
Abstract
Background
Species in the subfamily Aphidiinae from the Braconidae of Hymenoptera are endoparasitic wasps that exclusively utilize aphids as hosts. Some Aphidiinae species are widely used as biological agents. However, there were only one species with determined complete mitochondrial genome from this subfamily.
Methods and results
In this study, we sequenced and annotated the mitochondrial genome (mitogenome) of Binodoxys acalephae, which was 15,116 bp in size and contained 37 genes. The start codon of 13 protein-coding genes was ATN, and the complete stop codon TAA and TAG was widely assigned to 11 protein-coding genes. The lrRNA contains 43 stem-loop structures, and srRNA contains 25 stem-loop structures. Translocation and inversion of tRNA genes was found to be dominant in B. acalephae. In contrast to Aphidius gifuensis from the same subfamily Aphidiinae, inverted tRNALeu1 was translocated to the gene cluster between tRNALeu2 and COX2, and the control region between tRNAIle and tRNAMet was deleted in the mitogenome of B. acalephae. Within Braconidae, gene clusters tRNATrp-tRNACys-tRNATyr and CR-tRNAIle-tRNAGln-tRNAMet were hotspots for gene rearrangement. Phylogenetic analysis showed that both Bayesian and maximum-likelihood methods recovered the monophyly of Aphidiinae and suggested that Aphidiinae formed sister clades with the remaining subfamilies. The phylogenetic analyses of nine subfamilies supported the monophyly of Cyclostomes and Noncyclostomes in Braconidae.
Conclusion
The arrangement of mitochondrial genes and the phylogenetic relationships among nine Braconidae subfamilies were constructed better to understand the diversity and evolution of Aphidiinae mitogenomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Insect mitochondrial genome (mitogenome) is a double-stranded circular DNA molecule, which measures approximately 14–20 kb in size and contains 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes, and a control region (CR) [1, 2]. Due to its small size, simple structure, and rapid evolutionary rate, mitogenome has been extensively studied and widely used for species identification, molecular phylogeny, phylogeography, and molecular evolutionary research [3,4,5]. Although the arrangement of the mitochondrial genes is generally conserved across most inset orders [1, 6], gene rearrangement is also quite common in some highly derived groups [4, 7,8,9,10]. Hymenoptera (bees, wasps, hornets, sawflies, and ants) is one of the most important groups for studying mitogenome rearrangements in insects, with at least one translocated tRNA in each hymenopteran mitogenome [11, 12]. The mitogenomes of Hymenoptera are characterized by high A + T content [13], high substitution rates [14] and strong base composition bias [15], which may accelerate the rate of gene rearrangement. Mitochondrial gene rearrangement patterns were commonly used to revise the phylogeny in the species-rich family Braconidae, which was traditionally divided into Cyclostomes and Noncyclostomes in previous studies [16,17,18,19,20].
The species from the subfamily Aphidiinae (Hymenoptera: Braconidae) are endoparasitic wasps that exclusively utilize aphids (Hemiptera) as hosts. The adults of Aphidiinae lay eggs inside the aphids, and the larva gradually consumes the soft tissue of the hosts and spin a cocoon within them, forming a characteristic “mummy” [21]. Therefore, some species of wasps are widely used as biological control agents of aphids [22, 23]. This subfamily currently includes 56 genera and more than 500 species worldwide [24]. For a long time, Aphidiinae was considered to be a separate family “Aphidiidae”, located between the Cyclostomes and Noncyclostomes [20, 25, 26]. However, recent studies supported that Aphidiinae was a subfamily placed within the complex Cyclostomes [17, 27, 28]. Up to now, the complete mitogenome of only one species (Aphidius gifuensis) has been sequenced and annotated in the Aphidiinae [18]. In this study, the mitogenome of Binodoxys acalephae was sequenced and described, and the phylogenetic relationships among major Braconidae subfamilies were constructed to better understand the diversity and evolution of Aphidiinae mitogenomes.
Materials and methods
Sample collection and DNA extraction
Adult specimen of B. acalephae was collected from Kunming, Yunnan Province. The specimen was preserved in anhydrous ethanol and stored in a refrigerator at -20℃ before DNA extraction. After being identified based on morphological characters, the genomic DNA was extracted using DNeasy Blood and Tissue kit (Qiagen, Germany) on the basis of the manufacturer’s protocol, and the voucher specimen was stored at Entomological Museum of China Agricultural University.
