
Abstract
Background
Hepatocellular carcinoma (HCC) is a common cancer worldwide, and sorafenib is a first-line drug for the treatment of advanced liver cancer. Resistance to sorafenib has become a major challenge in the treatment of hepatocellular carcinoma, however, studies have shown that metformin can promote ferroptosis and sorafenib sensitivity. Therefore, the aim of this study was to investigate the promotion of ferroptosis and sorafenib sensitivity by metformin via ATF4/STAT3 in hepatocellular carcinoma cells.
Methods
Hepatocellular carcinoma cells Huh7 and Hep3B and induced sorafenib resistance (SR) Huh7/SR and Hep3B/SR cells were used as in vitro cell models. Cells were injected subcutaneously to establish a drug-resistant mouse model. CCK-8 was used to detect cell viability and sorafenib IC50. Western blotting was used to detect the expression of relevant proteins. BODIPY staining was used to analyze the lipid peroxidation level in cells. A scratch assay was used to detect cell migration. Transwell assays were used to detect cell invasion. Immunofluorescence was used to localize the expression of ATF4 and STAT3.
Results
Metformin promoted ferroptosis in hepatocellular carcinoma cells through ATF4/STAT3, decreased sorafenib IC50, increased ROS and lipid peroxidation levels, decreased cell migration and invasion, inhibited the expression of the drug-resistant proteins ABCG2 and P-GP in hepatocellular carcinoma cells, and thus inhibited sorafenib resistance in hepatocellular carcinoma cells. Downregulating ATF4 inhibited the phosphorylated nuclear translocation of STAT3, promoted ferroptosis, and increased the sensitivity of Huh7 cells to sorafenib. Metformin was also shown in animal models to promote ferroptosis and sorafenib sensitivity in vivo via ATF4/STAT3.
Conclusion
Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3, and it inhibits HCC progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide, and its incidence continues to rise, with approximately 110,000 deaths from liver cancer in China each year [1]. At present, there is no ideal treatment for HCC [2]. Surgical resection and transplantation are mainly used to treat early-stage liver cancer. Sorafenib, a first-line drug for the treatment of advanced liver cancer, is beneficial for progressive cases or patients who are not suitable for surgery, but many patients with liver cancer respond poorly to sorafenib or develop resistance to it after several months [3, 4]. More appropriate treatments or changes in treatment strategies are therefore needed to achieve better outcomes.
Metformin is a first-line drug for the treatment of type 2 diabetes, and the drug has received attention for its pleiotropic properties and reduced morbidity after treatment [5]. Metformin exerts its antitumor effects by inhibiting the survival and proliferation of cancer cells and inducing cell cycle arrest, autophagy and apoptosis [6,7,8]. Recent studies have identified potential modulatory effects of metformin on nonapoptotic forms of cell death (including ferroptosis, pyroptosis, and necroptosis) as a potentially promising therapeutic strategy for cancer treatment [9]. Metformin has been shown to exert tumor suppressive effects in liver cancer and other cancers [10]. A number of in vivo and in vitro studies support the clinical use of metformin for the treatment of HCC [11]. Studies have shown that a combination of metformin and sorafenib inhibits cell proliferation and promotes apoptosis, inhibits the epithelial-mesenchymal transition process in vitro and in vivo, and promotes sorafenib sensitivity, which reduces postoperative recurrence and pulmonary metastasis in HCC [12]. In addition, the combination of metformin and sorafenib has been found to enhance the anticancer efficacy of sorafenib by promoting autophagy in HCC cells to induce apoptosis [13]. However, the mechanism of action by which metformin promotes sorafenib sensitivity to inhibit HCC progression is not known.
Ferroptosis is a form of iron-dependent cell death characterized by reactive oxygen species (ROS) generation and iron overload, leading to abnormal accumulation of lethal levels of lipid peroxides [14]. Ferroptosis is closely associated with the development and progression of various diseases, including degenerative diseases, neoplastic diseases, and ischemic injury [15]. The liver is the main site of iron storage in the body, and ferroptosis plays a crucial role in hepatocellular carcinoma. Studies have found that metformin can induce ferroptosis as a potential anticancer agent [16]. Metformin also activates AMPK pathways through the induction of transcription factor 4 (ATF4), which produces anticancer effects [17]. ATF4 is a protective gene for the endoplasmic reticulum and oxidative stress and can activate genes required for various forms of cell stress, thus regulating the adaptive function of cells. Studies have shown that ATF4 affects the antioxidant or pro-oxidation index to influence ferroptosis during HCC [18]. Signal transduction and transcription activator 3 (STAT3) can be activated by ER stress and is involved in regulating ER stress and cell drug resistance [19, 20]. Studies have found that STAT3 knockdown can promote sorafenib-induced ER stress-induced HCC apoptosis [21]. In addition, sorafenib has been found to be a contributor to ferroptosis [22]. The depletion of intracellular iron reserves prevents the induction of oxidative stress by sorafenib in HCC cells, suggesting that ferroptosis may be a target for HCC drug therapy, thereby optimizing the therapeutic effect of sorafenib [23]. The use of metformin to induce ferroptosis may enhance the efficacy of sorafenib, which has important implications for exploring treatment strategies for hepatocellular carcinoma. We speculate that metformin affects hepatocellular carcinoma cells sensitivity to sorafenib through the ATF4/STAT3 molecular pathway.
