
Abstract
By the end of 2019, COVID-19 was reported in Wuhan city of China, and through human-human transmission, this virus spread worldwide and became a pandemic. Initial symptoms of the disease include fever, cough, loss of smell, taste, and shortness of breath, but a decrease in the oxygen levels in the body leads, and pneumonia may ultimately lead to the patient’s death. However, the symptoms vary from patient to patient. To understand COVID-19 disease pathogenesis, researchers have tried to understand the cellular pathways that could be targeted to suppress viral replication. Thus, this article reviews the markers that could be targeted to inhibit viral replication by inhibiting the translational initiation complex/regulatory kinases and upregulating host autophagic flux that may lead to a reduction in the viral load. The article also highlights that mTOR inhibitors may act as potential inhibitors of viral replication. mTOR inhibitors such as metformin may inhibit the interaction of SARS-CoV-2 Nsp’s and ORFs with mTORC1, LARP1, and 4E-BP. They may also increase autophagic flux by decreasing protein degradation via inhibition of Skp2, further promoting viral cell death. These events result in cell cycle arrest at G1 by p27, ultimately causing cell death.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
SARS-CoV-2 is caused by the virus belonging to the Coronaviridae family. The International Committee of Virus Taxonomy (ICVT) [1] named the pandemic COVID-19 as it came into existence in late 2019. By the end of May 2020, a report confirms approx. 3.6 million COVID-19 cases and 2.5 thousand death reports, making COVID-19 disease a pandemic [2]. The genomic structure of coronavirus is made up of positive-sense RNA (Single-stranded), about 26–32 kilobases in size. Humans are the primary targets of coronavirus who, when infected, experience symptoms such as cold and fever to an extreme level of acute respiratory syndrome, including pneumonia and bronchitis [3]. Recent research shows that human coronavirus infection may spread from the upper respiratory tract (URTIs) to the lower respiratory tract (LRTIs) [4]. Recent studies indicate that SARS-CoV-2 has the capability of intrahuman transmission.
Inter-connecting traits of SARS-CoV, MERS-CoV, & SARS-CoV-2
Epidemics related to coronaviruses have constantly been occurring over the last 20 years as “Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV or SARS-CoV-1)”. However, only the outbreak of SARS-CoV-2 turned into a pandemic [5]. Interestingly, the SARS- CoV-2 share a cross-link of origin with the other two strains of CoVs, and their infecting mechanisms are still unknown. The primary animal host of SARS-CoV-2 coronavirus is bats, MERS-CoV is Dromedary camel, and for SARS-CoV is Himalayan palm civet before moving to the secondary host, i.e., humans [6].
The SARS epidemic emerged in early 2003 in Asia. In 2012, the MERS-CoV epidemic originated in Saudi Arabia, whose primary host was bats and dromedary camels (Camelus dromedarius) as its intermediate host. It had a fatality rate of approximately 34% and 10% [6]. All the previous CoV epidemics, including COVID-19, share common zoonotic origins and are now confirmed with person-to-person transmission [7]. All CoV epidemic causes common symptoms like severe acute respiratory illness, including cough, fever, and shortness of breath. Researchers are now trying to decipher how the transmission of SARS-CoV-2 occurs from bats to humans [8].
The origin, structure & genomic composition of SARS-CoV-2
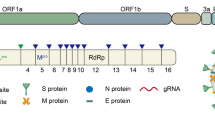
CoVs have a unique structural composition of the outer crown-like envelope of spike glycoprotein. Its genomic composition comprises positive sense, approximately 29.9 kb long single-stranded, polycistronic RNA, which codes for 9860 amino acids, and 29891 nucleotides. The CoVs genomic mRNA includes 6–11 open reading frames (ORFs). It encodes many non-structural proteins, such as ORF1a and ORF1b, which get transformed into 15 NSP proteins. The CoV mRNA also encodes for four structural proteins, namely, the surface protein (S, spike glycoprotein), a viral envelope protein (E), matrix protein (M), and nucleocapsid protein (N). It also consists of several accessory proteins from ORF9 to ORF10 at 3’-end, expressed to function as RNA polymerase, helicase, and various proteases [9]. All these proteins provide structural stability to the virus particle and interplay their regulatory role during viral replication.