Mitogenome sequencing and assembly
The Illumina TruSeq library was prepared with an average insert size of 350 bp and sequenced with the paired-end reads length of 150 bp on Illumina NovaSeq 6000 platform (Berry Genomic, Beijing, China). A total of 6 Gb raw data was obtained, and use Prinseq version 0.20.4 [29] to remove short and low-quality reads with the poly-Ns > 15 bp, or > 75 bp bases with quality score < 3. The remaining reads were de novo assembled using IDBA-UD [30], with minimum and maximum k values of 41 and 141 bp, respectively. The COX1 fragment (~ 610 bp) was amplified by PCR reaction (forward primer, 5’-3’: GGTCAACAAATCATAAAGATATTGG; reverse primer, 5’-3’: TAAACTTCAGGGTGACCAAAAAATCA). PCR cycling condition was: 95℃ for 1 min, 40 cycles of 95℃ for 20s, 50℃ for 50s, and 68℃ for 1.5 min, followed by 72℃ for 5 min. The PCR products were sequenced by Sanger sequencing at Tsingke Biotechnology (Beijing, China). To identify the corresponding mitogenome assemblies, the assembled contigs were searched with COX1 sequence using BLAST with at least 98% similarity. To investigate the assembled accuracy and sequencing depth, clean reads were mapped using Geneious version 10.1.3 [31]. Finally, we obtained a complete circular mitogenome with an average coverage depth of 676×.
Mitogenome annotation and nucleotide composition analysis
The gene sequences were preliminarily annotated by MitoZ [32] and accurately corrected in Geneious. The locations and secondary structures of tRNA genes were mainly determined by tRNAscan-SE search server [33]and ARWEN version 1.2 [34]. PCGs and rRNA genes were identified by alignment with homologous genes of other Braconidae species, and the secondary structure of rRNAs was predicted by RNAfold WebServer Version 2.4.18 [35] online platform. The gene rearrangement of B. acalephae was compared with the putative ancestral type of Drosophila melanogaster and 15 other published Braconidae species (Table 1). The nucleotide composition of mitogenome and synonymous codon usage (RSCU) of PCGs were analyzed by MEGA version 7.0 [36]. Nucleotide compositional differences were calculated by the formulae: AT skew = (A – T) / (A + T) and GC skew = (G - C) / (G + C) [37].
Phylogenetic analysis
The phylogenetic analysis was based on newly sequenced mitogenome of B. acalephae together with 15 mitogenomes published in GenBank from nine main subfamilies of Braconidae. Diadegma semiclausum and Enicospilus sp. from Ichneumonidae were selected as outgroups (Table 1).
The 13 PCGs of each species were aligned separately using the L-INS-I strategy of the MAFFT algorithm [38] implemented in TranslatorX [39]. Two rRNA genes were aligned individually using the G-INS-I strategy of MAFFT version 7.0 online server [40]. All alignments were checked manually in MEGA [36]. Gene fragments were imported into Geneious for concatenation into two datasets: (1) the PCGRNA matrix with 10,782 nucleotides including 13 PCGs and two rRNA genes (srRNA and lrRNA); (2) the PCG12RNA matrix with 7,430 nucleotides including the first and second codon positions of the 13 PCGs and two rRNA genes.
These two datasets were analyzed under the strategy of Bayesian inference (BI) and Maximum-likelihood (ML). The BI phylogenetic trees were recovered using PhyloBayes MPI version 1.5a [41] under the site-heterogeneous mixture CAT + GTR model [42, 43]. The Markov Chain Monte Carlo chains were run independently after removing constant sites from the alignment and were stopped after the two runs had satisfactorily converged (maxdiff < 0.1). A consensus tree was yielded from the remaining trees after discarding initial 25% trees of each run as burn-in. The ML phylogenetic trees were recovered using IQ-TREE web server [44], with automatic model prediction and1000 SH-aLRT replicates.