This study was designed to investigate the promotion of ferroptosis and sorafenib sensitivity by metformin in hepatocellular carcinoma cells via ATF4/STAT3. A metformin-sorafenib drug combination may enhance the therapeutic efficacy of sorafenib in hepatocellular carcinoma, providing a new and promising strategy for the treatment of hepatocellular carcinoma.
Materials and methods
Cell culture and processing
The hepatoma cell lines Huh7 and Hep3B were obtained from Procell Life Science & Technology Co., Ltd. (Wuhan, China). Cells were cultured in DMEM (Gibco, USA) containing 10% fetal bovine serum and 1% penicillin/streptomycin in a 37 °C incubator supplemented with 5% CO2. Sorafenib (Santa Cruz Biotechnology) was used to establish sorafenib-resistant (SR) Huh7/SR and Hep3B/SR cell models. Cells were transfected at approximately 90% cell fusion using an ATF4 overexpression plasmid (oe-ATF4), siRNA against ATF4 (si-ATF4), and a negative control (oe-NC/si-NC), according to the Lipofectamine 3000 (Thermo Fisher Scientific, USA) instructions.
Mouse liver cancer model
Tumor cells at the logarithmic growth stage were digested, centrifuged, and placed in DMEM for use. Nude mice (SPF grade thymus-free Balb/C nude mice, male, 4–6 weeks old, 16–18 g body weight) were anesthetized with 3% sodium pentobarbital, and the cells were injected subcutaneously into the right abdomen of the mice. Four weeks later, the mice were euthanised and dissected to observe liver tumor growth.
CCK-8
Cell proliferation viability was assayed using a CCK-8 kit (Solarbio, Beijing, China). Cell suspensions were inoculated in 96-well plates at a dose of 100 µL per well and incubated at 37 °C for 24 h. Different groups of cells were treated with metformin or sorafenib and incubated for the appropriate times. Ten microliters of CCK-8 solution was added to each well at the corresponding time points and incubated for 2 h. Absorbance at 450 nm was measured using an enzyme marker.
Western blot
The collected cells were lysed using lysis buffer, and the supernatant was extracted following centrifugation. Protein concentrations were measured using a BCA reaction kit (Thermo Scientific, USA). Target proteins were then separated by 10% SDS polyacrylamide gel electrophoresis, transferred to PVDF membranes and blocked at room temperature for 3 h using 5% skim milk. Prediluted primary antibodies against ABCG2, P-GP, GPX4, ACSL4, ATF4, STAT3, p-STAT3, STAT1, and p-STAT1 (Abcam, UK) were added, and cells were incubated overnight at 4 °C on a shaker with slow shaking, then incubated with the corresponding HRP-coupled secondary antibody for 1 h at room temperature. Immunoreactive bands were detected by the ECL system using an image reader, and densitometric analysis was performed with ImageJ.
Determination of the Fe2+ content
The Fe2+ content of the cells was assayed using an iron assay kit (Sigma‒Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions and as previously described (Wang et al., 2019).
Determination of ROS content
Logarithmic growth phase cells were collected, digested, and centrifuged, inoculated in 6-well plates, and cultured for 24 h. Different groups of cells were treated, collected after 24 h, and incubated with diluted DCFH-DA at 37 °C in a 5% CO2 incubator for 1.5 h. Each group of cells was harvested, and the fluorescence of DCF was detected using flow cytometry.
Lipid peroxidation assay
Lipid peroxidation levels in cells were analyzed by BODIPY staining. Cells were treated with BODIPY for 30 min, harvested, and resuspended in PBS. The fluorescence of BODIPY was detected using fluorescence microscope.
Scratch detection of cell migration
Cells were inoculated in 6-well plates and incubated for 24 h before being scribed with a 20 µL gun tip. After scribing was complete, cells were washed 3 times using sterile PBS to remove the scribed cells (so that the remaining gaps were clearly visible to the naked eye), then placed in fresh, serum-free medium and incubated at 37 °C in a 5% CO2 incubator. Cells were removed after appropriate time points (0, 6, 12, 24 h), observed under the microscope and photographed.
Transwell assays for cell invasion
Logarithmic growth phase cells were digested with trypsin and resuspended in serum-free medium. A layer of diluted Matrigel was applied to the upper surface of the PET film, left at 37 °C for 3 h, and dried overnight. Then, 600 µL of medium containing 10% serum was added to the lower chamber, and 100 µL of cell suspension was added to the upper chamber and incubated for 24 h. The lower surface was immersed in 70% methanol solution, fixed for 30 min, stained with crystal violet, microscopically examined, and the number of cells on the lower surface of the PET membrane was counted. The average for the middle and five surrounding fields of view was calculated.
Immunofluorescence
Cells were fixed using 4% paraformaldehyde for 15 min and washed three times in PBS. Diluted primary antibodies against ATF4 and STAT3 (Abcam, UK) were added dropwise for 30 min at 37 °C, and then cells were washed three times in PBS. Fluorescently labeled goat anti-mouse secondary antibody (Abcam, USA) was added dropwise for 30 min at 37 °C, and the cells were then restained using DAPI (Sigma‒Aldrich, USA). Images were collected with a fluorescence microscope.
Immunoprecipitation
Cells were collected and lysed using IP lysis buffer, and the supernatant was extracted following centrifugation. ATF4 antibody (Abcam, USA) was added and cells were incubated overnight at 4 °C with slow shaking. Pretreated protein A agarose beads were added and cells were incubated for 3 h at 4 °C with slow shaking to couple the antibody to the protein A agarose beads. The beads were collected by centrifugation and then washed three times with lysis buffer. Immunoprecipitates were analyzed by immunoblotting.
