different bodies in the early solar system, with thermal models demonstrating that size, rate of accretion, composition, fO2 and heliocentric distance all have a significant effect on segregation processes within planetesimals (e.g. Elkins-Tanton et al. 2011; Lichtenberg et al. 2019; Lichtenberg et al. 2021; Dodds et al. 2021). They might also represent various stages in the thermal evolution of planetary bodies, with scenarios 1, 3 and 4 occurring with increasing temperature, or represent processes occurring in different regions of large, heterogeneous bodies. Using data from a range of experimental studies, the effects of chemical exchange between different components within each scenario can be explored.

Core formation models. 1 FeS-rich liquid (black) segregates from Fe,Ni-rich solid/liquid (grey) above the Fe, Ni-FeS eutectic temperature. Under more oxidising conditions, FeS-rich liquid can wet silicate (white) grain boundaries and segregate. Alternatively, core-forming liquids form interconnected melt networks at high melt fractions (metal-rich or highly reduced bodies), or aided by deformation at variable melt fractions. 2 Tomkins et al. (2013, 2020) model where impact processes result in heating and segregation of FeS-rich liquid (black) from metal (grey), and chondrite fracturing (2a) followed by FeS-rich liquid injection (2b). This results in rapid separation of FeS-rich liquid from solid Fe,Ni metal. Repeated processes drive accumulation of solid Fe,Ni metal, which is not in full chemical equilibrium with silicate. During later silicate melting (2c; light grey is approximately 20% silicate melt), large accumulations of now liquid Fe,Ni-rich metal (dark grey) rapidly segregate to form a protocore. 3 protocore formation by segregation of metallic liquid(s) after low-degree silicate melting. Early-formed FeS-rich liquids melts cannot efficiently segregate 3a. However, metallic melt fraction increases during prolonged heating, and once silicate melting is initiated, metallic liquids separate to form a protocore. 4 Extensive silicate melting (magma ocean/magma mush model). Effective segregation of metallic liquid (dark droplets) only occurs after substantial silicate melting. Immiscible metallic liquid(s) rapidly segregate from silicate melt (light grey)
6 Chemical Equilibria in Fe-rich Systems
Parent bodies of metallic meteorites are inferred to have low to intermediate S contents, up to 19 wt%. In contrast, experimental studies of percolation typically involve observation of liquids close to FeS in composition, and partial melting experiments imply immiscibility of metallic portion into Fe-rich/S-poor and FeS-rich components. In this section, data from experimental studies are used to explore partitioning of major and trace elements between FeS-rich and Fe-rich phases, and to determine constraints on the composition of core-forming liquids inferred in various models.
6.1 Major Element Composition of FeS-Rich and Fe-Rich Phases
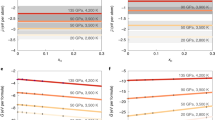
Although there have been numerous studies of melt relations in chondritic systems only a small proportion provide geochemical data on metallic phases. Figure 2 shows S contents of FeS-rich phases, quenched from liquid, from experiments and eutectic melt compositions in the Fe-S system. Partial melting experiments consistently demonstrate that immiscible FeS-rich liquid has S contents which significantly exceed those of eutectic melts in simplified systems, are generally between 35 and 40 wt% S, and decrease slightly with increasing pressure. Stoichiometrically, these FeS-rich liquids are close to ideal FeS in composition. In contrast, coexisting Fe-rich solids/liquids have S contents below 2 wt%. Also shown on Fig. 2 are FeS-rich experiments from higher temperature liquid sulfide/liquid silicate partitioning experiments. Bulk compositions for these studies are typically based on enstatite chondrite compositions with an additional, substantive metallic/sulfide component. Despite differences in bulk chemistry, data from these studies are consistent with partial melting experiments in chondritic compositions. FeS-rich liquid S content and S partitioning between FeS-rich and Fe-rich phases (Fig. 3) are not substantially modified by increasing degrees of silicate melting or by melting of Fe-rich components, reinforcing the fact that FeS-rich liquids in complex systems are consistently S-rich compared to simple eutectic systems. S readily partitions into metallic liquids over silicate liquids under conditions relevant to core segregation in planetesimals (Bercovici et al. 2022 and references therein), so it is not expected that the onset of silicate melting has a major influence on FeS-rich liquid S content. Lower S-content liquids in Fig. 2A. are mainly due to closure of the metal-sulfide immiscibility gap towards higher pressures, as evident from Fig. 2B. fO2 appears to have only a moderate impact on FeS-rich liquid S concentrations across the dataset, with higher S contents at very low fO2, likely related to a decrease in O solubility in FeS-rich liquid under very reducing conditions, and possibly also due to increase in the Fe:Ni ratio.
Derived FeS-rich/Fe-rich partition coefficients decrease with increasing temperature (Fig. 3A), although this partly reflects higher temperatures used in higher pressure experiments. In contrast, fO2 has little discernible influence on S partitioning, which instead appears to vary more as a function of bulk composition. Similarly there is no clear dependence of Ni partitioning on anything other than bulk composition, with Fe-rich phases tending to be 5 to 10 times richer in Ni than FeS-rich liquids across all studies. Slight decreases in FeS-rich liquid S content in Fig. 2C are likely due to the effect of fO2 on FeS-rich liquid/silicate melt partitioning. In an FeS-rich system at more oxidising conditions where Fe-rich phases were absent, Gaetani and Grove (1997) noted that sulfide S contents decreased at fO2 several log units above the IW buffer, continuing the broad trend seen in Fig. 2C. They also noted that fO2/fS2 has some control on sulfide Fe:Ni ratios. Similarly, silicate liquid/sulfide partitioning experiments show a control of fO2 on sulfide compositions due to sulfide-silicate exchange (e.g. Boujibar et al. 2020), as explored below.

Composition of FeS-rich liquids in equilibrium with Fe-rich phase and silicate as a function of run temperature (A), run pressure (B), and fO2 (C). Black/grey data points are from partial melting studies using chondritic starting compositions: unfilled squares: McCoy et al. (1999) for an enstatite chondrite (EH4) bulk composition, ambient pressure and log fO2 below IW – 4. Circles: Berthet et al. (2009), EH4 composition, 1 GPa, log fO2 = IW-1.2 to IW-2.8 (unfilled circles) and fO2 = IW – 4.2 to IW – 4.8 (grey circles). Grey diamonds: Ford et al. (2008), H6 ordinary chondrite composition, ambient pressure, log fO2 = IW – 2.8 to IW – 5.8; higher S contents represent experiments in sealed silica tubes, minimising volatile loss, at slightly more reducing conditions. Small grey squares: eutectic Fe-S melt compositions from 0–10 GPa in the Fe-S system from Usselman (1975). Filled black squares: sulfide melt compositions from Corgne et al. (2008) who constrained core-melt immiscibility in a model chondrite + Fe + Ni + FeS system. Red data points are from FeS-rich liquid/silicate liquid partitioning experiments. Filled red squares: Steenstra et al. (2020a), model reduced basalt + sulfide system. Open red squares: Boujibar et al. (2019), 50:50 silicate/metal starting composition similar to EH4 enstatite chondrite but with reduced O content and silicate/metal ratio. Open red circles: Malavergne et al. (2014), analogue Mercurian composition based on CI silicate glass, FeS and Si metal. Filled red circles: Steenstra et al. (2020b), basaltic composition +Fe-S and Fe-Si components. Open red diamonds: Boujibar et al. (2020), mixed silicate-Fe-FeS-Ni-Si/Fe2O3 starting compositions, with a silicate component ‘similar’ to CI carbonaceous and EH4 enstatite chondrite. Data from Gaetani and Grove (1997) not shown, although their data indicate that reduction in sulfide S content could continue to higher fO2 (up to 2.5 log units above IW).

Metal-sulfide partitioning of S (A. and B.) and Ni (C. and D.) as a function of temperature and fO2 in partial melting (grey/black) and partitioning (red) experiments. Same key as Fig. 2
It is evident that immiscibility between FeS-rich and Fe-rich phases remains up to 6–8 GPa, exceeding the range of conditions in large asteroids and planetesimals. Extensive immiscibility is also noted at very high temperatures where there is complete melting of silicate (e.g. Malavergne et al. 2014; Boujibar et al. 2019, 2020; Steenstra et al. 2020a, b). This is in accordance with Corgne et al. (2008), who demonstrated immiscibility of metallic (S-poor, C-rich, O-poor) and sulfide (S-rich, C-poor, O-rich) liquids in a metal-rich, C-saturated system up to 8 GPa. However, in chondritic compositions the S content of FeS-rich liquid is generally higher at comparable pressures. Solubility of other metallic elements into FeS-rich liquids may act to increase S weight fraction, with sulfide liquids in partial melting experiments also containing appreciable Ti, Cr, and Mn and reduced Ni (McCoy et al. 1997, 1999; Ford et al. 2008; Berthet et al. 2009). Although C and O contents of FeS-rich and Fe-rich phases are not reported in many studies, available data can be used to constrain minor element composition of core-forming melts in complex systems. In order to allow comparison between data sets, reported FeS-rich and Fe-rich phase compositions were converted to molar proportions, and then moles of Fe assigned successively to end-member compositions FeO, Fe2Si, Fe3C and FeS, based on moles of each anion, with remaining Fe assigned to pure Fe. Within the system FeS-Fe-Fe3C, compositions are in good agreement with Corgne et al. (2008), with the presence of a large miscibility gap between FeS-rich and Fe-Fe3C-rich liquids which shrinks at higher pressures (Fig. 4A). C contents were not determined in many studies, although as studies typically used graphite capsules they are expected to be C-saturated, similar to Corgne et al. (2008). Data indicate that Fe-rich phases are generally S-poor under lower pressure conditions of core formation in planetesimals, but can readily incorporate C, in accordance with Zhang et al. (2018) who reported C solubilities of 4–6 wt% in S-poor Fe-Ni-S liquid. FeS-rich liquids are generally close to FeS in composition. C solubility is markedly reduced in S-rich metallic liquids (typically < 1 wt%, Zhang et al. 2018) although immiscible FeS-rich liquids can contain several wt% C under very reducing conditions (Boujibar et al. 2020). Fe-rich liquids also readily incorporate Si, consistent with observations from the meteoritical record, for example compositions of Fe-metal grains in enstatite chondrites and achondrites (Harris and Bischoff, 2020). In contrast, FeS-rich liquids are Si-poor. From limited available data (Fig. 4B) it is clear that FeS-rich liquids can incorporate significant O, in contrast to Fe-rich liquids which tend to be very O poor. There is insufficient data to determine whether the notable solubility of O in FeS-rich liquid is pressure dependent in chondritic systems, although in their percolation experiments Terasaki et al. (2005; 2008) noted that the O content of FeS-rich liquids in equilibrium with olivine decreases markedly with increasing pressure, with high O contents requiring log fO2 significantly exceeding the IW buffer. Similarly, at much higher temperatures, Boujibar et al. (2020) noted that O contents of FeS-rich liquid can be considerable, at least under relative oxidising conditions, again above the IW buffer, and are controlled by FeO exchange between FeS-rich liquid and silicate melt. They further noted that O solubility in FeS-rich liquid is enhanced by the presence of alkalis, as had previously been observed by Murthy et al. (2003) and Steenstra et al. (2018), but that O solubility is reduced in Ni-rich systems, in accordance with the findings of Kiseeva and Wood (2015).