Results
Genome organization and base composition
We obtained a nearly complete mitogenome of B. acalephae (GenBank accession number OP612694), which was 15,116 bp in size and included all the 37 protein-coding genes (13 PCGs, 22 tRNAs and two rRNAs) and a partial control region with the exact number of the tandem repeated units undetermined (Fig. 1). Four PCGs (ND5, ND4, ND4L and ND1), nine tRNA genes (tRNAGln, tRNATyr, tRNACys, tRNAPhe, tRNAHis, tRNAPro, tRNAVal, tRNAIle and tRNAMet) and two rRNA genes (srRNA and lrRNA) were encoded on the minority strand (N-strand) while the other 22 genes were encoded on the majority strand (J-strand). In the mitogenome of B. acalephae, 14 gene overlap regions with a total of 48 bp were observed, ranging from 1 to 11 bp in size, and the longest gene overlap region was between ATP6 and COX3. Apart from the control region, we identified 13 non-coding regions (NCRs) comprising a total of 127 bp, with the longest NCR (56 bp) located between ND1 and lrRNA (Table S1).

The mitochondrial genome of Binodoxys acalephae. Orientation of gene transcription is indicated by the arrows. Protein-coding genes (PCGs) are shown as yellow arrows, transfer RNA (tRNA) genes as pink arrows, ribosomal RNA (rRNA) genes as red arrows. The partial control region (in grey) contains incomplete repeated region. The green line in the circle shows the A + T content, and the blue shows the G + C content
The A + T content of the whole mitogenome of B. acalephae was 82.7% and the mitogenome of B. acalephae shows a negative AT skew (-0.115) and a positive GC skew (0.179), indicating that the nucleotide composition was significantly biased toward T (Table S2).
PCGs and codon usage
All 13 PCGs were arranged in putative ancestral gene order in the mitogenome of B. acalephae. The total length of these PCGs was 11,138 bp, and the A + T content of PCGs (81.0%) was slightly lower than that of the whole mitogenome (82.7%). We analyzed the nucleotide composition of each codon position in PCGs and found that A or T are overwhelmingly overrepresented at the third codon position on each strand (A + T %= 86.2%). The negative AT skew was consistent in all three codons, while the positive GC skew only existed in the first (0.212) and third (0.029) codons (Table S2).
The bias toward A and T of B. acalephae mitogenome was also well documented in the table of codon usage (Table S3). We calculated the relative synonymous codon usage (RSCU) (Fig S1), and found the six most prevalent codons, UUA, GCU, CGU, ACU, GUU and UCU were mainly composed by T and/or A. At the third codon position, A or T were overrepresented in PCGs.
All 13 PCGs were initiated with ATN as the start codon (five with ATT, six with ATG, and two with ATA) (Table S1). The complete stop codon TAA and TAG was most widely assigned to ten PCGs and CYTB, respectively, while COX2 and ND3 used a single T residue as an incomplete stop codon.
tRNA genes and rRNA genes
Twenty-two tRNAs were detected in the mitogenome of B. acalephae, ranging from 62 bp (tRNACys and tRNAThr) to 71 bp (tRNALys) in size. Different from the ancestral tRNA arrangement, there were 13 tRNAs encoded by J-strand and nine encoded by N-strand. Among all tRNAs, 21 tRNAs of B. acalephae could be folded into the typical cloverleaf secondary structures except for tRNASer(AGN) (Fig S2). The dihydrouridine (DHU) arm of tRNASer(AGN) was a simple loop. Unlike the variable DHU-stem (3–4 bp) and TΨC‐stem (3–6 bp), the anticodon stem (5 bp) and amino-acid acceptor stem (7 bp) are relatively conservative in size, except for tRNAGln with amino-acid acceptor (8 bp) stem and tRNASer(AGN) with anticodon stem (6 bp). A total of 23 mismatched base pairs were detected in 15 tRNAs with 15 G‐U pairs and 8 U‐U pair.
lrRNA and srRNA were 1,283 bp and 745 bp in size, respectively, with an average A + T content of 86.2% (Table S2). We predicted the secondary structures for rRNAs using the RNAfold WebServer online platform and homologous species alignment. We identified that lrRNA has six domains (Fig. 2 A), among which the IV and V domains are relatively conserved, and the III domain only has a short sequence compared with other species. There were three domains in srRNA (Fig. 2B), and the III domain was more conserved than the I and II domains. Overall, lrRNA contains 43 stem-loop structures, and srRNA contains 25 stem-loop structures.