Immunohistochemistry
Sections were dewaxed in xylene, washed in buffer, and incubated dropwise with diluted primary antibodies against KI-67, ATF4, and STAT3 (Abcam, UK) overnight at 4 °C. The sections were washed in buffer, horseradish-labeled secondary antibody was added dropwise, and the sections were incubated for 2 h at 37 °C. The sections were developed with DAB-H2O2, followed by Mayer hematoxylin staining, washing in water, alcohol fractionation in hydrochloric acid, and dehydration. Neutral gum was used to seal the slices for photography.
Determination of MDA and GSH content
The levels of the oxidative stress markers MDA and GSH were determined with an MDA assay kit (Beyotime, Shanghai, China) and a GSH assay kit (Solarbio, Beijing, China), according to the manufacturer’s instructions.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, USA). All experiments were set up in three parallel groups and replicated three times. The results are expressed as the mean ± standard deviation. Differences between groups were compared by t test, and differences between multiple groups were compared by one-way ANOVA. p < 0.05 was considered statistically significant, and p < 0.01 was considered extremely statistically significant.
Results
Metformin promotes ferroptosis in hepatocellular carcinoma cells
First, the drug-resistant cell lines Huh7/SR and Hep3B/SR were established using sorafenib induction. Huh7 and Hep3B cells were cultured to develop sorafenib resistance by gradually increasing the concentration of sorafenib. The CCK-8 assay results showed a significant increase in sorafenib IC50 values in drug-resistant Huh7/SR and Hep3B/SR cells (Fig. 1A). Western blot assays showed high expression of the ABCG2 and P-GP drug resistance-associated proteins in Huh7/SR and Hep3B/SR cells (Fig. 1B). The above results indicated that drug-resistant cells were successfully established. Next, drug-resistant cells were treated with metformin to explore the effect of metformin on ferroptosis in sorafenib-resistant cells. The expression of the ferroptosis-associated protein GPX4 was upregulated and that of ACSL4 was downregulated in Huh7/SR and Hep3B/SR cells, as detected by Western blotting (Fig. 1C). Fe2+ levels in drug-resistant cells were decreased and significantly increased after metformin treatment (Fig. 1D). Flow cytometry results showed that ROS levels were reduced in drug-resistant cells and that the addition of metformin significantly promoted ROS production (Fig. 1E). Reduced fluorescence intensity of lipid peroxides in drug-resistant cells was observed by BODIPY staining, while fluorescence intensity was significantly enhanced by metformin treatment (Fig. 1F). The above results suggest that sorafenib-resistant cells inhibit ferroptosis and that treatment with metformin promotes ferroptosis in HCC cells.

Metformin promotes ferroptosis in hepatocellular carcinoma cells. (A) CCK-8 for cell proliferation and sorafenib IC50; (B) Western blot for expression of drug resistance-related proteins ABCG2 and P-GP in cells; (C) Western blot for expression of ferroptosis-related proteins GPX4 and ACSL4; (D) Fe2+ levels; (E) Flow cytometry for ROS; (F) BODIPY staining to determine lipid peroxidation levels. *P < 0.05, **P < 0.01, ***P < 0.001
Metformin reverses sorafenib resistance in hepatocellular carcinoma cells
It has been established that metformin promotes ferroptosis in sorafenib-resistant HCC cells. To further investigate the effect of metformin on sorafenib resistance in hepatocellular carcinoma cells, we treated sorafenib-resistant Huh7/SR and Hep3B/SR cells with metformin. The CCK-8 assay showed that compared with the control group, metformin-treated Huh7/SR and Hep3B/SR cells had reduced proliferation and viability, and sorafenib IC50 values were significantly lower (Fig. 2A). Western blotting showed that the expression of the drug resistance-associated proteins ABCG2 and P-GP in the cells was significantly reduced after the addition of metformin (Fig. 2B). The results of the scratch assay showed that the migration of drug-resistant cells was significantly reduced after metformin treatment (Fig. 2C). Cell invasion was detected by Transwell assays, and metformin treatment significantly inhibited drug-resistant cell invasion (Fig. 2D). These results indicated that metformin was able to reduce the IC50 value of sorafenib, inhibit the resistance of sorafenib-resistant cells, and alleviate cell migration and invasion to reverse sorafenib resistance in hepatocellular carcinoma cells.