Composition of FeS-rich liquids (filled symbols) and Fe-rich solids/liquids (open symbols) from experimental studies based on partial melting of chondritic compositions (squares) and silicate liquid-metal liquid partitioning studies (circles). (A) Ternary plots which share the compositional join Fe-FeS. In (B) the component Fe + Fe2Si + Fe3C is used to readily separate metallic (FeS-poor) and sulfide (FeS-rich) components. Blue squares: McCoy et al. (1999) (1 atm), green squares: Berthet et al. (2009) (1 GPa), red squares: Ford et al. (2008) (1 atm), green circles: Boujibar et al. (2020) (1–8 GPa, with most experiments 3-3.5 GPa), blue circles: Boujibar et al. (2019) (1–5 GPa,mostly 1 GPa), black squares: Steenstra et al. (2020a) (1 GPa), red circles: Steenstra et al. (2020b) (1 GPa), and grey circles: Malavergne et al. (2014) (1 GPa). Yellow squares are sulfide compositions from olivine-sulfide percolation experiments of Terasaki et al. (2008). Darker yellow squares are from experiments where low dihedral angles imply percolation of sulfide, and lighter squares are from more reducing/higher pressure experiments where higher dihedral angles inhibit percolation. Shaded region on the FeS-Fe-Fe3C ternary shows melt immiscibilities from Corgne et al. (2008)
To summarise, all systems are characterised by immiscibility between O-bearing, Si- and C-poor FeS-rich liquid and Si-rich, C-rich, O-poor Fe-rich solids/liquids. The miscibility gap is reduced towards higher pressure and fO2 plays an important role in terms of phase chemistry. There is no evidence of closure of the immiscibility gap at high temperatures. Other minor elements are also expected to be fractionated during separation of FeS-rich and Fe-rich liquids, including P, which partitions readily into Fe-rich liquid (McCoy et al. 1999; Ford et al. 2008). C, S and O are important constituents in chondrites, and a significant consequence of immiscibility is that light element composition of core-forming liquids is strongly dependent on the mechanism for melt segregation. O, C and Si (+ P) content of core-forming liquids, as well as Fe/Ni ratio, may depend significantly on core-segregation process and on the timing and conditions of FeS-rich/Fe-rich phase segregation. However, immiscibility will be a key feature in all models of core formation based on chondritic compositions, including high temperature magma ocean models. Incorporation of O into FeS-rich liquids can have a significant effect on lowering sulfide/silicate mineral dihedral angles, increasing the likelihood of percolation of FeS-rich liquids.
6.2 Proportions of FeS-Rich and Fe-Rich Components in Chondritic Compositions
Some percolation models invoke segregation of FeS-rich liquid at the onset of melting, or following substantial melting of the metallic component of planetesimals. The proportion of FeS-rich liquid formed is an important control on whether interconnected melt networks can form due to characteristically high dihedral angles. Aside from conditions of melting, both bulk composition and immiscibility in Fe-rich systems will control volume proportions of liquids formed. McCoy et al. (1999) reported proportions of FeS-rich liquid during melting of EH4 enstatite chondrite compositions varying from 19 to 0% by volume (vol%), from 1000 to 1500 °C at ambient pressure. Over this temperature range the proportion of silicate melt increased, but proportion of Fe-rich liquid remained relatively constant. However, in higher temperature runs the authors noted significant pooling of metal at the edge of sample volumes, and reported proportions must, therefore, be treated with caution. At 1 GPa for the same composition, Berthet et al. (2009) reported FeS-rich liquid proportions of 2.3 to 6.5 vol%, with no discernible influence of temperature. Similarly, Ford et al. (2008) reported FeS-rich liquid proportions of 0.8 to 3.2 vol% during partial melting of an H-type ordinary chondrite composition. Some S is also dissolved into silicate liquids depending on run conditions, and thus the extent of silicate melting influences the proportion of sulfide at higher temperatures. Berthet et al. (2009) also noted that S becomes increasingly lithophile below log fO2 ≈ IW-3, with silicate melts dissolving several wt% S, in accordance with subsequent studies by Wood and Kiseeva (2015) and Steenstra et al. (2020a). At high temperatures, therefore, proportion of FeS-rich liquid also depends on fO2.
Proportions of FeS-rich liquid produced during complete melting of chondritic compositions can also be estimated based on chemical exchange between liquid silicate and liquid metal components. For eight ordinary chondritic bulk compositions Bercovici et al. (2022) calculated core compositions and volume fractions in fully molten planetesimals. For their models they calculated fO2 based on reported Fe and FeO proportions for each composition, and then modelled core composition and size based on parameterisations of S partitioning between Fe-rich and silicate liquids. Due to the limited effect of pressure and temperature on S partitioning, they demonstrated that bulk composition, and by consequence fO2, have a dominate influence on silicate-metal segregation. Bercovici et al. (2022) modelled single phase Fe-Ni-S core compositions, assuming complete miscibility, ranging from 9.4 to 35 wt% S. This correlates with fO2 for each composition from log fO2 = IW−1.4 to IW + 2.8, i.e. more S-rich cores in more oxidised bodies, although the authors noted that correlation is simply due to the control of bulk composition on fO2. Calculated core size also varies significantly in this model. Building on the approach of Bercovici et al. (2022), and assuming complete segregation of their modelled core compositions into troilite (FeS, zero pressure density of g/cm3) and taenite (7.5 wt% Ni, 94 wt% Fe, density of 7.81 g/cm3), their data imply variation in the calculated proportion of FeS liquid in parent bodies of ≈ 2 vol% to ≈ 10 vol%. The lack of reported data on FeO in enstatite chondrites, and the increase in solubility of S in silicate under very reducing conditions, mean that no similar estimate of core composition, size and proportion of FeS can be made for enstatite chondrite compositions (Bercovici et al. 2022). However, this range of vol% of FeS is broadly consistent with estimations from partial melting experiments in enstatite chondrite-based compositions, Fig. 5, and with phase proportions in an ordinary chondrite composition reported by Ford et al. (2008). For a percolating FeS-rich melt, the critical melt threshold required to ensure melt connectivity is estimated to range from 5 to 17 vol%. As such, in chondritic bulk compositions, percolation of FeS-rich liquid is not expected unless (1) the critical threshold is dramatically reduced by deformation-aided segregation (Berg et al. 2017), a mechanism presumably only applicable to larger bodies, and which may only be enabled following onset of silicate melting, (2) bodies are highly reduced (log fO2 = < IW−4), due to reduction of FeO in silicate and an increase in the proportion of metallic liquid, (3) partitioning of O into sulfide melts is sufficient to lower dihedral angles enough to facilitate percolation at low melt fractions (log fO2 > IW + 1.3) or (4) migration of moderate fraction silicate melt results in an effective increase in proportion of FeS-rich liquid. An increase in overall proportion of core-forming liquids due to presence of both Fe-rich and FeS-rich liquids might also promote melt segregation, although there is a lack of experimental data on percolation in systems with multiple Fe-rich liquids. Core formation by percolation might otherwise be limited to more Fe-rich bodies, i.e. planetesimals which accrete with a higher proportion of metallic components, due to higher inherent volume proportions of core-forming liquids produced once bodies exceed the eutectic temperatures.

Proportion of FeS-rich liquid reported in partial melting experiments in chondritic compositions as a function of temperature (A) and fO2 (B); same key as Fig. 2. B also shows (large green squares) FeS volume% in eight chondritic compositions calculated using data from Bercovici et al. (2022), assuming full planetesimal melting (see text of details). Labels refer to chondritic bulk compositions
6.3 Trace Element Partitioning Between FeS-rich and Fe-rich Phases
The onset of FeS-rich melting is well constrained in a range of compositions, although there is less certainty regarding the temperature window over which S-poor, Fe-rich components melt. FeS-rich liquids will be in equilibrium with a solid Fe-rich phase at low temperature and a liquid Fe-rich phase at higher temperature. Any process of FeS-rich liquid segregation results in variable ‘liquid FeS-solid Fe and ‘liquid FeS-liquid Fe’ element partitioning. The method of Chabot and Jones (2003), Chabot et al. (2017) allows estimation of solid/liquid Fe-rich metal partition coefficients. This parameterisation is based on the concept of ‘Fe domains’, with element partitioning into FeS-rich liquid controlled by interaction with Fe. The model accurately predicts partitioning of siderophile elements, and is variably successful at predicting non-siderophile element behaviour (Chabot and Jones 2003). Development, and most applications of this model pertain to fractional crystallisation during slow cooling of parent bodies to metallic meteorites. The applicability of this model to higher pressure conditions of core formation is limited by the ambient pressure experimental data on which it is based, and the effect of other non-metallic elements such as O on element partitioning. Importantly, the model is also not specifically designed to investigate partitioning in S-rich systems.
Figure 6A shows molar solid (Fe-rich)/liquid (FeS-rich), i.e. metal/sulfide, partition coefficients calculated from Fe-S liquid compositions reported in chondrite partial melting experiments. Only ranges in calculated partition coefficients are shown for clarity. For several FeS-rich liquid compositions, modelled partition coefficients cannot be obtained as high S contents imply the absence of Fe domains. Similarly, calculated partition coefficients for some elements range to extreme values. As expected, low partition coefficients, i.e. high concentrations in FeS-rich liquid, and low concentrations in Fe-rich solid, are calculated for the chalcophile elements Cu, Zn, Ag, Pb and Bi. Low values are also noted for V and Cr, which are variably lithophile and somewhat siderophile, although the model predicts partitioning of other variably lithophile/siderophile elements P, Ga and Ge into Fe-rich solid. The chalcophile elements Sn and Sb are predicted to have near-unity to moderate partition coefficients, implying slight partitioning into Fe-rich solid over FeS-rich liquid. Siderophile elements are predicted to variably, and sometimes extremely, partition into Fe-rich solid. In Fig. 6B calculated coefficients are compared to coefficients calculated directly using data from chondrite partial melting and sulfide-silicate partitioning studies. Partition coefficients for the latter are all Fe-rich liquid/FeS-rich liquid, and for the former, vary from Fe-rich solid/FeS-rich liquid to Fe-rich liquid/FeS-rich liquid. Directly determined partition coefficients are generally much closer to unity than modelled values which (1) implies that the model cannot be applied to S-rich liquids for many elements, and (2) makes it difficult to assess any effect of Fe-rich phase melting on element partitioning. Chabot et al. (2017) noted that fits for their model for chalcophile elements are less robust than other elements. For S-rich liquid compositions used here, over and underestimation of chalcophile element partitioning is comparable to that for elements that are variable lithophile and only weakly chalcophile. In contrast, limited data for siderophile elements suggests that the Chabot and Jones (2003), Chabot et al. (2017) model does provide useful predictions.