Predicted secondary structures of the lrRNA (A) and srRNA (B) of Binodoxys acalephae. Short lines indicate Watson–Crick base-pairing and dots indicate noncanonical G-U pairs
Gene rearrangement
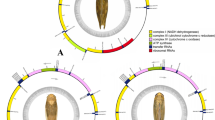
Gene rearrangements could be classified into translocations, local inversions (inverted in the local position), gene shuffling (local translocation) and remote inversions (translocated and inverted) [12]. Compared with the putative ancestral gene arrangement of D. melanogaster, there were at least seven tRNA rearrangement events in B. acalephae. These gene rearrangements were mainly from the tRNA clusters of tRNATrp(W)-tRNACys(C)-tRNATyr(Y) and CR-tRNAIle(I)-tRNAGln(Q)-tRNAMet(M) (Fig. 3). The gene shuffling of tRNACys(C) and tRNATyr(Y), and remote inversions of tRNAIle(I), tRNAMet(M) and tRNALeu1(L1) were found in the mitogenome of B. acalephae.

Mitochondrial genome organization in Braconidae referenced with the ancestral insect mitochondrial genomes. The topology is derived from the following phylogenetic analyses. The control region is abbreviated as CR and tRNA genes are denoted by a one-letter symbol according to the IPUC-IUB single-letter amino acid codes. Genes with underlines indicate that the gene is encoded on the minority strand. Rearranged protein-coding genes are marked in grey, and each of the rearranged tRNAs is marked with a different color
Phylogenetic analyses
Phylogenetic studies were conducted on 16 mitogenomes of Braconidae by using two species of Ichnummonidae as outgroups. PhyloBayes and ML analyses based on the two datasets (PCGRNA and PCG12RNA) yield identical topology with high node support values among subfamilies in Braconidae. Our analyses supported the monophyly of the subfamily Aphidiinae with high Bayesian posterior probabilities (BPP = 1) and ML bootstrap values (BSV = 100). The subfamily Aphidiinae formed sister clades with the remaining subfamilies (BPP = 0.91 and BSV = 100) (Fig. 4).

Phylogenetic trees inferred from Bayesian inference and maximum-likelihood analyses of the PCGRNA and PCG12RNA datasets. Values at corresponding nodes are ML bootstrap values (BSV) for PCGRNA, PCG12RNA and Bayesian posterior probabilities (BPP) for PCGRNA, PCG12RNA from left to right, respectively
Discussion
The mitogenome of B. acalephae was highly conserved in terms of gene content, gene size, base composition, PCG codon usage, and RNA secondary structures. In terms of base composition, the high A + T content of B. acalephae was corresponding with that of hymenopteran mitogenomes [14, 45, 46]. In terms of PCG codon usage, the most prevalent codons were mainly composed by T and/or A, which is consistent with other hymenopteran mitogenomes [4, 15, 16, 47]. And a single T residue as an incomplete stop codon has been noticed in several other hymenopteran mitogenomes [48,49,50]. In terms of RNA secondary structures, the simple loop of DHU arm of tRNASer(AGN) was found in many other insects [2]. Compared with A. gifuensis from the same subfamily Aphidiinae, the mitochondrial gene of B. acalephae undergone two rearrangement events. The inverted tRNALeu1 was translocated to the gene cluster between tRNALeu2 and COX2, and the CR between tRNAIle and tRNAMet was then deleted in the mitogenome of B. acalephae. Based on the topology derived from our phylogenetic analysis, we conclude five tRNA rearrangement events (R1 to R5) in Cyclostomes. The putative ancestral type rearranged to the gene arrangement of A. gifuensis by the tandem duplication-random loss [51] of gene block CR-tRNAIle-tRNAGln-tRNAMet (R1). The CR between tRNAIle and tRNAMet was lost in the remaining Cyclostomes species during evolution (R2). Then, tRNAAsp was inverted and translocated upstream of tRNALys (R3) and tRNAHis was remote inverted to form a gene cluster tRNAAsp-tRNAHis-tRNALys (R4). Last, tRNAThr and tRNAPro translocated in some species of the genus Psyttalia (R5). Furthermore, the ancestral gene arrangement of PCGs was present in all mitogenomes of Braconidae except for the Cotesia vestalis [16], suggesting heterogeneity in gene rearrangement rates among different Braconidae lineages. Gene clusters tRNATrp-tRNACys-tRNATyr and CR-tRNAIle-tRNAGln-tRNAMet were found to be the gene rearrangement hotspots, in which remote translocation of tRNAIle and tRNAMet may be synapomorphic rearrangements in Braconidae. The local inversion was reported to be the major type of gene rearrangement in Hymenoptera [11], while remote inversion of tRNA genes was found to be dominant in B. acalephae. This phenomenon was also found in other species of Braconidae and was thought to be the result of two independent recombination events [16].