Metformin reverses sorafenib resistance in hepatocellular carcinoma cells. (A) CCK-8 for cell proliferation and sorafenib IC50; (B) Western blot for expression of drug resistance-associated proteins ABCG2 and P-GP in cells; (C) Scratch assay for cell migration; (D) Transwell assay for cell invasion. *P < 0.05, **P < 0.01, ***P < 0.001

Adding ferroptosis inhibitors weakens the effect of metformin on Huh7 sorafenib resistance. (A) CCK-8 for cell proliferation and sorafenib IC50; (B) Western blot for expression of drug resistance-related proteins ABCG2 and P-GP in cells; (C) Scratch assay for cell migration; (D) Transwell assay for cell invasion; (E) Western blot for expression of signaling pathway-related proteins ATF4, STAT3, p-STAT3, STAT1, and p-STAT1. *P < 0.05, **P < 0.01, ***P < 0.001, ns: no significant difference
The addition of ferroptosis inhibitors impairs the effect of metformin on Huh7 sorafenib resistance
To investigate the effect of ferroptosis promotion by metformin on sorafenib resistance, we treated sorafenib-resistant Huh7/SR cells with Fer-1, a ferroptosis inhibitor. We determined the cellular value-added viability and IC50 of sorafenib by CCK-8, which showed that metformin inhibited cell proliferation and decreased the IC50 of sorafenib and that the addition of Fer-1 reversed the effect of metformin and increased the IC50 of sorafenib (Fig. 3A). Western blot assays showed that metformin inhibited the expression of the drug resistance-associated proteins ABCG2 and P-GP in the cells, and Fer-1 treatment restored the expression of ABCG2 and P-GP (Fig. 3B). The scratch assay showed that metformin inhibited the migration of drug-resistant cells and that the migration of cells increased after the addition of Fer-1 (Fig. 3C). Inhibition of drug-resistant cell invasion by metformin was detected by Transwell assays, and treatment with Fer-1 reversed the inhibitory effect of metformin and increased cell invasion (Fig. 3D). The expression of the signaling pathway-related proteins ATF4, STAT3, p-STAT3, STAT1, and p-STAT1 was detected by Western blotting, which showed that ATF4, P-STAT3, and P-STAT1 were highly expressed in Huh7/SR cells and that metformin treatment inhibited the expression of P-STAT3 and P-STAT1. The addition of Fer-1 reversed the effect of metformin, and the expression of P-STAT3 and P-STAT1 increased (Fig. 3E). These results suggest that the addition of Fer-1, a ferroptosis inhibitor, can attenuate the effect of metformin on Huh7 sorafenib resistance.
Metformin alleviates sorafenib resistance via ATF4-induced ferroptosis
To further investigate the mechanism by which metformin-induced ferroptosis alleviates sorafenib resistance, we transfected metformin-treated sorafenib-resistant Huh7/SR cells with oe-ATF4 and added the ferroptosis inducer erastin. We detected a significant increase in ATF4 expression in cells transfected with oe-ATF4 (Fig. 4A), indicating that the transfection was successful. Next, Western blotting showed that metformin inhibited the expression of the intracellular ferroptosis-related protein GPX4 and promoted the expression of ACSL4; transfection with oe-ATF4 reversed the effect of metformin, and the expression of GPX4 was upregulated and ACSL4 was downregulated, whereas metformin treatment followed by transfection with oe-ATF4 and the addition of erastin resulted in decreased expression of GPX4 and increased expression of ACSL4 (Fig. 4B). We found that metformin promoted intracellular Fe2+ accumulation and that the Fe2+ content was reduced in metformin-treated ATF4-overexpressing cells, whereas the Fe2+ content was increased again in metformin-treated ATF4-overexpressing and erastin-treated cells (Fig. 4C). Metformin treatment increased intracellular ROS levels as detected by flow cytometry, while ROS levels decreased in transfected oe-ATF4 cells after metformin treatment and increased again in transfected oe-ATF4 and erastin-treated cells after metformin treatment (Fig. 4D). BODIPY staining showed that metformin increased intracellular lipid peroxidation. The level of lipid peroxidation was inhibited by transfection with oe-ATF4, and the level of lipid peroxidation was reduced, while transfection with oe-ATF4 and the addition of erastin after metformin treatment led to another increase in lipid peroxidation levels (Fig. 4E). Transwell cell invasion results showed that metformin inhibited cell invasion. After overexpressing ATF4, cell invasion was increased, while in metformin treatment with overexpression of ATF4 and addition of erastin, cell invasion was reduced again (Fig. 4F). Cell migration was examined by a scratch assay, and the results showed that metformin inhibited cell migration. After overexpressing ATF4, the migration of cells was increased, whereas the migration of cells overexpressing ATF4 and treated with erastin was reduced again (Fig. 4G).These results suggest that metformin promotes ferroptosis and inhibits migration and invasion in drug-resistant cells, whereas overexpression of ATF4 reverses the effects of metformin, and then the addition of erastin restores cellular ferroptosis and inhibits migration and invasion. Thus, metformin alleviates sorafenib resistance by inhibiting ATF4-induced ferroptosis.

Metformin alleviates sorafenib resistance through ATF4-induced ferroptosis. (A) Western blot for transfection efficiency; (B) Western blot for expression of ferroptosis-related proteins GPX4 and ACSL4; (C) Fe2+ content; (D) Flow cytometry for ROS; (E) BODIPY staining for lipid peroxidation levels; (F) Transwell assay for cell invasion; (G) Scratch assay for cell migration. *P < 0.05, **P < 0.01, ***P < 0.001, ns: no significant difference
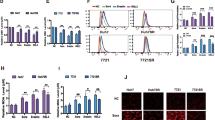
Interaction between ATF4 and STAT3
By immunofluorescence localization, we found that ATF4 was expressed in the nucleus, while STAT3 was expressed in the cytoplasm (Fig. 5A), suggesting that the regulatory interaction between ATF4 and STAT3 does not occur at the transcriptional level. Immunoprecipitation results showed that enrichment of STAT3 was detected in the ATF4 antibody pull-down complex (Fig. 5B), suggesting a protein interaction between ATF4 and STAT3. The above results suggested that the interaction between ATF4 and STAT3 was likely to have occurred with nuclear translocation. Next, we used ZDOCK 3.0.2 to perform protein binding raw signal prediction for ATF4 with STAT3 and obtained the mode of interaction of ATF4 with the STAT3 protein. The ATF4 protein is shown in beige and the STAT3 protein is shown in blue. The ATF4 protein is seen in the cartoon diagram interacting with the active structural domain SH2 in the head of the STAT3 protein as a beta-sheet interacting with the helix, indicating that ATF4 may prevent SH2 from undergoing phosphorylation. As can be observed from the interaction on the left, V341, I338, E330, and Y333 on the ATF4 protein form hydrogen bonds with N64, Q644, Q635, and K658 on the STAT3 protein (Fig. 5C).