Molar Fe-rich/FeS-rich (i.e. metal/sulfide) partition coefficients for various elements in chondritic systems. A Ranges in modelled coefficients after the method of Chabot et al. (2017) for FeS-rich liquid compositions from experiments with a coexisting Fe-rich phase: McCoy et al. (1999) (blue), Berthet et al. (2009) (red), and Ford et al. (2008) (black). Arrows indicate elements for which calculated values exceed plot range. B Molar partition coefficients calculated directly using data from partial melting experiments (black/grey) and partitioning experiments (red). Same key as Fig. 2, additionally with solid, black diamonds from Borisov and Palme (1995). Grey boxes represent (truncated) ranges in calculated molar partition coefficients from A. Black circles at bottom of figure denote typically chalcophile elements, and open circles, highly siderophile elements. Arrows indicate the effect of highly reducing conditions (log fO2 ≈ IW−4) which result in silicate reduction by FeS and formation of Ca-, Mg-, and Mn-bearing sulfides (Berthet et al. 2009)
As expected, experimental data similarly show that most chalcophile elements tend to partition into FeS-rich liquid (Bi, Cd, Cu, In, Pb, Sb, Se, Te, Zn), although for some of these elements coefficients are close to unity. Data also suggests partitioning of the chalcophile element Tl into Fe-rich phases. Limited data for Ga and Ge also suggest partitioning into the Fe-rich phase, although less than predicted, there is only limited partitioning of V into the FeS-rich phase, Co can partition into either phase depending on composition and conditions, and the extent of W partitioning into the Fe-rich phase is much lower than predicted and very variable. However, data for the variably siderophile elements Au, Ir and Pd from chondrite partial melting experiments are consistent with modelled behaviour. The limited dataset shows no clear variation in partitioning of any element as a function of temperature. Similarly, no discernible differences in element partitioning are noted between partial melting and higher temperature partitioning data sets, and there is no clear control of fO2 on behaviour of multi-valent elements. Instead, differences in bulk composition within datasets appear to have a more significant effect on element partitioning. An exception to this is certain elements (Mg, Mn, Cr and Ca, possibly also Ti) which readily form sulfides under very reducing conditions (fO2 = IW−4) (Berthet et al. 2009).
To summarise, there is only limited data on element partitioning between FeS-rich and Fe-rich phases. Light element composition of both phases is contrasting. However, many trace/minor elements are either not strongly partitioned between FeS-rich and Fe-rich phases, or partition coefficients are variable and probably dependent on numerous factors, including bulk composition. Fe-rich phases are likely enriched in highly siderophile elements as expected, although there is very limited data to support this, and partitioning of W is surprisingly variable. FeS-rich liquids are enriched in most chalcophile elements, although the extent of partitioning is moderate and variable. Sulfide chemistry can vary significantly under very reducing conditions due to silicate reduction, and presumably at much more oxidising conditions (IW + 2 or higher), Fe oxidation will eventually destabilise a metallic component. The role of O, which is readily incorporated into sulfide liquids under more oxidising conditions, is also likely to have a significant effect on element partitioning. Importantly, within constraints of the limited data set, there is no clear evidence for a strong change in element partitioning at temperatures above/below the melting point of the Fe-rich phase, or during progressive melting of silicate. Data does indicate that segregation of FeS-rich and Fe-rich components can influence chalcophile/siderophile compositions. Limited data also suggest that U, and possibly Th, partition into FeS-rich liquid, which may be important in terms of heat production in fully segregated bodies.
Finally, modelling based on FeS-rich liquid compositions in experimental studies indicates a strong control of liquid composition on element partitioning, even for chalcophile and siderophile elements. The extent of immisicibility and liquid compositions are strongly pressure dependent. As such, conditions of segregation are predicted to have a strong control on trace element partitioning, and it might be possible to differentiate the effects of low pressure sulfide/metal segregation in small bodies, from later segregation of FeS-rich and Fe-rich liquids during unmixing in the cores of larger planetesimals, especially during subsequent cooling and crystallisation.
7 Interaction of FeS-Rich and Fe-Rich Liquids with Crystalline Non-Metallic Phases
7.1 Chemical Equilibrium Between Metallic Liquids and Crystalline Silicates/Oxides
Interaction between metallic liquids and solid silicate phases can be considered in terms of Fe exchange via the redox reaction 2Feliquid + O2 = 2FeOsilicate. Under more oxidising conditions this reaction is driven to the right, which increases Fe/(Fe + Mg) ratios in silicates and modifies phase proportions. Progressive changes in silicate composition with fO2 are consistently shown in experimental studies. Berthet et al. (2009) noted FeO contents of pyroxene from 6.3 to 11.52 wt% at reducing conditions (log fO2 = IW-1.2 to IW-2.8), and 0.5 to 3.54 wt% at highly reducing experiments (log fO2 = IW−4.2 to IW−4.8). In both sets of experiments, pyroxene and olivine FeO content also decreased with increasing temperature, corresponding to a change in fO2, and increase in silicate melt fraction. Figure 7 shows molar olivine/liquid partition coefficients for both Fe-rich and FeS-rich liquids calculated from literature data. Expected decreases in DFeolivine/liquid with decreasing fO2 are clear within datasets (e.g. Gaetani and Grove 1997), although scatter in a broader dataset also demonstrates compositional controls on Fe partitioning. At very low fO2 olivine breaks down to form Ca-poor pyroxene-dominated assemblages, e.g. Fe + FeSiO3 + 1/2O2 = Fe2SiO4. Again, stability depends on conditions and composition. Berthet et al. (2009) noted that olivine breaks down to pyroxene and a free silica phase at log fO2 below IW-2 to IW-3 (at 1 GPa, in an EH4 chondritic bulk composition at 1 GPa, with fO2 controlled by choice of starting materials), while Ford et al. (2008) observed olivine in their experiments down to 6 log units below IW (at ambient pressure in an H6 ordinary chondrite composition, using both CO-CO2 gas mixes and graphite-buffered sealed silica tubes).

Molar partition coefficients for Fe, Ni, Cr, and Mn for olivine and FeS-rich liquid (black), and olivine and Fe-rich liquid (red). Squares: olivine-silicate melt-sulfide partitioning experiments of Gaetani and Grove (1997), in synthetic komatiite + sulfide and analogue olivine chondrule + sulfide systems at ambient pressure; circles: 1 GPa partial melting experiments of Berthet et al. (2009); diamonds: ambient pressure partial melting experiments of Ford et al. (2008). For Ni one data point is not shown, but its position indicated with an arrow. High Ni contents in metal and sulfide in this low temperature experiment and anomalous D value for Ni might indicate disequilibrium
There have been numerous studies of Fe:Ni exchange between olivine and sulfide, typically expressed by the exchange reaction: KD=(XNiS/XFeS)liquid/(XNiSi12O2/XFeSi12O2)olivine, where X is mole fraction. Brenan (2003) determined the fO2 dependence of this exchange over a range of conditions, down to several log units above the IW buffer. Although they observed the previously noted dependence of KD on temperature at more oxidising conditions, they noted little dependence on temperature close to the IW buffer. This change in Fe:Ni exchange behaviour close to the IW buffer implies that most studies of olivine-sulfide exchange, conducted under more oxidising conditions, are not applicable to studies of core formation in planetesimals. Extracted molar DNiolivine/liquid coefficients for olivine and Fe-rich or FeS-rich liquid from more reduced experiments (Fig. 7) verify that (1) Ni is generally very incompatible in olivine, that (2) at more reducing conditions, DNiol/sulfide is further reduced, notably in the experiments of Gaetani and Grove (1997) but also, possibly, in experiments of Ford et al. (2008). Ni is more incompatible in olivine than Fe, and compatibilities of Fe and Ni in orthopyroxene (Fig. 8) are similar. Interaction of core forming liquids with solid silicate can, therefore, lead to modification of Fe:Ni ratios in liquids, and also a reduction in Ni content of silicate phases, depending on fO2 and temperature.
There are limited data on partitioning of many trace elements between olivine and core-forming melts, partly because olivine is only able to accommodate a small range of elements. Gaetani and Grove (1997) noted that olivine-sulfide partition coefficients for Cu, Cr and Mn all decrease towards lower fO2 and the IW buffer. Cu is highly incompatible in olivine, although Cr and Mn partition coefficients can exceed unity. Within a wider data set (Fig. 7) there appears to be some control of fO2 and temperature on D-Cr (olivine/FeS-rich liquid), with Cr more incompatible at lower temperatures and under more reducing conditions. Limited data for Mn suggests that olivine/FeS-rich liquid partition coefficients scatter about unity. Similar behaviour is noted for orthopyroxene/FeS-rich liquid and orthopyroxene/Fe-rich liquid partitioning (Fig. 8). Although considerable uncertainty remains regarding formation of pallasites, chemical gradients in olivine rims record olivine/Fe-rich metal interaction, allowing comparison with experimentally derived partition coefficients. Donohue et al. (2018) note that olivine rims are variably depleted in Cr, Co and Ni, while exhibit minimal gradients in Fe and Mn, broadly consistent with experimental partitioning data from more reducing experiments (Fig. 8), although caution that samples likely record the effect of complex histories of crystal/melt interaction.
For multivalent elements, fO2 can have a considerable influence on silicate/FeS-rich liquid and silicate/Fe-rich liquid exchange. Figure 9 shows approximate fO2 ranges over which important valence changes in metal-oxide systems occur. For example, oxidation of Ni to NiO occurs mainly around 4 log units above the IW buffer, consistent with a decrease in DNiolivine/liquid for FeS-rich liquids in many experimental studies. Likewise, changes in Mo and Co valence close to the IW buffer are expected to control partitioning over core-forming conditions. The proportion of Cr2+/Cr3+ can affect partitioning behaviour, and below log fO2 = IW−4, further reduction of Cr should reduce partition coefficients considerably. For multivalent elements, progressive, small changes in average valence at fO2 several log units difference from major valence changes can result in progressive changes in element partitioning, especially where different species have markedly different compatibilities (e.g. Bromiley 2021). As such, progressive reduction in, for example, Cr partition coefficients would be expected at less reducing conditions as well. Similarly, reduction of Ti4+ to Ti3+ may result in increases or decreases in Ti partitioning, dependent on site chemistry. Complex changes in V average valence are also expected to affect V partitioning, and progressive changes in V valence may result in changes in partitioning.
For most phases stable in solid, silicate portions of planetesimals there is little to no data on element partitioning under core-forming conditions, which limits any deeper consideration of the legacy of percolation processes. The dominant effect of equilibration/reaction between core-forming melts and silicate matrices will be in controlling phase/melt proportions, sulfide O content, and Fe:Ni exchange. Incompatible element concentrations in olivine and orthopyroxene may also record evidence of reaction with Fe-rich and FeS-rich liquids. Limited experimental data and simple consideration of valence changes, for example, indicate that highly reducing conditions should result in pyroxene-dominated, Fe-poor silicates. Mineral-melt partitioning data suggests that Cr and Mn will partitioning into sulfide melts once the metallic component starts to melt, whilst Mo is preferentially retained in metallic solid (Fig. 6). However, sulfide melt could be further modified by interaction the silicate matrix through which sulfides percolates depending on fO2. For example, highly reducing conditions can enhance Cr and Ni partitioning into core-forming liquids. It is also clear from Figs. 7 and 8 that there is a compositional control on partitioning behaviour, and an effect of temperature. As such, dedicated studies of element partitioning are required to characterise signatures of core formation via percolation through a silicate matrix, including for phases such as clinopyroxene and feldspar which are important components in chondritic systems.