In our analyses of nine subfamilies, the phylogeny supported the monophyly of Cyclostomes and Noncyclostomes. Phylogenetic analysis recovered the monophyly of Aphidiinae and suggested that Aphidiinae was the earliest branching lineage of Cyclostomes. The results were consistent with previous studies based on both mitochondrial and nuclear genes [20, 21]. Within the Noncyclostomes lineage, the sister relationship between Euphorinae and the remaining subfamilies of Noncyclostomes was recovered (Fig. 4).
Data availability
The data that support the findings of this study will be available in GenBank at https://www.ncbi.nlm.nih.gov/, with accession number OP612694.
References
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780. https://doi.org/10.1093/nar/27.8.1767
Cameron SL (2014) Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol 59:95–117. https://doi.org/10.1146/annurev-ento-011613-162007
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc Lond B 270:313–321. https://doi.org/10.1098/rspb.2002.2218
Song SN, Tang P, Wei SJ, Chen XX (2016) Comparative and phylogenetic analysis of the mitochondrial genomes in basal hymenopterans. Sci Rep 6:20972–20970. https://doi.org/10.1038/srep20972
Du Z, Hasegawa H, Cooley JR, Simon C, Yoshimura J, Cai W, Sota T, Li H (2019) Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol Biol Evol 36:1187–1200. https://doi.org/10.1093/molbev/msz051
Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8(6):668–674. https://doi.org/10.1016/S0959-437X(98)80035-X
Dowton M, Campbell NJH (2001) Intramitochondrial recombination-is it why some mitochondrial genes sleep around? Trends Ecol Evol 16(6):269–271. https://doi.org/10.1016/s0169-5347(01)02182-6
Shao R, Kirkness EF, Barker SC (2009) The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res 19:904–912. https://doi.org/10.1101/gr.083188.108
Cameron SL, Johnson KP, Whiting MF (2007) The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): Effects of extensive gene rearrangements on the evolution of the genome. J Mol Evol 65:589–604. https://doi.org/10.1007/s00239-007-9042-8
Mao M, Austin AD, Johnson NF, Dowton M (2014) Coexistence of minicircular and a highly rearranged mtDNA molecule suggests that recombination shapes mitochondrial genome organization. Mol Biol Evol 31(3):636–644. https://doi.org/10.1093/molbev/mst255
Dowton M, Austin AD (1999) Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol Biol Evol 16:298–309. https://doi.org/10.1093/OXFORDJOURNALS.MOLBEV.A026111
Dowton M, Castro LR, Campbell SL, Bargon SD, Austin AD (2003) Frequent mitochondrial gene rearrangements at the hymenopteran nad3-nad5 junction. J Mol Evol 56:517–526. https://doi.org/10.1007/s00239-002-2420-3
Zheng BY, Cao LJ, Tang P, van Achterberg K, Hoffmann AA, Chen HY, Chen XX, Wei SJ (2018) Gene arrangement and sequence of mitochondrial genomes yield insights into the phylogeny and evolution of bees and sphecid wasps (Hymenoptera: Apoidea). Mol Phylogenet Evol 124:1–9. https://doi.org/10.1016/j.ympev.2018.02.028
Oliveira D, Raychoudhury R, Lavrov DV, Werren JH (2008) Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae). Mol Biol Evol 25:2167–2180. https://doi.org/10.1093/molbev/msn159
Wei SJ, Li Q, van Achterberg C, Chen XX (2014) Two mitochondrial genomes from the families Bethylidae and Mutillidae: independent rearrangement of protein-coding genes and higher‐level phylogeny of the Hymenoptera. Mol Phylogenet Evol 77:1–10. https://doi.org/10.1016/j.ympev.2014.03.023
Wei SJ, Shi M, Sharkey MJ, van Achterberg C, Chen XX (2010) Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics 11:371. https://doi.org/10.1186/1471-2164-11-371