The Coronaviridae family infects an extensive range of vertebrates. The CoVs are divided into four major genera such as α-CoVs (alpha-coronavirus), β-CoVs (beta-coronavirus), γ- CoVs (gamma-coronavirus), δ-CoVs (delta-coronavirus). A study suggests that genomic isolates from several patients suffering from human CoV and having symptoms like severe pneumonia possess 89% genomic similarity to bat-like SARS-CoVZXC21, whereas 82% similarity to human SARS-CoV. A comparison study between SARS-CoV-2 and the β-CoV RaTG133 bats (Rhinolophus affinis) shows a high genomic identity of 96% between them [10].
SARS-CoV-2 pathogenic viral proteins and their major functions
The spike glycoprotein (S) is vital in the pathogenesis of SARS-CoV-2 as it mediates the viral entry into the host cell. It provides subsequent attachment and cell-cell fusion with the host cell surface receptors and S protein [11]. The virus is said to spread directly between the host cells by subverting antibodies produced to neutralize the viruses resulting in the giant formation of multinucleated cells or syncytia. The nucleocapsid protein (N) is responsible for viral genomic replication processes, regulation of cellular responses to viral infections [12], and nucleocapsid formation [13]. Interestingly, in assembly and budding, localization of the N protein inside the endoplasmic reticulum (ER)-Golgi region also plays a significant role in viral replication inside the host cell [14]. The expression of the N protein is linked to an increase in the replication number of some coronaviruses and virus-like particles (VLPs) [15]. The viral Membrane protein (M) is a highly abundant structural protein responsible for determining the shape of the viral envelope [16]. It interacts with and assembles the larger structural subunits of CoV [17]. The homotypic interactions between the M and other accessory proteins aid in forming virion envelopes [16]. The Envelope protein (E) is an important structural protein involved in virus replication. The infected host cells are rich in E protein due to their enhanced expression during the replication cycle and are linked with the virion envelope [18]. The E protein is also significant in the maturation of viruses [19].
Interaction of M-S proteins
The interaction between M-S proteins is essential to stabilize the S protein, the ER-Golgi intermediate compartment (ERGIC), and its transformation into a new virion particle [20].
Interaction of M-N proteins
The binding of M protein with nucleocapsid protein provides additional stability to the N protein-RNA complex and the center complexes of virions. The N protein aids in the formation of viral assembly [21].
Interaction of M-E proteins
M protein, in combination with E protein, helps form the viral envelope and generate and release new virus-like particles [22].
The viral capsid protein: insights into SARS-CoV-2 disease etiology, structure, and composition of N protein
An increase in virus replication causes severe cell inflammation and injury to lung tissue. Thus, understanding the mechanism behind its replication is essential to develop effective therapeutic and preventive measures against this SARS-CoV-2 [23]. A study reported the role of the N protein in viral growth, making it a target to suppress viral growth. N protein is a 46 kDa protein comprising ~ 422 amino acids [24]. The N protein is encoded by the transcription of sequences on the 9th ORF of SARS- CoVs genomic transcript. It also codes for other accessory proteins (ORF9b) whose function is still unknown. 5′ORF9 of SARS-CoV-2 mRNA consists of moieties responsible for mediating attachments with the mRNA assembly of the host cells. The C-terminus region starting from 370–390 amino acids, is found to be highly rich in amino acid lysine; this composition of the C terminus aids in nuclear localization signaling. ORF9 comprises two structural domains: the 5′ end for putative RNA binding, and the second at the 3′ end is responsible for self-association [24].
Various studies interlink the interaction of the M-protein with human cellular hnRNPA1 protein that is possible due to the N-protein domain. Such interactions serve as a hot spot for the phosphorylation of N protein [25]. Moreover, this nucleocapsid protein divides into three distinct regions, each having its role during various stages of the viral replication cycle.