Molar mineral/melt partition coefficients for various elements between olivine, orthopyroxene and spinel, and ether FeS-rich liquid (sulfide) or Fe-rich liquid (metallic), calculated using data from Gaetani and Grove (1997) (olivine-sulfide system, open squares), and Cartier et al. (2014) (orthopyroxene-metallic melt in an enstatite chondrite system, black circles), and from partial melting experiments of Berthet et al. (2009) (open circles for unbuffered, reduced samples, and filled grey circles for highly reduced samples) and Ford et al. (2008) (open diamonds for experiments performed in a free gas stream, and filled, grey diamonds for experiments performed under slightly more reducing conditions in sealed silica tubes). For clarity, only full ranges are shown

At low silicate melt fractions, several minor phases may have a significant influence on element redistribution during FeS-rich/Fe-rich liquid migration, including oxides and phosphates. Presumably, stability of phosphates directly affects the P content of metallic melts, which can be considerable. Spinel group minerals are also important as they can be stable up to at least low degrees of silicate melting. Ford et al. (2008) noted that percolating Fe-rich liquids in their experiments sometimes reacted with chromite to form Mn-rich sulfide. Additional sulfide liquid phases are noted in other experimental studies, although this may in part be due to difficulties in attaining chemical equilibration across sample volumes in small melt fraction experiments (e.g. Jurewicz et al. 1995; McCoy et al. 1999; Ford et al. 2008). Berthet et al. (2009) and Jurewicz et al. (1993) noted complex, variable FeS-rich liquid compositions in run products equilibrated at different temperature and fO2, again consistent with local reaction between FeS-rich liquid and spinel/chromite. Spinel/FeS-rich liquid/ and spinel/Fe-rich liquid partition coefficients extracted from available experimental data are shown in Fig. 8. Spinel in many chondrite-based experimental run products is dominantly chromite in composition, and DCrspinel/liquid for both FeS-rich liquid and Fe-rich liquid are typically around 102. Mn also strongly partitions into spinel. Once again, fO2 likely has an important control. It is noted (Jurewicz et al. 1991) that spinels in run products are more Cr-rich at log fO2 < IW, and more Al-rich under oxidising conditions. The stability of spinel also depends on bulk composition and extent of silicate melting. At 1 GPa in an EH4 composition, Berthet et al. (2009) noted that spinel is not present under highly reducing conditions, or following the onset of silicate melting. In contrast, at ambient pressure in an H6 composition, Ford et al. (2008) noted the presence of minor chromite up to 10% silicate melting, evidence of chromite-sulfide element exchange, and progressive changes in chromite composition. Jurewicz et al. (1993) noted that in CV and CM compositions at ambient pressure, spinel is only stable at low silicate melt fractions at fO2 below the IW buffer, but under more oxidising conditions above IW, is volumetrically more important and stable to higher temperatures/melt fractions. Jurewicz et al. (1993) also observed that spinel is an important host for Cr and Ti. For some bodies, therefore, the presence of spinel up to low-degree silicate melting could influence composition of core forming melts, especially Cr, Mn (Fig. 8) and Ti, with element exchange likely to be strongly dependent on fO2. Notably, spinel/chromite is present in some pallasites, with variable Fe/Mg/Ti/Al/Cr/Mn contents which may reflect equilibrium or partial disequilibrium exchange between olivine, spinel and silicate or metallic liquids, in addition to compositional changes during cooling (Boesenberg et al. 2012). In partial melting experiments, spinel is rapidly consumed at higher temperatures/melt fractions, implying a change in sulfide/metallic liquid composition. In such systems, the presence of silicate melt will have a further complicating influence on composition of core-forming melts. As noted below, Cr tends to partition into FeS-rich liquid over silicate liquid, and thus the retention of Cr in the silicate portion of differentiating planetesimals might be dependent on the stability of spinel, with Cr readily partitioning into S-bearing liquids at higher melt fractions once spinel is no longer stable.
7.2 Kinetic Controls on the Interaction of Core-Forming Liquids with Crystalline Phases
The extent of reaction between percolating core-forming melts and the solid matrix through which they migrate will depend on the kinetics of exchange between melt and solid phases. This can be readily assessed using simple diffusion modelling based on available data, assuming that flux of species in solid phases is the limiting factor for melt/solid chemical exchange. This is reasonable in a system where (1) percolating melt forms small channels or thin sheets, typically < 0.1 mm, in a matrix with > mm sized crystals, and (2) melt migration velocities exceed element diffusivities. Here, the extent of any kinetic control on matrix/liquid exchange is assessed using model scenarios, which are in part based on available data.
Details on the modelling approach used are outlined in supplementary material, and results are shown in Fig. S1. Using published diffusivity data, 3 scenarios are investigated: (1) Cr3+ flux in olivine, (2) Fe-Mg exchange in olivine, and (3) Cr3+ and Fe-Mg exchange in spinel. Diffusion models for scenario (1) imply that under most conditions, diffusion of trace elements in olivine, even less mobile trivalent cations, has a limited control on chemical exchange between olivine and core-forming liquids. For conditions of core-formation, Cr3+ flux is rapid, with diffusion fronts extending several mm into olivine crystals over 105 to 106 timescales of core-formation. At the lowest temperatures, i.e. at the onset of FeS-melting, Cr3+ flux is more limited, and as models likely provide an upper estimate for the extent of Cr3+ flux, diffusion within olivine may partially limit olivine/FeS-rich liquid chemical exchange. However, Cr3+ diffusivity is strongly temperature-dependent, and under most conditions, mm-sized olivine will rapidly re-equilibrate with core-forming liquids.
Scenario (2) tests the extent to which Fe mobility within olivine can constrain interaction between olivine and core-forming liquids. Fe-Mg exchange in olivine is considerably faster, and models imply full re-equilibration under all conditions and timescales for planetesimal core formation. In scenario (3), kinetic controls due to interaction with spinel are assessed. Cr3+ diffusion in spinel is sluggish, and at the lowest temperatures of core segregation, i.e. close to the Fe,Ni-FeS eutectic temperature, diffusion effectively minimises the extent of spinel-liquid exchange. However, Cr3+ diffusivity in spinel is also strongly temperature dependent, and in models where core segregation is initiated by melting of the silicate portion of planetesimals, rapid diffusion in spinel enables fast matrix-liquid re-equilibration well within timescales for core-formation. Fe diffusion in spinel is considerably faster, and rapid re-equilibration with core-forming liquids would be expected under all conditions.
Initial assessment of kinetic controls suggests, therefore, only a limited effect of diffusion in crystalline phases on chemical interaction between core-forming melts and silicate matrices. In models were core-segregation starts as soon as sulfide liquids are formed, diffusion of some species may partially mask signatures of matrix/core-forming liquid interaction. This effect may be limited to accessory minerals such as spinel, although as noted, these phases have a greater influence on core-forming liquid chemistry. At higher temperatures no such kinetic control is expected, and rapid re-equilibration between matrices and percolating liquids is expected. This implies no kinetic control in models where onset of silicate melting initiates core segregation. The stability of many accessory phases within the silicate portion of planetesimals is also limited at higher temperatures, again minimising any kinetic control.
8 Chemical Equilibria Between Core-Forming Liquids and Silicate melt
In models where segregation of core-forming liquids is only effective after the onset of silicate melting, composition of FeS-rich and Fe-rich liquids will additionally be influenced by chemical exchange with silicate liquid. There have been numerous studies of silicate liquid/FeS-rich liquid and silicate liquid/Fe-rich liquid element partitioning, and partitioning of many elements as a function of variables such as pressure, temperature, fO2 and composition has been parameterised. However, studies are typically conducted in fully molten systems relevant to segregation in magma oceans. In chondritic systems undergoing small to moderate degrees of melting, silicate melt compositions can differ significantly. Before testing model applicability, it is first necessary to compare compositions of low degree silicate melts in chondritic compositions to silicate liquid compositions used in most partitioning studies.
8.1 Compositions of Low-Fraction Silicate Melts in Chondritic Systems
Compositions of low fraction silicate melts in various bulk compositions are shown in Fig. 10. A systematic decrease in SiO2 content with increasing melt fraction is evident from the recent dataset of Collinet and Grove (2020b) who demonstrate that low fraction melts in chondritic bulk compositions are broadly andesitic/dacitic and alkali-rich. A detailed comparison of melt compositions in experimental studies with bulk compositions of various achondrites is given in Collinet and Grove (2020a, b). Alkali contents are highly variable across the broader dataset of experimental silicate melt compositions shown in Fig. 10, partly due to varying melt fractions, but possibly also due to alkali loss from experimental charges. As expected, bulk composition also has a strong influence on melt chemistry.
Here, the applicabilities of three models of silicate/FeS-rich liquid element partitioning to systems with low degree partial melts are explored. Models are based on lower pressure partitioning data in S-bearing systems, and thus more relevant to core-formation in small bodies. The Steenstra et al. (2020a) model is based on a large experimental dataset and builds on previous parameterisations of element partitioning. The model allows calculation of sulfide/silicate partition coefficients for a large number of elements as a function of temperature, fO2, FeO content of both sulfide and silicate liquids, and silicate S content. Steenstra et al. (2022) is an extension of this work to include highly siderophile elements (HSE). The model of Boujibar et al. (2019) provides another useful extension to the Steenstra et al. (2020a) model, especially for alkali elements, as it uses a similar parametrisation and is based on partitioning data in slightly different bulk compositions.
Compositions of silicate liquids used in the models of Steenstra et al. (2020a, 2022) and Boujibar et al. (2019) are also shown in Fig. 10. The SiO2 content of silicate liquids is an important control on element partitioning due to its effect on polymerisation and melt structure (e.g. Bromiley 2021). The range of silicate liquid SiO2 contents from the dataset used by Steenstra et al. (2020a) is large and mostly overlaps compositions of partial silicate melts in chondritic systems, although these can extend to more SiO2-rich compositions. In an extensive study of low degree partial melting in a range on bulk compositions Collinet and Grove (2020b) noted that low-degree partial melts in chondritic systems are typically SiO2- and alkali-rich. By comparison, silicate liquids in the dataset used by Steenstra et al. (2020a) are Na2O- and K2O-poor, and CaO-rich. The presence of alkali elements can modify melt structure as they behave as network modifying cations, possibly enhancing incorporation of various species into silicate melts, and can also increase O content of sulfide liquids (Murthy et al. 2003; Steenstra et al. 2018), both of which could influence element partitioning. However, the range of FeO contents in silicate liquids in the Steenstra et al. (2020a) dataset, an important variable in modelling element partitioning, is large and overlaps that of most low degree melts in various chondritic compositions in the Collinet and Grove (2020b) study. In contrast, the range of silicate melt SiO2 contents in the Boujibar et al. (2019) dataset is limited, although melts are comparatively alkali-rich. Once again, however, there are some notable differences between melt compositions used for modelling partitioning data and compositions of chondritic partial melts. FeO contents in the Boujibar et al. (2019) dataset are, for example, comparatively low, although do match those in a small subset of partial melting experiments. As such, the models of Steenstra et al. (2020a, 2022) and Boujibar et al. (2019) might be broadly applicable to partially molten chondritic systems, although differences in melt composition and structure could limit their application, especially at very low melt fractions.

Silicate glass compositions from datasets used in the partitioning models of Steenstra et al. (2020a) (open, black circles) and Boujibar et al. (2019) (open, red circles), compared to compositions from Berthet et al. (2009) (E4 chondrite composition at 1 GPa, log fO2 ≈ IW-5 to IW-1, filled black squares), Usui et al. (2015) (H chondrite at ambient pressure, log fO2 ≈ IW-1 to IW + 2, filled grey squares), Lunning et al. (2017) (chondrite at ambient pressure, log fO2 ≈ IW to IW + 1, filled blue squares), and Gaetani and Grove (1997) (olivine/silicate liquid/sulfide liquid partitioning in a simplified chondritic system at ambient pressure, log fO2 > IW, filled yellow squares). Filled blue circles are silicate glass compositions from low degree partial melt experiments of Collinet and Grove (2020b) at 0.1–13 MPa, log fO2 ≈ IW-2.5 to IW + 0.8 for a range of chondritic compositions (from darker to lighter blue: CV, CM, LL, H, CI).