Interaction between ATF4 and STAT3. (A) Immunofluorescence localization analysis of ATF4 and STAT3 expression; (B) Immunoprecipitation analysis of ATF4-STAT3 interaction; (C) ATF4-STAT3 binding force raw signal prediction
Metformin mediates ferroptosis and sorafenib resistance via ATF4/STAT3
Metformin alleviates sorafenib resistance through ATF4-induced ferroptosis, and a further interaction between ATF4 and STAT3 has been demonstrated. Therefore, to investigate metformin-mediated ferroptosis via ATF4/STAT3 in sorafenib resistance, we transfected sorafenib-resistant Huh7/SR cells with si-ATF4 and added the STAT3 activator Colivelin. We detected a significant reduction in ATF4 expression in cells transfected with si-ATF4 (Fig. 6A), indicating that si-ATF4 transfection was successful. The expression of the signaling pathway-related proteins STAT3, p-STAT3, STAT1, and p-STAT1 was detected by Western blotting, and the results showed that the expression of p-STAT3 and p-STAT1 was significantly reduced after transfection with si-ATF4, and the expression of p-STAT3 and p-STAT1 was increased again after transfection with si-ATF4 and the addition of Colivelin, whereas STAT3 and STAT1 did not change significantly (Fig. 6B). The results of the Western blot assay showed that transfection with si-ATF4 inhibited STAT3 phosphorylation and that STAT3 phosphorylation was restored by transfection with si-ATF4 and the addition of Colivelin (Fig. 6C). GPX4 expression was downregulated, ACSL4 expression was upregulated after transfection with si-ATF4, GPX4 expression increased, and ACSL4 expression decreased after inhibition of ATF4 expression and the addition of Colivelin (Fig. 6D). An increase in Fe2+ content occurred following the inhibition of ATF4 expression and a decrease in Fe2+ content occurred following the inhibition of ATF4 expression and the addition of Colivelin (Fig. 6E). ROS levels, detected by flow cytometry, increased after transfection with si-ATF4 and decreased after transfection with si-ATF4 and the addition of Colivelin (Fig. 6F). BODIPY staining showed that transfection with si-ATF4 promoted lipid peroxidation levels, and transfection with si-ATF4 and the addition of Colivelin led to a decrease in lipid peroxidation levels (Fig. 6G). Next, the IC50 of sorafenib was measured by CCK-8 and showed that transfection with si-ATF4 significantly decreased the IC50, and transfection with si-ATF4 and the addition of Colivelin increased the IC50 (Fig. 6H). These results indicated that transfection with si-ATF4 resulted in increased sensitivity of Huh7 to sorafenib, while transfection with si-ATF4 and the addition of Colivelin decreased sensitivity. Western blot assays showed that the expression of the drug resistance-associated proteins ABCG2 and P-GP in the cells decreased after inhibition of ATF4 expression and increased after inhibition of ATF4 expression and the addition of Colivelin (Fig. 6I). The above results suggest that downregulation of ATF4 inhibits the phosphorylated nuclear translocation of STAT3, promotes ferroptosis, and increases the sensitivity of Huh7 cells to sorafenib, while activation of STAT3 reverses the effect of ATF4 downregulation. Thus, the ATF4/STAT3 pathway mediates cellular ferroptosis to improve sorafenib resistance.

Metformin mediates ferroptosis and sorafenib resistance via ATF4/STAT3. (A) Western blot for transfection efficiency; (B) Western blot for expression of signal pathway-related proteins STAT3, p-STAT3, STAT1, and p-STAT1; (C) Western blot for STAT3 phosphorylated nuclear translocation; (D) Western blot for expression of ferroptosis-related proteins GPX4 and ACSL4; (E) Fe2+ levels; (F) Flow cytometry for ROS; (G) BODIPY staining for lipid peroxidation levels; (H) CCK-8 to detect cell proliferation and sorafenib IC50; (I) Western blot to detect expression of drug resistance-associated proteins ABCG2 and P-GP in cells. *P < 0.05, **P < 0.01, ***P < 0.001
Metformin mediates ferroptosis and sorafenib resistance via ATF4/STAT3 in vivo
At the animal level, we verified that metformin mediates ferroptosis and sorafenib resistance via ATF4/STAT3 by injecting metformin and transfecting oe-ATF4 or adding Colivelin. Immunohistochemical results showed that Ki-67 decreased after metformin injection and increased after metformin injection and transfection with oe-ATF4 or metformin injection and the addition of Colivelin (Fig. 7A). These results indicated that metformin inhibited tumor cell proliferation, while transfection with oe-ATF4 or the addition of Colivelin both reversed the effect of metformin. Immunohistochemistry showed that injection of metformin inhibited the expression of ATF4 and STAT3, while injection of metformin and transfection with oe-ATF4 resulted in increased expression of ATF4 and STAT3 and injection of metformin and the addition of Colivelin significantly inhibited the expression of ATF4 and had no significant effect on the expression of STAT3 (Fig. 7B). Western blot analysis showed that GPX4 decreased and ACSL4 expression increased after metformin injection, and GPX4 increased and ACSL4 expression decreased after metformin injection and oe-ATF4 transfection or metformin injection and Colivelin addition (Fig. 7C). Metformin promoted Fe2+ accumulation, whereas transfection with oe-ATF4 or the addition of Colivelin both reversed the effect of metformin (Fig. 7D). ROS levels were significantly increased by flow cytometry after metformin injection, whereas transfection with oe-ATF4 or the addition of Colivelin both reduced ROS production after metformin injection (Fig. 7E). MDA and GSH expression increased after metformin injection, whereas transfection with oe-ATF4 or the addition of Colivelin after metformin injection resulted in a decrease in MDA and GSH expression (Fig. 7F-G). The above results suggest that metformin mediates ferroptosis and sorafenib sensitivity via ATF4/STAT3 in vivo, which in turn inhibits hepatocellular carcinoma progression.