Li Q, Wei SJ, Tang P, Wu Q, Shi M, Sharkey MJ, Chen XX (2016) Multiple lines of evidence from mitochondrial genomes resolve phylogenetic relationships of parasitic wasps in Braconidae. Genome Biol Evol 8(9):2651–2662. https://doi.org/10.1093/gbe/evw184
Feng Z, Wu Y, Yang C, Gu X, Wilson JJ, Li H, Cai W, Yang H, Song F (2020) Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int J Biol Macromol 164:540–547. https://doi.org/10.1016/j.ijbiomac.2020.07.149
Chen XX, van Achterberg C (2019) Systematics, phylogeny, and evolution of Braconid wasps: 30 years of progress. Annu Rev Entomol 64:19.1–19.24. https://doi.org/10.1146/annurev-ento-011118-111856
Shi M, Chen XX, van Achterberg C (2005) Phylogenetic relationships among the Braconidae (Hymenoptera: Ichneumonoidea) inferred from partial 16S rDNA, 28S rDNA D2, 18S rDNA gene sequences and morphological characters. Mol Phylogenet Evol 37(1):104–116. https://doi.org/10.1016/j.ympev.2005.03.035
Belshaw R, Quicke DL (1997) A molecular phylogeny of the Aphidiinae (Hymenoptera: Braconidae). Mol Phylogenet Evol 7(3):281–293. https://doi.org/10.1006/mpev.1996.0400
Hawkins BA, Cornell HV (1994) Maximum parasitism rates and successful biological control. Science 266:1886. https://doi.org/10.1126/science.266.5192.1886
De Conti BF, Bueno VHP, Sampaio MV (2008) The parasitoid Praon volucre (Hymenoptera: Braconidae: Aphidiinae) as a potential biological control agent of the aphid Uroleucon ambrosiae (Hemiptera: Aphididae) on lettuce in Brazil. Eur J Entomol 105:485–487. https://doi.org/10.14411/eje.2008.062
Liu JJ, Yang JQ, Chen JH (2012) Phylogenetic relationships among 16 Aphidiidae species based on the V4 sequences of 18S rDNA (Insecta, Hymenoptera). J Fujian Agric Forestry Univ 41(01):7–12
Belshaw R, Fitton M, Herniou E, Gimeno C, Quicke DLJ (2002) A phylogenetic reconstruction of the Ichneumonoidea (Hymenoptera) based on the D2 variable region of 28S ribosomal RNA. Syst Entomol 23(2):109–123. https://doi.org/10.1046/j.1365-3113.1998.00046.x
Quicke DLJ, van Achterberg C (1990) Phylogeny of the subfamilies of the family Braconidae (Hymenoptera: Ichneumonidae). Verhandlungen des Zoologisch-Botanischen Vereins in Wien 258:1–95
Belshaw R, Dowton M, Quicke DLJ, Austin AD (2000) Estimating ancestral geographical distributions: a Gondwanan origin for aphid parasitoids? Proc Royal Soc B 267(1442):491–496. https://doi.org/10.1098/rspb.2000.1027
Sharanowski BJ, Dowling APG, Sharkey MJ (2011) Molecular phylogenetics of Braconidae (Hymenoptera: Ichneumonoidea), based on multiple nuclear genes, and implications for classification. Syst Entomol 36:549–572. https://doi.org/10.1111/j.1365-3113.2011.00580.x
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. https://doi.org/10.1093/bioinformatics/btr026
Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. https://doi.org/10.1093/bioinformatics/bts174
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Meng GL, Li YY, Yang CT, Liu SL (2019) MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res 47:e63. https://doi.org/10.1093/nar/gkz173
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. https://doi.org/10.1093/nar/25.5.955
Laslett D, Canbäck B (2008) ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24:172–175. https://doi.org/10.1093/bioinformatics/btm573
Lorenz R, Bernhart SH, Hoener zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA Package 2.0. Algorithm Mol Biol 6:26. https://doi.org/10.1186/1748-7188-6-26
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Perna NT, Kocher TD (1995) Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol 41:353–358. https://doi.org/10.1007/BF00186547
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518. https://doi.org/10.1093/nar/gki198