N protein: the genome encapsulating protein/viral capsid protein
The N protein aids in the formation of the protective viral envelope by packing the transcribed genomic RNA of viruses. The distinct characteristic properties of N protein to achieve complete viral structure includes: (i) identifying and associating with transcribed genomic RNA; (ii) being able to self-associate as an oligomer to form capsid. N-protein possesses the following properties: (1) Identification and interaction with the host genomic RNA and (2) Capsid formation [24].
In addition to being the virus capsid protein, the N-protein also serves as a regulatory protein as it modulates the host cell machinery during its viral life cycle. The heterologous expression of N-protein controls major cellular processes [26], such as (1) Host cell cycle deregulation, (2) Suppression of Interferon production, (3) Increase in levels of COX2 production, (4) AP1 activity up-regulation, (5) Triggering apoptosis, (6) Cross-regulation of the host cell proteins.
Inhibition of the SARS-CoV-2 replication and propagation in the host cell
To understand the inhibition of SARS-CoV-2 replication, this study further emphasizes the mechanisms as described as, (1) Directly: Inhibiting mTOR, thereby downregulating SARS-CoV-2 replication and growth and (2) Indirectly: Enhancement of autophagic pathway by Skp2 inhibition and Beclin1 (BECN1) phosphorylation.
Shapira et al. identified how mTORC1 could control Skp2 regulation [27]. Rapamycin, an mTOR inhibitor, decreased Skp2 levels by elevating the degradation of Skp2, whereas the mTORC1 also upregulates p27 levels in rapamycin-sensitive cells [28]. The mTORC1 interaction occurs directly with Skp2 in a phosphatase-independent approach. Further, Beclin 1 helps form the isolation membrane (double-membrane structure) at the early autophagy stage, which engulfs cytoplasmic material to form the mature autophagosomes [26].
Modulation of SARS-CoV-2 replication and propagation through mTOR and autophagy
Viruses cannot propagate without the machinery and metabolism of a host cell. The virus confirms its attachment to the cell membrane of the host cell during host infection and injects its RNA or DNA into the host to initiate infection [29]. However, the host cell engulfs any virus particle in a pinocytotic vacuole. Suppose the virus can evade the phagocytic machinery of the host cell. In that case, the infecting viral RNA forms messenger RNA (mRNA) for certain RNA viruses in a host cell. The viruses take advantage of the existing cellular structures of the host cell to replicate [29]. The de novo synthesis of coronaviruses’ genetic material and its transcribed proteins leads to the propagation of new virions by utilizing infectious host cellular machinery [30]. For effective adaptation into the host environment, the new virions co-opt the biological processes of the host cell to facilitate the generation of new infectious particles.
The mTORC1 and Autophagy
The mTOR (Ser/Thr kinase) consists of two accessory protein complexes, i.e., mTORC1 and mTORC2. Each mTOR complex has various regulatory properties and is responsible for various cellular activities [31]. The mTORC1 functions to suppress autophagy by activating protein synthesis assembly and RNA translation in nutrient-replete conditions to encourage cellular growth mechanisms and proliferation of host cells [32]. Meanwhile, mTORC2 participates in the upstream regulation of PI3K, which mainly links with increasing mTORC2-ribosomal binding [31]. Phosphorylation of the Thr308 motif stimulates Akt kinase protein primarily when the PI3K is recruited to the plasma membrane. The hydrophobic site ‘Ser473’ of Akt is also phosphorylated by mTORC2 [33].
Autophagy as a threat to Viral Propagation
Autophagy is the primary survival response towards cellular stress. It tends to sequester and degrade the intracellular constituents, including pathogens, foreign bodies, and damaged organelles of host cells [34]. Autophagy can work as a threat as it can cause viral protein degradation, while it is beneficial for other viruses and is required for replication. Viral-infectious cells appear to begin the stress stimuli by triggering autophagic proteins. This tends to the elimination of invading virus, causing cellular apoptosis and prevention of further viral propagation. Viruses are specialized with mechanisms to sustain their basal growth rate and regulate intracellular signaling pathways; induction of mTORC1 tends to neutralize stress-induced autophagy by preventing apoptosis.