Metformin mediates ferroptosis and sorafenib resistance via ATF4/STAT3 in vivo. (A) Ki-67 immunohistochemical staining; (B) immunohistochemical detection of ATF4 and STAT3; (C) Western blot for expression of ferroptosis-related proteins GPX4 and ACSL4; (D) Fe2+ content; (E) Flow cytometry for ROS; (F) expression of MDA and GSH. *P < 0.05, **P < 0.01, ***P < 0.001, ns: no significant difference
Discussion
Sorafenib is a first-line chemotherapeutic agent for advanced hepatocellular carcinoma. However, sorafenib resistance is becoming increasingly common and is a major cause of poor patient prognosis and a challenge to clinical care [24]. Ferroptosis has been reported to be strongly associated with sorafenib resistance in hepatocellular carcinoma [25]. A recent study reported that depletion of intracellular iron stores protects HCC cells from the cytotoxicity of sorafenib and prevents sorafenib from inducing oxidative stress in the cells [26]. In addition, sorafenib was found to induce ferroptosis in different cancer cell lines [27]. Sorafenib-induced ferroptosis may be a potent mechanism for the induction of HCC cell death [28]. Therefore, the induction of ferroptosis in hepatocellular carcinoma cells may be an effective way to enhance the anticancer activity of sorafenib. In the present study, we found that metformin promoted ferroptosis and sorafenib sensitivity in hepatocellular carcinoma cells via ATF4/STAT3.
Metformin is currently a first-line drug for the treatment of type 2 diabetes. Studies have shown that metformin can induce cell cycle arrest and promote cell death in hepatocellular carcinoma [29] and thus inhibit the progression of HCC. In Lai et al. [13], metformin was found to restore HCC cell sensitivity to sorafenib via AMPK-dependent autophagy activation. In addition, metformin enhanced sensitivity to sorafenib by reversing the EMT process in HepG2 cells and reducing the formation of cancer stem cells [30]. Consistent with these studies, we found that metformin reduced the IC50 of sorafenib in hepatocellular carcinoma cells, inhibited resistance in sorafenib-resistant cells, and mitigated cell migration and invasion to reverse sorafenib resistance in hepatocellular carcinoma cells. In earlier studies, metformin was also found to induce ferroptosis in an AMPK-independent manner to inhibit tumor growth [16, 31]. Here, we demonstrated that metformin promotes Fe2+ and lipid ROS levels and promotes ferroptosis in HCC cells. Using the ferroptosis inhibitor Fer-1, we found that the effect of metformin on Huh7 sorafenib resistance was attenuated, suggesting that metformin may alleviate HCC sorafenib resistance by promoting ferroptosis.
Metformin activation of ATF4 induces inhibition of the mitochondrial respiratory chain, which in turn affects tumor growth [32, 33]. There is growing evidence that ATF4 promotes the transcription of genes that support respiration and alleviate mitochondrial stress [34, 35]. In the present study, we found that upregulation of ATF4 reversed the effects of metformin, inhibited ROS production, reduced Fe2+ and lipid peroxidation levels, and inhibited ferroptosis in HCC cells. In addition, STAT3, a transcription factor important for HCC development [36], promotes hepatocellular carcinoma stem cell expansion and has important functions in tumorigenesis, progression, recurrence, and drug resistance in hepatocellular carcinoma [37, 38]. We found that ATF4 was expressed in the nucleus and STAT3 was expressed in the cytoplasm, indicating that the regulatory interaction between ATF4 and STAT3 did not occur at the transcriptional level; however, the immunoprecipitation results showed a protein interaction between ATF4 and STAT3. Previous studies have reported increased nuclear translocation of activated STAT3 in the mammary gland of transgenic ATF4 mice with upregulated expression of their target genes [39]. Using immunoblot analysis, we found that STAT3 undergoes phosphorylated nuclear translocation. In addition, using ZDOCK 3.0.2 for binding force raw letter prediction, we obtained the protein interaction mode of ATF4 with STAT3. We concluded that metformin mediates sorafenib sensitivity and ferroptosis via ATF4/STAT3.