Abascal F, Zardoya R, Telford MJ (2010) TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res 38:W7–W13. https://doi.org/10.1093/nar/gkq291
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Lartillot N, Rodrigue N, Stubbs D, Richer J (2013) PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol 62:611–615. https://doi.org/10.1093/sysbio/syt022
Lartillot N, Philippe HA (2004) Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21:1095–1109. https://doi.org/10.1093/molbev/msh112
Tavare S (1986) Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci 17:57–86
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast-online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44:W232–W235. https://doi.org/10.1093/nar/gkw256
Wei SJ, Shi M, He JH, Sharkey MJ, Chen XX (2009) The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome 52:308–319. https://doi.org/10.1139/g09-008
Carapelli A, Comandi S, Convey P, Nardi F, Frati F (2008) The complete mitochondrial genome of the Antarctic springtail Cryptopygus antarcticus (Hexapoda: Collembola). BMC Genomics 9:315. https://doi.org/10.1186/1471-2164-9-315
Ma Y, Zheng BY, Zhu JC, van Achterberg C, Tang P, Chen XX (2019) The first two mitochondrial genomes of wood wasps (Hymenoptera: Symphyta): novel gene rearrangements and higher-level phylogeny of the basal hymenopterans. Int J Biol Macromol 123:1189–1196. https://doi.org/10.1016/j.ijbiomac.2018.11.017
Wu YF, Yang HL, Feng ZB, Li BY, Zhou WB, Song F, Li H, Zhang LM, Cai WZ (2020) Novel gene rearrangement in the mitochondrial genome of Pachyneuron aphidis (Hymenoptera: Pteromalidae). Int J Biol Macromol 149:1207–1212. https://doi.org/10.1016/j.ijbiomac.2020.01.308
Xiao JH, Jia JG, Murphy RW, Huang DW (2011) Rapid evolution of the mitochondrial genome in Chalcidoid wasps (Hymenoptera: Chalcidoidea) driven by parasitic lifestyles. PLoS ONE 6:e26645. https://doi.org/10.1371/journal.pone.0026645
Yan ZC, Fang Q, Tian Y, Wang F, Chen XX, Werren JH, Ye GY (2019) Mitochondrial DNA and their nuclear copies in the parasitic wasp Pteromalus puparum: a comparative analysis in Chalcidoidea. Int J Biol Macromol 121:572–579. https://doi.org/10.1016/j.ijbiomac.2018.10.039
Boore JL (2000) The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. Comp Genomics 1:133–147. https://doi.org/10.1007/978-94-011-4309-7_13
Funding
This research was funded by the China National Tobacco Corporation of Science and Technology Major Projects (No. 110202001036[LS-05]), Major Science and Technique Programs in Yunnan Province (No. 202102AA310055), Young Elite Scientist Sponsorship Program by CAST (No. YESS20200106) and the Research and Development Program of Ningxia Hui Autonomous Region (No. 2021BEF03002).
Author information
Authors and Affiliations
Contributions
SX: methodology, formal analysis, investigation, data curation, writing—original draft preparation WL: methodology, writing—original draft preparation, writing—review and editing, funding acquisition QL: software, formal analysis, investigation, data curation, writing—original draft preparation YW: formal analysis, investigation, funding acquisition XL: software, investigation, funding acquisition XD: investigation, data curation JH: investigation, funding acquisition FS: conceptualization, methodology, software, writing—review and editing, funding acquisition. All authors read and approved the manuscript. Additional information: Shiwen Xu and Weiwei Li contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, S., Li, W., Liu, Q. et al. The mitochondrial genome of Binodoxys acalephae (Hymenoptera: Braconidae) with unique gene rearrangement and phylogenetic implications. Mol Biol Rep 50, 2641–2649 (2023). https://doi.org/10.1007/s11033-022-08232-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-08232-0