8.2 Sulfide-Silicate Melt Partitioning
Figure 11 shows sulfide/silicate partition coefficients calculated using parametrisations of Steenstra et al. (2020a, 2022) and Boujibar et al. (2019), based on compositional data from chondrite partial melting studies, and where available, partition coefficients determined directly from compositions of run products. There are currently several limitations in applying partitioning models such as these to systems with low degree partial melts: (1) Only a few studies report compositions of FeS-rich liquid demonstrably in equilibrium with low fraction silicate melt in chondritic compositions. In part this is due to difficulties in attaining equilibrium, and determining melt compositions in, systems which have undergone small amounts of melting. (2) Studies also do not report FeS-rich liquid O contents, even though sulfide liquids readily incorporate O at higher fO2. Here, O contents of sulfides in the experimental dataset are estimated based on element totals, which introduces a significant error as low totals are assumed to be due only to O incorporation. (3) Models of Steenstra et al. (2020a); Boujibar et al. (2019) do not account for changes in oxidation state of multivalent elements, although Steenstra et al. (2022) do provide partitioning models for different oxidation states for selected HSE. As the dataset used in these studies is largely from experiments performed under very reducing conditions, this implies limited applicability to segregation processes in more oxidised planetesimals. However, as discussed below, comparison of modelled partition coefficients and those determined from partial melting experiments, and the range of modelled partition coefficients with variations on silicate and sulfide FeO content, is instructive in reviewing variations in element behaviour as a function of liquid compositions and fO2.
Only limited data is available to directly test modelled partition coefficients. Both Steenstra and Boujibar models reasonably reproduce FeS-rich liquid/silicate liquid partition coefficients overall (Fig. 11A), although this is in part due to variations in liquid compositions in experimental studies at varying melt fractions and fO2, which result in large ranges in modelled partition coefficients, in some cases over 6 orders of magnitude. As expected, chalcophile elements are predicted to partition into FeS-rich liquid. Also as expected, the strongly siderophile elements Pd, Ag, Re, Ir, Pt and Au, modelled after Steenstra et al. (2022), partition strongly into FeS-rich liquid, although choice of oxidation state is important. HSE partitioning into FeS liquid is consistent with sulfide compositions in, and variable bulk HSE depletion from, achondrite meteorites (Dhaliwal et al. 2017; Day et al. 2019). The Steenstra et al. (2020a) model includes several moderately siderophile elements (Co, Ni, Mo, Sb etc.) which again strongly partition into S-rich liquid, although others (P, Ga, Mn) show a more variable behaviour, and may partition into silicate. Silicate/sulfide bulk partition coefficients calculated from a range of highly-reduced, enstatite-rich meteorites similarly show that both P and Ga are variably slightly chalcophile to lithophile (Wilbur et al. 2022). Presumably, at low S contents for metallic liquids, siderophile element partition coefficients will be higher. Partitioning between silicate and Fe-rich (i.e. low S) liquids is not directly modelled here due to limitations in the data set, which make it difficult to estimate low O contents in Fe-rich liquid data reported in the literature.
Elements such as Mg, Al, and the REE are commonly assumed to remain strongly lithophile under all conditions of core formation (e.g. Brennan et al. 2020; Faure et al. 2021). The REE and highly incompatible elements such as Zr will also readily partition into silicate melts but not into solid silicates which dominate chondritic systems, such as olivine and orthopyroxene. As such, these elements may be useful indicators of silicate melting prior to segregation of core-forming liquids. However, as noted by Steenstra et al. (2020a), higher sulfide O contents mean that some lithophile elements such as Ca, Rb and Li behave in an increasingly chalcophile manner. This is consistent with previous observations by Murthy et al. (2003) for K. Similarly, sulfide/silicate partition coefficients calculated from highly reduced (log fO2 = IW-2 to -6) enstatite-rich meteorites imply that both Ca and Na vary from strongly lithophile to chalcophile, in contrast to Mg which is always very strongly lithophile. Other elements considered moderately lithophile are predicted, and sometimes directly observed, to partition into either sulfide or silicate (Nb, Ta, U, Th) (Wohlers and Wood, 2017; Steenstra et al. 2020a). As such, direct experimental data is required to groundtruth and refine models of silicate/FeS-rich liquid element partitioning and to accurate predict the effect of silicate melting on core compositions.

A. FeS-rich liquid/silicate liquid partition coefficients, by weight. Small squares are modelled coefficients after the parameterisations of Steenstra et al. (2020a) and Steenstra et al. (2022) based on data from Berthet et al. (2009) (black squares) and McCoy et al. (1999) (red squares). Arrows at top of plot mark where calculated coefficients exceed range of figure. Red circles are modelled coefficients after the parameterisation of Boujibar et al. (2020), using data from Berthet et al. (2009). Larger data points are directly determined coefficients: blue circles: Gaetani and Grove (1997), coexisting olivine, silicate melt and sulfide melt under more oxidising conditions (fO2 > IW); green diamonds: Boujibar et al. (2019), experiments in an analogue Mercurian system under highly reducing conditions. Element valences are after Steenstra et al. (2020a) and not be strictly applicable to all data shown. Circles at the bottom of the figure signify chalcophile elements (black), highly siderophile elements (dark grey) moderately siderophile elements (open), slightly siderophile elements (black and white) and moderately/strongly lithophile elements (light grey circles). B. Subset of modelled coefficients based on Berthet et al. (2009) which highlight compositional controls. Black arrows show trends in calculated partition coefficients across a subset of experiments where temperature increases from 1500 to 1700 °C and log fO2 changes from IW-1.2 to IW-2.8. Over this temperature range silicate melting increases from 40 to 100%, and FeO content of silicate melt decreases from ≈ 16 to 2.6 wt% due to a change in fO2. Blue arrows are from a subset of more reducing experiments, log fO2 ≈ IW−4.3, where temperature increases from 1400 to 1700 °C, corresponding to an increase in silicate melting from 17 to 100%, while silicate melt FeO is constant at ≈ 7 ± 1 wt%
Some multivalent elements highlight limitations in the applicability of models to more oxidising planetesimals. Data from Gaetani and Grove (1997) indicate much stronger partitioning of V into silicate melt under more oxidising conditions than predicted by the Steenstra et al. (2020a) model, likely due to higher proportions of V4+ at log fO2 > IW. In contrast, data from Gaetani and Grove (1997) also indicate that the extent of Co partitioning into FeS-rich liquid is slightly lower at more oxidising conditions. Co is more metallic in nature below the IW buffer due to the equilibrium Co + 1/2O2 = CoO (Fig. 9), consistent with higher proportions of Co in FeS-rich liquids at more reducing conditions. However, in this instance modelled partitioning coefficients for Co from Steenstra et al. (2020a) are broadly consistent with experimental data. Comparison of partitioning data from the more oxidising experiments of Gaetani and Grove (1997) with Boujibar et al. (2019) indicates higher proportions of Cr in FeS-rich liquid under very reducing conditions, possibly due to a change in proportions of Cr3+ (the dominant oxidation state at fO2 > > IW in the Gaetani and Grove 1997, dataset) and Cr2+ (which dominates under much more reducing conditions). Sulfides in highly reduced enstatite-rich meteorites and other achondrites are similarly Cr-rich, consistent with a variably chalcophile behaviour (Wilbur et al. 2022; Goodrich et al. 2017). Conversely, calculated Cr2+ partition coefficients using the Steenstra et al. (2020a) model imply preferential incorporation of Cr into silicate melt, suggesting that Cr partitioning is also strongly dependent on the O content of FeS-rich liquids, which possibly has a more significant effect than valence changes, and on silicate FeO and S content. For other multivalent elements where significant changes in oxidation state are not noted close to the IW buffer (Ni, Cu, Ti, Mn) (Fig. 8), and/or where multiple oxidation states of species are readily incorporated into silicate liquids (Stokes et al. 2019), modelled and experimentally determined partition coefficients tend to be in close agreement. Cr, Mn and Ti are also interesting to note here as experimental data suggest that they more favourably partition into S-bearing melts over silicate melt. However, they may be retained within spinel within silicate portions of bodies (e.g. Stokes et al. 2018). Given that spinel is stable only at low silicate melt fractions, this suggests that further characterisation of element partitioning could be used to identify contrasting signatures of core formation before, and after substantial silicate melting.
Figure 11B shows general trends in modelled partition coefficients for subsets of data from Berthet et al. (2009). Black arrows correspond to a set of less reducing experiments over which there is a decrease in fO2 as well as large increase in temperature and melt fraction. For each element partition coefficients vary from ≈ 1 to 2 + orders of magnitude. The main driver for this variation is FeO content of silicate melt, which Berthet et al. (2009) note correlates with fO2, but will also vary as a function of bulk FeO content and FeO activity. Blue arrows indicate trends in modelled partition coefficients from a subset of experiments in Berthet et al. (2009) where fO2 is substantially lower, but constant around log fO2 = IW−4.3, and where temperature increases markedly, corresponding to low fraction to complete melting of silicate. There is some variation in silicate melt S content across the dataset, but little variation in melt FeO content. As a consequence, there are small variations in partition coefficients for some elements over this temperature range, although for many elements there is minimal scatter about average values. Comparison of the datasets again emphasises the dominant effect of fO2 and concomitant changes in FeO content of silicate melt and O content of sulfide, on modelled partition coefficients. However, exploration of input parameters within models suggest that S content of the metallic/sulfide liquid phase has a much weaker control on element partitioning.
9 Discussion
A proportion of planetesimals accreted soon enough after formation of first solids in the solar system, and grew rapidly enough, to retain sufficient heat to partially melt and segregate into metal-rich cores and silicate-rich mantle/crusts. Various lines of evidence suggest that when core formation did occur it was a rapid process, complete within a few Myr. Core formation may have been reliant on substantial silicate melting and formation of a ‘crystal mush’. Higher temperatures required for large fraction silicate melting significantly constrains the proportion and size of bodies which could have segregated. Alternatively, various lower temperature models of core formation have been proposed which invoke separation of liquid metal from solid silicate, or from a silicate portion which had undergone low-degree melting. A larger proportion of planetesimals would have retained sufficient heat to undergo low degree silicate melting, and especially to undergo melting of a metallic component only. Substantial differences in temperature between melting of metallic components and large-degree silicate melting, especially at higher pressures and under more reducing conditions, also raises the possibility that percolation-driven protocore formation made a substantial contribution to segregation in bodies which later underwent large-scale silicate melting. Low temperature core formation could have important implications for the compositions of metallic and silicic portions of differentiated bodies, and ultimately the compositions of planets to which planetesimals accreted. Furthermore, any lower temperature segregation process would also influence thermal history of differentiating bodies.