In conclusion, our study demonstrates that metformin promotes ferroptosis and sorafenib sensitivity in hepatocellular carcinoma cells via ATF4/STAT3. As metformin is a mitochondrial complex enzyme inhibitor and hepatocellular carcinoma cells have higher mitochondrial respiration efficiency [40], they will be more sensitive to treatment for HCC. Studies have shown few complications when metformin is given to cancer patients, including hepatocellular carcinoma patients, and although studies have suggested that metformin may impair cognitive function, a causal relationship between metformin prescription and cognitive impairment has not been confirmed. In conclusion, metformin is a safe drug for cancer patients [41]. The use of metformin may enhance the therapeutic efficacy of sorafenib in hepatocellular carcinoma. However, long-term exposure to metformin has been found to lead to metabolic adaptation and drug resistance in HCC cells, and high doses of metformin can also lead to lactic acidosis [42, 43]. At the same time, ATF4 and STAT3 inhibitors may have further applications in sorafenib-resistant liver cancer patients, and understanding the role of ATF4/STAT3 molecular pathways in HCC progression will contribute to the development of potential drugs for targeted treatment of liver cancer.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
COURI T (2019) Goals and targets for personalized therapy for HCC [J]. Hepatol Int 13(2):125–137
ZHAO B, LV X, ZHAO X et al (2022) Tumor-promoting actions of HNRNP A1 in HCC are Associated with Cell cycle, mitochondrial Dynamics, and necroptosis [J].Int J Mol Sci, 23(18)
XU J, JI L, LIANG Y et al (2020) CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1 [J]. Signal Transduct Target Ther 5(1):298
GRANDHI MS, KIM A K, RONNEKLEIV-KELLY S M et al (2016) Hepatocellular carcinoma: from diagnosis to treatment [J]. Surg Oncol 25(2):74–85
JUNG W J JANGS, CHOI W J et al (2022) Metformin administration is associated with enhanced response to transarterial chemoembolization for hepatocellular carcinoma in type 2 diabetes patients [J]. Sci Rep 12(1):14482
PODHORECKA M, IBANEZ B (2017) Metformin - its potential anti-cancer and anti-aging effects [J]. Postepy Hig Med Dosw (Online) 71(0):170–175
MALLIK R, CHOWDHURY TA (2018) Metformin in cancer [J]. Diabetes Res Clin Pract 143:409–419
COYLE C, CAFFERTY F H, VALE C et al (2016) Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis [J]. Ann Oncol 27(12):2184–2195
HSU S K, CHENG K C, MGBEAHURUIKE M O et al (2021) New Insight into the Effects of Metformin on Diabetic Retinopathy, Aging and Cancer: nonapoptotic cell death, immunosuppression, and Effects beyond the AMPK pathway [J].Int J Mol Sci, 22(17)
ZHANG Y, WANG H (2021) XIAO H. Metformin actions on the liver: Protection Mechanisms Emerging in Hepatocytes and Immune cells against NASH-Related HCC [J].Int J Mol Sci, 22(9)
DE OLIVEIRA S, HOUSERIGHT R A, GRAVES A L et al (2019) Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish [J]. J Hepatol 70(4):710–721
YOU A, CAO M, GUO Z et al (2016) Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models [J]. J Hematol Oncol 9:20
LAI H Y, TSAI H H, YEN C J et al (2020) Metformin Resensitizes Sorafenib-Resistant HCC cells through AMPK-Dependent Autophagy activation [J]. Front Cell Dev Biol 8:596655
WANG H, LIU C, ZHAO Y et al (2020) Mitochondria regulation in ferroptosis [J]. Eur J Cell Biol 99(1):151058
WU X, LI Y, ZHANG S et al (2021) Ferroptosis as a novel therapeutic target for cardiovascular disease [J]. Theranostics 11(7):3052–3059
HOU Y, CAI S (2021) Metformin induces ferroptosis by targeting miR-324-3p/GPX4 axis in breast cancer [J]. Acta Biochim Biophys Sin (Shanghai) 53(3):333–341
JANG S K, HONG S E, LEE D H et al (2021) Inhibition of mTORC1 through ATF4-induced REDD1 and Sestrin2 expression by metformin [J]. BMC Cancer 21(1):803
GUAN L, WANG F, WANG M et al (2022) Downregulation of HULC Induces Ferroptosis in Hepatocellular Carcinoma via Targeting of the miR-3200-5p/ATF4 Axis [J]. Oxid Med Cell Longev, 2022: 9613095
CHEN Y, WANG J J, LI J et al (2012) Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes [J]. Diabetologia 55(9):2533–2545
WANG MAL (2022) JAK2/STAT3 inhibitor reduced 5-FU resistance and autophagy through ATF6-mediated ER stress [J]. J Recept Signal Transduct Res 42(2):206–213
WANG X, HU R, SONG Z et al (2022) Sorafenib combined with STAT3 knockdown triggers ER stress-induced HCC apoptosis and cGAS-STING-mediated anti-tumor immunity [J]. Cancer Lett 547:215880
CAPELLETTI M M, MANCEAU H, PUY H et al (2020) Ferroptosis in Liver Diseases: an overview [J].Int J Mol Sci, 21(14)
GAO R, KALATHUR R K R, COTO-LLERENA M et al (2021) YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis [J]. EMBO Mol Med 13(12):e14351
YANG X D, KONG F E QIL et al (2021) PARP inhibitor Olaparib overcomes Sorafenib resistance through reshaping the pluripotent transcriptome in hepatocellular carcinoma [J]. Mol Cancer 20(1):20
SUN X, NIU X, CHEN R et al (2016) Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis [J]. Hepatology 64(2):488–500
NIE J, LIN B, ZHOU M et al (2018) Role of ferroptosis in hepatocellular carcinoma [J]. J Cancer Res Clin Oncol 144(12):2329–2337
CHEN X, KANG R, KROEMER G et al (2021) Broadening horizons: the role of ferroptosis in cancer [J]. Nat Rev Clin Oncol 18(5):280–296
TANG W, CHEN Z, ZHANG W et al (2020) The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects [J]. Signal Transduct Target Ther 5(1):87
CHEN H P, SHIEH J J, CHANG C C et al (2013) Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: population-based and in vitro studies [J]. Gut 62(4):606–615
FENG Y, GUO X, HUANG X et al (2018) Metformin reverses stem cell–like HepG2 sphere formation and resistance to sorafenib by attenuating epithelial–mesenchymal transformation [J]. Mol Med Rep 18(4):3866–3872
YANG J, ZHOU Y, XIE S et al (2021) Metformin induces ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer [J]. J Exp Clin Cancer Res 40(1):206
KIM K H, JEONG Y T, KIM S H et al (2013) Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation [J]. Biochem Biophys Res Commun 440(1):76–81
KRALL A S, MULLEN P J, SURJONO F et al (2021) Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth [J]. Cell Metab 33(5):1013–26e6
BALSA E, SOUSTEK M S THOMASA et al (2019) ER and nutrient stress promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis [J].Mol Cell, 74(5): 877 – 90.e6.