Relation between SARS-CoV-2 and Autophagy through Skp2 inhibition (S-phase kinase-associated protein 2) and BECN1 (Beclin1) phosphorylation
BECN1 is a central regulatory element of phosphatidylinositol 3-kinase (PI3K) which mediates the lysine-48-linked polyubiquitination process by interlinking with S-phase kinase-associated protein 2 (Skp2) to facilitate proteasomal degradation. BECN1 functions as a key regulator by interacting with ATG14 located on autophagosomes to enhance the initial nucleation phases of autophagy. Hence, the BECNI1-ATG14 complex matures the autophagosomes and ultimately aids infusion with lysosomes to cause cell death [35]. The downregulation of Skp2, either by genetic or pharmacological agents/ inhibitors, leads to the inhibition of BECN1 ubiquitination leading to the enhancement of autophagic flux. Recent studies also report that the multiplication of MERS-CoV decreases BECN1 levels and blocks autophagosomes and lysosome fusion. Skp2 not only functions as an agonist for SARS-CoV-2 by enhancing autophagy but also reduces MERS-CoV replication by up to 28,000 times [6]. However, Skp2 can also be regulated by the antagonist for autophagy i.e., mTOR (Fig. 1).

Protein-Protein interaction between the regulatory components of autophagy and endoplasmic reticulum-associated degradation (ERAD). Essential targets include BECN1 (Beclin1), SKP2 (S-phase kinase-associated protein 2), and USP10 (Ubiquitin Specific Peptidase 10) (STRING: Functional Protein Association Networks, n.d.)
Skp2 regulation by mTORC1: a hypothesis against SARS-CoV-2 replication
Skp2
E3 Ubiquitin-protein-ligase complex comprises of substrate recognition component known as Skp2. Skp2 is a primary mediator of the ubiquitination and proteasomal degradation of specific proteins target, which interconnects with the cell cycle progression (mainly p27 proteolysis), signal transduction, and transcription. p27 degradation by SCF ((Skp1/Cul1/F-box protein)-Skp2 is done through phosphorylation of p27 [36]. Skp2, or S-phase kinase linkage to the ubiquitin-proteasome system (UPS), regulates the timely turnover of new proteins of different cellular processes[37]. Moreover, Skp2 also causes p27-independent proteolysis by phosphorylation at the Thr187 site of p27 [38].
Skp2 inhibition and BECN1 phosphorylation
Skp2 inhibition increases BECN1 levels and improves lysosomal SNAP receptor protein assembly and autophagy. Further, it restricts MERS-CoV replication. The FKBP51 subunit tends to initiate the linkage of Skp2 to BECN1 (inactive form) markedly due to its association with AKT1 kinase. Likewise, BECN1 phosphorylation increases FKBP51-directed protein interactions, and decreases AKT1 phosphorylation. FKBP51 affects at least two pathways interconnecting with the phosphorylation and regulation of E3 ligase-Skp2 and BECN1; both pathways include AKT1 [39].
mTORC1, a key regulator of Skp2 protein
The interaction of mTORC1 at Ser64 position of Skp2 and its pSkp2 can consequently protect Skp2 from degradation by Ub-Proteasomal pathway [40]. Jin et al. reported that Skp2 regulates the negative feedback to trigger the activation of mTORC1 signaling, which aids in recruiting Skp2 to RagA [41]. Hence, the activation of mTORC1 works by influencing the activity of Skp2 and mTORC1-mediated Skp2 phosphorylation. The two effector protein subunits, namely (i) 4E-binding protein 1 (4EBP1) and (ii) S6 kinase 1 (S6K1), participate in cellular growth, metabolism, protein synthesis, and initiation of angiogenesis; hence the activation of mTORC1 is an essential step [42,43,44]. Thus, mTORC1 works as a key regulator of the Skp2 protein.