Experimental evidence for the viability and efficiency of percolation-driven core formation is mixed, as are estimates of metallic melt migration velocities. In the small fraction of more oxidised planetesimals, i.e. fO2 significantly exceeding that of the IW buffer, percolation of O-rich, FeS-rich liquids could have contributed to core formation at the onset of metallic melting. However, in other bodies core-forming liquids would only have formed interconnected melt networks at volume fractions which likely exceed those expected based on compositions of undifferentiated meteorites. This implies that low-temperature core-formation would be limited in less oxidised bodies to those which had inherently high metal fractions, or those which had undergone small to moderate degree silicate melting. It is possible that deformation promoted segregation of non-wetting metallic liquids, either in terms of shock processes or large-scale planetesimal deformation, although again, the viability of these processes remains unclear. Segregation of low fraction silicate melts can increase proportion of metallic liquids, although larger melt fractions are required to drive substantive changes in metallic liquid proportions. Hotter, partially molten bodies may allow more ready silicate deformation and operation of processes such as deformation-aided metallic liquid segregation. Onset of silicate melting and reduction of the viscosity of silicate matrices may also be required to allow large-scale metallic liquid segregation, which is only locally redistributed via percolation at lower temperatures.
Key constraints on the viability of percolation models are (1) initial formation of melt networks, (2) efficiency of large-scale metallic liquid migration, which also relies of deformation of the silicate portion, and (3) timescales for melt migration. Once formed, and as long as deformation of silicate portions was able to accommodate significant melt migration, percolation is likely effective in draining metallic liquid to much lower fractions than required to initiate network formation. However, processes in the latter stages of segregation, and segregation velocities, remain unclear. Scaling issues also make it difficult to extrapolate from laboratory measurements of percolation processes.
A key feature of low-temperature percolation models is that they imply variable extents of segregation and reaction between FeS-rich liquids, Fe-rich liquids and solids, crystalline phases in silicate portions, and low- to moderate-fraction silicate melts. This contrasts ‘magma ocean’ or ‘magma mush’ models where full equilibrium between metallic liquid and silicate liquid can be assumed. Data from a range of experimental studies demonstrates that conditions of early planetesimal interiors, assuming chondritic compositions not extensively modified by degassing, are always characterised by immiscibility of FeS-rich liquid and Fe-rich (i.e. S-poor) solids and liquids. FeS-rich components initially represent the first liquids formed during melting, are close to FeS in composition, and can incorporate significant O under more oxidising conditions. Under highly reducing conditions (log fO2 at around IW−4) they may also incorporate nominally lithophile elements such as Mg, Mn and Ca. Melting of Fe-rich components is not well constrained. Fe-rich liquids, in comparison to FeS-rich liquids, are O-poor but can readily incorporate minor components such as C, Si and P. FeS-rich and Fe-rich liquid compositions are dependent on fO2 and bulk composition. Pressure results in closure of the miscibility gap, although light element composition of liquids remains poorly constrained, as does the dependence of composition on temperature and fO2. Light element composition of both, in turn, has a significant effect on element partitioning between metallic liquids and other phases.
Magmatic iron meteorites evidence segregation, and at least localised accumulation, of ‘core-forming’ liquids within parent bodies. Modelled S contents of parent bodies range from 1 to 19 wt% S (Hilton et al. 2022). In contrast, immiscible Fe-rich liquids across multiple systems have S contents ranging up to 2 wt% at low pressure, and FeS-rich liquids have S contents of roughly 30 wt% S or higher. Experimental data suggests that the immiscibility gap between FeS-rich and Fe-rich liquids in chondritic compositions is only significantly closed at high pressure, although the exact role of carbon in promoting immiscibility in experimental studies remains unclear. Modelled magmatic iron parent body S contents appear consistent with segregation of Fe-rich liquids for a subset of S-poor meteorites. However, S contents of 2–19 wt% imply a parental liquid which can only be formed in chondritic compositions, at least on available data, at pressures of around 5 GPa (Corgne et al. 2008). This is comparable to pressures formed at the centre of Earth’s Moon, and greatly exceeds maximum attainable pressure in planetesimals.
There are various explanations for this ‘S discrepancy’. S could have been variably lost from planetesimals or meteorite parent bodies by degassing, especially magmatic iron parent bodies which had protracted histories and may have degassed extensively as liquid iron bodies in space (e.g. Steenstra et al. 2023; Grewal and Asimow 2023). The miscibility gap in metallic systems demonstrates that no degree of S loss is sufficient to explain intermediate S contents in modelled parental liquids at low pressure (Fig. 4), although extensive degassing and loss of C from magmatic iron parent bodies (Grewal and Asimow 2023) may have been an important factor in reducing the extent of immiscibility. Parental bodies could have formed via segregation and accumulation of both FeS-rich and Fe-rich liquids which were subsequently mixed. However, these liquids should have remained immiscible, so it is unclear how a mixture of both could be a single phase parent to magmatic iron meteorites. Alternatively, the assumption that immiscible FeS-rich and Fe-rich liquids were in equilibrium may be incorrect. Tomkins et al. (2013) proposed a shock-impact driven mechanism for non-equilibrium segregation of metallic components within planetesimals which could result in localised accumulation, and then protocore formation, of intermediate S-content liquids. However, latter stages of core segregation in such models would still result in immiscibility of Fe-rich and FeS-rich liquids, which have marked differences in density.
It is also possible that approaches used to model parental bodies to magmatic irons require refinement, as solutions to modelled parent body compositions are non-unique. Modelling approaches used parameterised solid/liquid partition coefficients (Chabot et al. 2017; Chabot and Jones 2003). In S-rich systems, these models overestimate partition coefficients compared to those obtained from direct experimental studies (Fig. 6). However, models do successfully reproduce, at least based on limited available data, HSE partitioning during chondrite-melting, and it is perhaps unlikely that FeS-rich liquids could be parental to Fe-rich meteorites. Experimental data does, however, highlight a key role of light elements in metallic liquid immiscibility. The O content of FeS-liquids, for example, can be considerable (Fig. 6), is fO2 dependent, and likely influences partitioning of other elements. Whether the presence of light elements, fO2 or pressure could significantly alter HSE partitioning, is, however, unclear.
Immiscibility between FeS-rich and Fe-rich liquids is noted during large fraction silicate melting, implying that it is important regardless of mechanism for core formation. However, conditions under which separation of FeS-rich and Fe-rich components occurred will influence light element composition. FeS-rich liquids are Ni-poor and variably O-bearing dependent on fO2. Under very reducing conditions, substantial Ti, Mg, Mn and Ca will also be incorporated into FeS-rich liquids. Fe-rich liquids are 5 to 10 times more Ni rich in chondritic bulk compositions (Fig. 3C), O-poor but variably C-rich and Si-rich (Fig. 4). Some low-temperature core formation models imply segregation of FeS-rich and Fe-rich liquids throughout planetesimal interiors. At lower pressure, immiscibility of liquids is greatest (Corgne et al. 2008). Alternatively, magma ocean models might imply segregation of, and/or chemical equilibration between, Fe-rich and FeS-rich liquids under higher pressure conditions of planetesimal cores where the miscibility gap between compositions is smaller. As such, light element compositions of cores may be sensitive, and could record, conditions of segregation. For most trace elements there is only very limited data on partitioning between Fe-rich phases and FeS-rich liquid in chondritic systems. As expected, chalcophile elements should partition into FeS-rich liquids, and siderophile elements into Fe-rich liquids. Variation in derived partition coefficients demonstrates that bulk composition has a considerable influence on element partitioning, which may in part reflect changes in light element content of both liquids. fO2 also has a clear influence on partitioning of a number of multivalent elements (Fig. 6).
Percolation of core-forming liquids also implies reaction between FeS-rich and Fe-rich liquids and solid phases within silicate portions of differentiating bodies. There is unlikely to be a significant kinetic control on the extent of reaction between solid phases and percolating metallic liquids, especially if low degree silicate melting is required to initiate significant segregation. The most significant effect of exchange is in terms of Fe/FeO redox and Fe partitioning between liquids and phase such as olivine and orthopyroxene, which are depleted in Fe under more reducing conditions. This modifies metallic compositions in terms of S contents and Fe:Ni ratios. Fe:Ni ratios in phases such as olivine will also be modified. There is, again, limited data on element partitioning. Key differences in incompatible lithophile element partitioning are expected for subsolidus and supersolidus silicate melting. The influence of minor phases stable up to low to moderate degrees of silicate melting may be considerable. Breakdown of phosphates at low degrees of silicate melting will result in marked increases in metallic P content. Stability of spinel results in retention of Cr and Mn in the silicate portion of differentiating bodies, dependent upon fO2. Behaviour of P here is notable as magmatic irons typically contain P contents of 0.01-2 wt% (Scott 2020), and modelled parent bodies (Hilton et al. 2022) 0.1-3 wt%. High P contents are consistent with at least low-degree silicate melting during differentiation. However, partitioning of P between Fe-rich and FeS-rich liquids during segregation is less clear. Although somewhat chalcophile in nature, data from some experimental studies (Fig. 6) suggests that P can partition into either phase. In contrast, Chabot and Drake (2000) demonstrated that in simple metallic systems P is preferentially incorporated in immiscible Fe-rich liquid over FeS-rich liquid, and comparison to the similarly moderately compatible As also indicates that P should also partition into Fe-rich phases (Fig. 6). As such, P contents of core-forming liquids are also dependent on equilibrium between metallic components.
Low degree silicate melts in chondritic systems are SiO2-rich and alkali-rich (Collinet and Grove 2020b). Silicate melting should result in marked changes in trace element partitioning. Existing models based on parameterisation of silicate-sulfide partition coefficients provide reasonable estimates, and also demonstrate that partition coefficients for many elements vary most significantly with fO2, concomitant with marked changes in silicate FeO content, FeO activity and, therefore, the O content of sulfide liquid. This appears to override the effects of changing silicate melt composition as a function of temperature and melt fraction. Changing silicate melt fraction, which drives small changes in S content, instead appears to result in only small changes in modelled element partitioning. An important caveat here is that the effect of temperature and extent of silicate melting on the FeO content of metal/sulfide liquids remains poorly constrained. Variations in sulfide O content will drive changes in element partitioning, although probably to a much lesser degree than large changes in silicate melt FeO. Both factors are, however, to an extent correlated. Changes in element oxidation state may limit application of models of silicate/sulfide partitioning, although the influence of fO2 and element valence on partitioning can be usefully assessed. For example, as demonstrated by Kiseeva and Wood (2013, 2015), elemental partition coefficients between sulfide and silicate liquids depend on element valence (N), with an ideal behaviour yielding a slope of N/2 for most elements. This implies that partitioning of high valence elements (W, Mo) should be much more dependent on silicate composition compared to low valence elements such as Ni and Co, at least under more reducing conditions. However, as also demonstrated by Kiseeva and Wood (2013, 2015), low or high O concentrations in sulfide melt result in ‘non-ideal’ behaviour for some elements, and other discrepancies between modelled and experimentally determined element partitioning can be considered in terms of valence changes over a range of fO2. Parameterisations for alkali elements are less reliable in chondritic systems, likely due to marked differences in silicate melt compositions. Overall, fO2 has a dominant influence on variations on element partitioning, both by controlling Fe redox and FeO partitioning, but also in terms of behaviour of a number of multivalent elements. Partitioning under more oxidised systems (fO2 > IW) for many elements is less well constrained. Future studies are required to constrain compositions of silicate, FeS-rich and Fe-rich liquids, and to test and refine portioning models for realistic low fraction silicate melt compositions.