QUIRóS PM, PRADO M A, ZAMBONI N et al (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals [J]. J Cell Biol 216(7):2027–2045
MAKINO Y, HIKITA H (2023) STAT3 is activated by CTGF-mediated tumor-stroma Cross Talk to Promote HCC progression [J]. Cell Mol Gastroenterol Hepatol 15(1):99–119
YANG C, CAI W C, DONG Z T et al (2019) lncARSR promotes liver cancer stem cells expansion via STAT3 pathway [J]. Gene 687:73–81
WANG D, ZHENG X (2019) Hepatectomy promotes recurrence of liver cancer by enhancing IL-11-STAT3 signaling [J]. EBioMedicine 46:119–132
BAGHERI-YARMAND R, VADLAMUDI R K, KUMAR R (2003) Activating transcription factor 4 overexpression inhibits proliferation and differentiation of mammary epithelium resulting in impaired lactation and accelerated involution [J]. J Biol Chem 278(19):17421–17429
HSU C C, WU L C, HSIA C Y et al (2015) Energy metabolism determines the sensitivity of human hepatocellular carcinoma cells to mitochondrial inhibitors and biguanide drugs [J]. Oncol Rep 34(3):1620–1628
FUJITA K, IWAMA H, MIYOSHI H et al (2016) Diabetes mellitus and metformin in hepatocellular carcinoma [J]. World J Gastroenterol 22(27):6100–6113
JIN P, JIANG J (2022) Disrupting metformin adaptation of liver cancer cells by targeting the TOMM34/ATP5B axis [J]. EMBO Mol Med 14(12):e16082
SIDDHARTH S, KUPPUSAMY P, WU Q et al (2022) Metformin enhances the anti-cancer efficacy of Sorafenib via suppressing MAPK/ERK/Stat3 Axis in Hepatocellular Carcinoma [J].Int J Mol Sci, 23(15)
Acknowledgements
Not applicable.
Funding
This study was supported by National Natural Science Foundation of China, Regional Science Fund Project (82060436); Young and Middle-aged Academic Technology Leaders in Yunnan Province (202205AC160089); Medical Academic leader in Yunnan Province (D–2018032); Key school level projects of the 14th five years plan of Kunming Medical University (J11301854); Spring City Plan: the High-level Talent Promotion and Training Project of Kunming (2022SCP002); Expert Work station of Ceshi Chen in Yunnan (Kunming) (YSZ-JFZZ-2020025); Expert Work station of Xiao Xu in Yunnan Province (202205AF150064); The third batch of “Spring City Plan” high-level talents - Spring City Famous medical special project; The third batch of “Yun Lin scholars”; Major Science and Technology Project of Yunnar Provincial Science and Technology Department (Biomedical Special) - Liver and Kidney Organ Transplantation Research and clinical application of key technology (202302AA310018).
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to this manuscript. Zongqiang Hu, Yingpeng Zhao and Laibang Li performed the majority of experiments and data analysis. Jie Jiang, Wang Li and Yuanyi Mang contributed to carrying out the experiments and interpreted the results. Yang Gao, Yun Dong and Jiashun Zhu helped with project design. Zongqiang Hu, Jianghua Ran, Li Li and Shengning Zhang wrote the manuscript. Zongqiang Hu and Chaomin Yang contributed to designing the project. Jie Jiang and Yuanyi Mang helped with sample preparation. Shengning Zhang and Zongqiang Hu revised the manuscript and designed and conducted the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experimental protocols were approved by the Laboratory Animal Welfare Ethics Committee of Kunming Medical University (approval number kmmu20221859), and the animal procedures adhered to the ARRIVE guidelines 2.0.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All the authors agreed on the publication of this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, Z., Zhao, Y., Li, L. et al. Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3. Mol Biol Rep 50, 6399–6413 (2023). https://doi.org/10.1007/s11033-023-08492-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08492-4