Modelled FeS-rich liquid/silicate liquid partition coefficients based on experimental data from low degree partial melt experiments show expected trends in chalcophile and siderophile enrichment into FeS-rich liquids and lithophile element retention in silicate melts. For some elements there are marked changes in behaviour which highlight the importance of changes in silicate and sulfide liquid chemistry. For example, modelled P FeS-rich liquid/silicate liquid partition coefficients vary over 6 orders of magnitude, driven by changes in silicate FeO content. This is in accordance with Kiseeva and Wood (2013, 2015), as the dominant 5 + valence of P implies a value N/2 = 2.5, and thus a strong dependence of partitioning on silicate melt FeO content. As such, the nominally siderophile, P strongly partitions into silicate melt at log fO2 close to the IW buffer, as verified from limited experimental data, but partitions into FeS-rich liquids under very reducing conditions (below log fO2 = IW−4). Coupled changes in liquid composition and fO2 may mask variations in element partitioning with increasing temperature and increasing melt fraction, and also drive important changes in element behaviour. The scale and influence of variations in FeS-rich liquid FeO content is poorly constrained. Extension of partitioning models to more oxidising conditions, and to a greater range of melt compositions, might allow signatures of low vs. high fraction silicate melting to be distinguished. As noted, behaviour of incompatible lithophile elements such as Zr and the rare-Earths (REE) may allow the effects of Fe-liquid segregation before and after the onset of silicate melting to be ascertained. For other elements, complexity of melt processes in meteorite parent bodies and a lack of experimental data limit this at present. For example, some nominally lithophile elements become more chalcophile at either high or very low sulfide FeO contents. For example, Mg, Ca, Li, Rb and Cs partitioning is increased both at very low silicate FeO and very high silicate FeO contents, driven by changes in O content of sulfide melt and S content of silicate melt (Fig. 11 and Steenstra et al. 2020a). For other elements, the effects of increasing S in silicate melt at very low FeO content of silicate melt is more significant than the positive O effect at very high FeO content of silicate melt or vice versa. As such, given complexities in elemental behaviour, it remains possible that trace element concentrations in core-forming liquids do not readily evidence differences due to segregation during low degree silicate melting vs. high degree melting and formation of a magma mush.
There are several areas of uncertainty regarding the geochemical consequences of different mechanisms for core-formation which require redress. (1) One key uncertainty is light element (C, O, Si, P) composition of immiscible FeS-rich and Fe-rich liquids. fO2 has a strong control on Fe redox, on silicate FeO content, on Fe/Ni/S contents of metallic liquids and on FeS-rich liquid O content. Highly reducing conditions also result in several additional elements such as Mg and Mn readily incorporating into FeS-rich liquid. FeS-rich and Fe-rich liquids are immiscible and have contrasting light element compositions. Increasing pressure reduces the extent of immiscibility, but 2 liquids remain stable under all conditions of core formation in planetesimals, unless compositions are substantially modified by degassing and volatile loss. Constraining compositions of liquids over variable pressure/temperature/ fO2 would be instructive in refining models for core formation. Light element composition of liquids in turn has a significant influence on partitioning of other elements. (2) Melt relations for Fe-rich (S-poor) components remain poorly constrained in many systems. Melting of Fe-rich components appears to occur prior to significant silicate melting, even at very low pressures, although this remains to be determined. (3) Current parameterisations of metallic solid/liquid trace element partitioning are not designed to predict S-rich systems, or partitioning following Fe-rich melting, and likely overestimate the extent of element partitioning. fO2 probably has a significant role in trace element partitioning, as does minor element composition. In theory, rigorous partitioning data could provide valuable insight into separation of metallic phases during planetesimal evolution, including during conditions and mechanisms for core formation and later processes such as immiscibility within protocores. This data might also be useful in more fully exploring later crystallisation of parent bodies to magmatic iron meteorites. (4) Minor phase stability could have a strong influence on composition of core-forming liquids, and may provide a ready means for determining whether onset of silicate melting is required to initiate large-scale segregation. However, due to small melt and phase fractions in chondritic systems, there is limited data on phase stability and evolution of FeS-rich and Fe-rich liquid compositions before and after the onset of silicate melting. Finally, although models for FeS-rich liquid/silicate liquid partitioning provide reasonable predictions (5) the key role of fO2 in terms of phase/melt relations and element partitioning, and element partitioning in low melt-fraction systems, needs to be better constrained. This would facilitate identification of key signatures of segregation following low fraction silicate melting (percolation-driven) vs. segregation following large degree silicate melting (magma ocean/mush model).
Preliminary data analysis does, however, highlight numerous ways in which signatures of planetesimal differentiation depend on segregation processes. Planetesimal differentiation is important in understanding not just the evolution of small bodies, but also the composition of terrestrial planets to which they accreted. More widely, exploration of low temperature models of metal segregation provides insight into planetary processes in extrasolar systems. In our solar system, thermal models suggest that breakdown of the short-lived radioisotope Al26 provided sufficient heat to promote melting in smaller bodies. However, Al26 concentration is probably a consequence of the supernovae processes which promoted initial gravitational collapse of material which would form our solar system. In the absence of sufficient concentrations of this radioisotope, solar system differentiation and planetary evolution may have been very different, with large-scale segregation confined to much larger bodies which could more efficiently retain heat from energetic impacts. As a consequence, understanding conditions under which core formation can occur, and constraints of large-scale differentiation, is insightful in exploring planetary evolution beyond our solar system.
Change history
16 September 2023
The Google Scholar link for reference Berg et al. 2018 has been updated.
References
M.D. Abramoff, P.J. Magelhaes, S.J. Ram, Biophotonics Int. 117, 36 (2004)
C.B. Agee, J. Geophys. Res. 98, 5419 (1993)
C.B. Agee, J. LI, M.C. Shannon, S. Circone, J. Geophys. Res. Earth. 100, 17725 (1995)
N. Bagdassarov, G.J. Golabek, G. Solferino, M.W. Schmidt, Phys. Earth Planet. Inter. 177, 139 (2009a)
N. Bagdassarov, G. Solferino, G.J. Golabek, M.W. Schmidt, Earth Planet. Sci. Lett. 288, 84 (2009b)
H.L. Bercovici, L.T. Elkins-Tanton, J.G. O’Rourke, L. Schaefer, Icarus. 380, 114976 (2022)
M.T.L. Berg, G.D. Bromiley, I.B. Butler, M. Frost, R. Bradley, J. Carr, Y. Le Godec, L.G.J. Montési, W. Zhu, K. Miller, J.-P. Perrillat, E. Mariani, D. Tatham, S.A.T. Redfern, Phys. Earth Planet. Inter 263, 38 (2017)
M.T.L. Berg, G.D. Bromiley, Y. Le Godec, J. Philippe, M. Mezouar, J.-P. Perrillat, N.J. Potts, Front. Earth Sci. 6, 77 (2018)
S. Berthet, V. Malavergne, K. Righter, Geochim. Cosmochim. Acta. 73, 6402 (2009)
J.S. Boesenberg, J.S. Delaney, R.H. Hewins, Geochim. Cosmochim. Acta. 89, 134 (2012)
A.A. Borisov, Petrology. 21, 305 (2013)
A. Borisov, H. Palme, in Lunar Planet. Sci. Conf. (1995), p. 147
A. Boujibar, M. Habermann, K. Righter, D.K. Ross, K. Pando, M. Righter, B.A. Chidester, L.R. Danielson, Am. Mineral. 104, 1221 (2019)
A. Boujibar, K. Righter, E.S. Bullock, Z. Du, Y. Fei, Geochim. Cosmochim. Acta. 269, 622 (2020)
J.M. Brenan, Geochim. Cosmochim. Acta. 67, 2663 (2003)
M.C. Brennan, R.A. Fischer, J.C.E. Irving, Earth Planet. Sci. Lett. 530, 115923 (2020)
G.D. Bromiley, Lithos. 384–385, 105900 (2021)
D. Bruhn, N. Groebner, D.L. Kohlstedt, Nature. 403, 883 (2000)
C. Cartier, T. Hammouda, R. Doucelance, M. Boyet, J.-L. Devidal, B. Moine, Geochim. Cosmochim. Acta. 130, 167 (2014)
V. Cerantola, N.P. Walte, D.C. Rubie, Earth Planet. Sci. Lett. 417, 67 (2015)
N.L. Chabot, Geochim. Cosmochim. Acta. 68, 3607 (2004)
N.L. Chabot, M.J. Drake, Meteorit Planet. Sci. 35, 807 (2000)
N.L. Chabot, J.H. Jones, Meteorit Planet. Sci. 38, 1425 (2003)
N.L. Chabot, E.A. Wollack, W.F. McDonough, R.D. Ash, S.A. Saslow, Meteorit Planet. Sci. 52, 1133 (2017)
M. Collinet, T.L. Grove, Geochim. Cosmochim. Acta. 277, 358 (2020a)
M. Collinet, T.L. Grove, Geochim. Cosmochim. Acta. 277, 334 (2020b)
A. Corgne, B.J. Wood, Y. Fei, Geochim. Cosmochim. Acta. 72, 2409 (2008)
J.M.D. Day, C.A. Corder, N. Assayag, P. Cartigny, Geochim. Cosmochim. Acta. 266, 544 (2019)
J.K. Dhaliwal, J.M.D. Day, C.A. Corder, K.T. Tait, K. Marti, N. Assayag, P. Cartigny, D. III Rumble, L.A. Taylor, Geochim. Cosmochim. Acta. 216, 115 (2017)
J.K. Dhaliwal, J.M.D. Day, C.A. Corder, K.T. Tait, K. Marti, N. Assayag, P. Cartigny, D. III Rumble, L.A. Taylor, Geochim. Cosmochim. Acta. 254, 310 (2019)
K. Dodds, J.F.J. Bryson, J. Neufeld, R.J. Harrison, J. Geophys. Res. Planets. 126, e2020JE006704 (2021)
P.H. Donohue, E. Hill, G.R. Huss, Geochim. Cosmochim. Acta. 222, 305 (2018)
L.T. Elkins-Tanton, B.P. Weiss, M.T. Zuber, Earth Planet. Sci. Lett. 305, 1 (2011)
P. Faure, M. Boyet, M.A. Bouhifd, G. Manthilake, T. Hammouda, J.-L. Devidal, Earth Planet. Sci. Lett. 554, 116644 (2021)
Y. Fei, C.M. Bertka, L.W. Finger, Science. 275, 1621 (1997)
R.L. Ford, G.K. Benedix, T.J. McCoy, T. Rushmer, Meteorit Planet. Sci. 43, 1399 (2008)
G.A. Gaetani, T.L. Grove, Geochim. Cosmochim. Acta. 61, 1829 (1997)
S. Ghanbarzadeh, M.A. Hesse, M. Prodanovic, Proc. Natl. Acad. Sci. 114, 13406 (2017)
C.A. Goodrich, N.T. Kita, Q.-Z. Yin, N.E. Sanborn, C.D. Williams, D. Nakashima, M.D. Lane, S. Boyle, Geochim. Cosmochim. Acta. 203, 381 (2017)
R.C. Greenwood, I.A. Franchi, A. Jambon, P.C. Buchanan, Nature. 435, 916 (2005)
D.S. Grewal, P.D. Asimow, Geochim. Cosmochim. Acta. 344, 146 (2023)
N. Groebner, D.L. Kohstedt, Earth Planet. Sci. Lett. 245, 571 (2006)
C.D. Hilton, R.D. Ash, R.J. Walker, Geochim. Cosmochim. Acta. 318, 112 (2022)
M.M. Hirschmann, E.A. Bergin, G.A. Blake, F.J. Ciesla, J. Li, Proc. Natl. Acad. Sci. 11, e2026779118 (2021)
A. Holzheid, H.S. O’Neill, Geochim. Cosmochim. Acta. 59, 475 (1995)
A. Holzheid, M.D. Schmitz, T.L. Grove, J. Geophys. Res. Earth. 105, 13555 (2000)
T. Hopp, T. Kleine, Geochim. Cosmochim. Acta. 302, 46 (2021)
J.W. Hustoft, D.L. Kohlstedt, Geochem. Geophys Geosyst. 7, 11 (2006)
J.H. Jones, M.J. Drake, Nature. 322, 221 (1986)
A.J.G. Jurewicz, D.W. Mittlefehldt, J.H. Jones, Science. 252, 695 (1991)
A.J.G. Jurewicz, D.W. Mittlefehldt, J.H. Jones, Geochim. Cosmochim. Acta. 57, 2123 (1993)
A.J.G. Jurewicz, D.W. Mittlefehldt, J.H. Jones, Geochim. Cosmochim. Acta. 59, 391 (1995)
E.S. Kiseeva, B.J. Wood, Earth Planet. Sci. Lett. 383, 68 (2013)
E.S. Kiseeva, B.J. Wood, Earth Planet. Sci. Lett. 424, 280 (2015)
T. Kleine, M. Wadhwa, in Planetesimals Early Differ. Consequences Planets, edited by L. Elikins-Tanton and B. Weiss (Cambridge University Press, Cambridge, UK, 2017), pp. 224–244
T. Kleine, C. Munker, K. Mezger, H. Palme, Nature. 418, 952 (2002)
T.S. Kruijer, M. Touboul, M. Fischer-Godde, K.R. Bermingham, R.J. Walker, T. Kleine, Science. 344, 1150 (2014)
T.S. Kruijer, C. Burkhardt, G. Budde, T. Kleine, Proc. Natl. Acad. Sci. 114, 6712 (2017)
T. Lichtenberg, T. Keller, R.E. Katz, G.J. Golabek, T.V. Gerya, Earth Planet. Sci. Lett. 507, 154 (2019)
T. Lichtenberg, J. Drazkowska, M. Schonbachler, G.J. Golabekt, T.O. Hands, Science. 371, 365 (2021)
N.G. Lunning, K.G. Gardner-Vandy, E.S. Sosa, T.J. Mccoy, E.S. Bullock, C.M. Corrigan, Geochim. Cosmochim. Acta. 214, 73 (2017)
C. Ma, J.R. Beckett, Meteorit. & Planet. Sci. 56, 96 (2021)
V. Malavergne, P. Cordier, K. Righter, F. Brunet, B. Zanda, A. Addad, T. Smith, H. Bureau, S. Surble, C. Raepsaet, E. Charon, R.H. Hewins, Earth Planet. Sci. Lett. 394, 186 (2014)
E.R. Mare, A.G. Tomkins, B.M. Godel, Meteorit Planet. Sci. 49, 636 (2014)
C. Maurel, J.F.J. Bryson, R.J. Lyons, M.R. Ball, R. Chopdekar, V.A. Scholl, F.J. Ciesla, W.F. Bottke, B.P. Weiss, Sci. Adv. 6, (2020)
T.J. McCoy, K. Keil, D.W. Muenow, L. Wilson, Geochim. Cosmochim. Acta. 61, 639 (1997)
T.J. McCoy, T.L. Dickinson, G.E. Lofgren, Meteorit. & Planet. Sci. 34, 735 (1999)
T.J. McCoy, W.D. Carlson, L.R. Nittler, R.M. Stroud, D.D. Bogard, D.H. Garrison, Geochim. Cosmochim. Acta. 70, 516 (2006a)
T.J. McCoy, D.W. Mittlefehldt, L. Wilson, in Meteorites Early Sol. Syst. II, edited by S. L. Dante and H. Y. McSween (University of Arizona Press, 2006b)
S.J. McKibbin, L. Pittarello, C. Makarona, C. Hamann, L. Hecht, S.M. Chernonozhkin, S. Goderis, P. Claeys, Meteorit Planet. Sci. 54, 2814 (2019)
D.W. Mittlefehldt, M.M. Lindstrom, D.D. Bogard, D.H. Garrison, S.W. Field, Geochim. Cosmochim. Acta. 60, 867 (1996)
V.R. Murthy, W. van Westrenen, Y. Fei, Nature. 423, 163 (2003)
A. Neri, J. Guignard, M. Monnereau, M.J. Toplis, G. Quitte, Earth Planet. Sci. Lett. 518, 40 (2019)
P. Ni, N.L. Chabot, C.J. Ryan, A. Shahar, Nat. Geosci. 13, 611 (2020)
H.S. O’Neill, Am. Mineral. 73, 470 (1988)
H.S. O’Neill, M.I. Pownceby, Contrib. Mineral. Petrol. 114, 296 (1993a)
H.S. O’Neill, M.I. Pownceby, Contrib. Mineral. Petrol. 114, 315 (1993b)
K. Righter, Annu. Rev. Earth Planet. Sci. 31, 135 (2003)
K. Righter, M.J. Drake, Icarus. 124, 513 (1996)
K. Righter, M. Drake, E. Scott, Meteorites Early Sol. Syst. II (2006)
J.J. Roberts, J.H. Kinney, J. Siebert, F.J. Ryerson, Geophys. Res. Lett. 34, L14306 (2007)
J.F. Rudge, T. Kleine, B. Bourdon, Nat. Geosci. 3, 439 (2010)
T. Rushmer, N. Petford, Geochem. Geophys Geosyst (2011). https://doi.org/10.1029/2010GC003413
T. Rushmer, W. Minarik, G. Taylor, in Orig. Earth Moon, edited by R. M. Canup and K. Righter (University of Arizona Press, Tuscon, 2000), pp. 227–243
T. Rushmer, N. Petford, M. Humayun, A.J. Campbell, Earth Planet. Sci. Lett. 239, 185 (2005)
A. Scheinberg, R.R. Fu, L.T. Elkins-Tanton, B.P. Weiss, in (2015)
E.R.D. Scott, Oxford Research Encyclopedia of Planetary Science (Oxford Univeristy Press, 2020). https://doi.org/10.1093/acrefore/9780190647926.013.206
M.C. Shannon, C.B. Agee, Science. 280, 1059 (1998)
C.Y. Shi, L. Zhang, W.G. Yang, Y.J. Liu, J.Y. Wang, Y. Meng, J.C. Andrews, W.L. Mao, Nat. Geosci. 6, 971 (2013)
G.F.D. Solferino, P.R. Thomson, S. Hier-Majumder, Front. Earth Sci. (2020). https://doi.org/10.3389/feart.2020.00339
E.S. Steenstra, N. Agmon, J. Berndt, S. Klemme, S. Matveev, W. van Westrenen, Sci. Rep. 8, 7053 (2018)
E.S. Steenstra, E. Kelderman, J. Berndt, S. Klemme, E.S. Bullock, W. van Westrenen, Geochim. Cosmochim. Acta. 286, 248 (2020a)
E.S. Steenstra, V.T. Trautner, J. Berndt, S. Klemme, W. van Westrenen, Icarus 335, (2020b)
E.S. Steenstra, J. Berndt, A. Rohrbach, E.S. Bullcok, W. van Westernen, S. Klemme, M.J. Walter, Geochim. Cosmochim. Acta. 336, 15 (2022)
E.S. Steenstra, C.J. Renggli, J. Berndt, S. Klemme, 54th Lunar Planet. Sci. Conf. 54, 142 (2023)
T.N. Stokes, G.D. Bromiley, G.D. Gatta, N. Rotiroti, N.J. Potts, K. Saunders, Mineral. Mag. 82, 975 (2018)
T.N. Stokes, G.D. Bromiley, N.J. Potts, K.E. Saunders, A.J. Miles, Chem. Geol. 506, 162 (2019)
S.R. Taylor, M.D. Norman, in Orig. Earth, edited by H. Newsom and J. Jones (Oxford University Press, New York, 1990), pp. 29–43
G.J. Taylor, K. Keil, T. McCoy, H. Haack, E.R.D. Scott, Meteoritics. 28, 34 (1993)
H. Terasaki, D.J. Frost, D.C. Rubie, F. Langenhorst, Earth Planet. Sci. Lett. 232, 379 (2005)
H. Terasaki, D.J. Frost, D.C. Rubie, F. Langenhorst, Earth Planet. Sci. Lett. 273, 132 (2008)
K.A. Todd, H.C. Watson, T. Yu, Y.B. Wang, Am. Mineral. 101, 1996 (2016)
A.G. Tomkins, Meteorit Planet. Sci. 44, 1133 (2009)
A.G. Tomkins, R.F. Weinberg, B.F. Schaefer, A. Langendam, Geochim. Cosmochim. Acta. 100, 41 (2013)
A.G. Tomkins, T.E. Johnson, J.T. Mitchell, Meteorit Planet. Sci. 55, 857 (2020)
T.M. Usselman, Am. J. Sci. 275, 278 (1975)
T. Usui, J.H. Jones, D.W. Mittlefehldt, Meteorit. & Planet. Sci. 50, 759 (2015)
N.P. Walte, J.K. Becker, P.D. Bons, D.C. Ruble, D.J. Frost, Earth Planet. Sci. Lett. 262, 517 (2007)
N.P. Walte, D.C. Rubie, P.D. Bons, D.J. Frost, Earth Planet. Sci. Lett. 305, 124 (2011)
K. Wang, J.M.D. Day, R.L. Korotev, R.A. Zeigler, F. Moynier, Earth Planet. Sci. Lett. 392, 124 (2014)
H.C. Watson, J.J. Roberts, Phys. Earth Planet. Inter. 186, 172 (2011)
Z.E. Wilbur, A. Udry, F.M. McCubbin, K.E. Vander Kaaden, C. Defelice, K. Ziegler, D.K. Ross, T.J. McCoy, J. Gross, J.J. Barnes, N. Dygert, R.A. Zeigler, B.D. Turrin, C. McCoy, Meteorit Planet. Sci. 57, 1387 (2022)
A. Wohlers and B. J. Wood, Geochim. Cosmochim. Acta 205, 226 (2017)
B.J. Wood, Philos. Trans. R Soc. a-Mathematical Phys. Eng. Sci. 366, 4339 (2008)
B.J. Wood, E.S. Kiseeva, Am. Mineral. 100, 2371 (2015)
B.J. Wood, M.J. Walter, J. Wade, Nature. 441, 825 (2006)
T. Yoshino, M.J. Walter, T. Katsura, Nature. 422, 154 (2003)
Z. Zhang, P. Hastings, A. Von der Handt, M.M. Hisrchmann, Geochim. Cosmochim. Acta. 225, 66 (2018)
Acknowledgements
GDB would like to thank two anonymous reviewers, whose comments and suggestions improved this manuscript.
Author information
Authors and Affiliations
Contributions
All results included in the manuscript are original and solely the work of GB. GB also prepared the manuscript in full.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bromiley, G.D. The Geochemical Legacy of Low-Temperature, Percolation-Driven Core Formation in Planetesimals. Earth Moon Planets 127, 4 (2023). https://doi.org/10.1007/s11038-023-09552-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11038-023-09552-2