Pharmacological interventions toward antiviral therapy
The siRNA and mTOR inhibitor rapamycin or metformin can regulate the expression of Skp2. Skp2 inhibitors act in the following ways: (i) by inducing cell cycle arrest via Skp2 inhibition, (ii) by the interaction of p27 to attenuate Skp2, and (iii) by reduction of p27 ubiquitination [40, 45, 46]. Other autophagy regulator includes: the HSP90 chaperone as well as more recently found FK506 binding protein (FKBP)512 [46]. CoV engages differentially with autophagic pathway components. Thus, autophagy modulation may alter virus replication [47]. It is shown that substances inhibiting the generation of DMVs are broadly reactive against in vitro CoV replication especially emerging paradigmatic viruses of various genera, including MERS-CoV [48]. A MERS-CoV-infected cellular genome study indicates significant phosphorylation changes affiliating with several regulatory kinase proteins such as AKT1 and mTOR. Other studies have correlated autophagy with mTOR and AKT1in other research studies [49, 50].
According to early research, the enhancement of PI3K following ADV infection causes viral protein production and proliferation [51]. Epstein-Barr virus (EBV) encodes for the latency protein, i.e., LMP2A and the G protein-coupled receptor (vGPCR). Herpesviruses, and Kaposi’s sarcoma herpesvirus (KSHV), trigger the PI3K/Akt pathway upstream of mTORC1 [52, 53]. The mTOR activity rises after pharmacological suppression of the PI3K pathway, as shown by a negative effect on West Nile Virus (WNV) development and replication [54, 55]. As a result, WNV is predicted to target mTORC1 initiation and delay apoptosis to maintain its translational machinery [55, 56]. PI3K-dependent apoptosis blocking interferes with DENV and JEV entry, although pharmacological targeting of PI3K inhibition does not affect virus replication, unlike WNV [57].
Viruses compete directly with host machinery to oppose the stress response for their survival. Therefore, modulating the mTOR and autophagy pathway in the host cell may inhibit virus replication and play a role in antiviral therapy [49].
Potential mTOR inhibitors
mTOR inhibition can act as one of the strategies to inhibit virus replication. Furthermore, different studies signify mTORC1 as a primary regulator of viral replication, including the Andes orthohantavirus and coronavirus [58]. Sirolimus, the most potent inhibitor of the mTOR pathway, effectively blocks the translation of viral proteins and new virions. Earlier studies indicate that sirolimus was used to treat patients with H1N1 infection and those suffering from pneumonia and acute respiratory failure [59]. Another study revealed that sirolimus reduced MERS-CoV replication by 60% [60]. mTOR signaling plays an essential role in the progress of MERS-CoV infection [49].
Another study indicates that using 1 µg/ml dactinomycin effectively inhibits feline enteric CoV development [61]. Another study predicts that combining sirolimus and dactinomycin can target HCoV. They synergistically target the complementary exposure pattern of the HCoV-related host protein subnetwork. In particular, sirolimus and dactinomycin efficiently inhibit the mTOR signaling and viral mRNA synthesis in HCoV-infected cells [62].
Another mTOR inhibitor, i.e., metformin, inhibits the virus by enhancing insulin sensitivity [63, 64]. It can also serve to inhibit mTOR signaling by associating with protein-protein aggregates. Thus, metformin may serve as a potential drug against SARS-CoV-2 to prevent viral replication and pathogenesis [65, 66].
Possible replication mechanism of SARS-CoV-2 and its modulation
mTOR and Skp2 may act as potential targets to inhibit the replication of SARS-CoV-2 [31]. The SARS-CoV-2 baits communicate with the accessory components of virus-hosts signaling systems, including respiratory complex 1 by Nsp7, Nsp12, Orf9c, leucine importer B, Nsp6, and LARP1 [67]. Protein-Protein interaction studies suggest direct signaling of human proteins regulation by the mTORC1 pathway-associated proteins like LARP1, some regulatory kinases, and FKBP7. These proteins further interact with the viral N and Orf8 proteins. Hence, these regulatory pathways can control virus propagation, replication, and inhibition of target proteins and can also lead to the degradation of infected cells by enhancing autophagy. Thus we propose the indirect modulation of the mTORC1 protein complex through metformin. (Fig. 2)

Potential replication mechanism of SARS-CoV-2 and its modulation. Direct and Indirect Co-regulation of mTORC1 and Skp2 via Metformin enhances the autophagic flux and inhibits SARS-CoV-2 protein translational assembly by targeting LARP1, Nsps 1–6, Orf 3–10, and 4E-BP proteins
As a proof of concept, we performed a molecular interaction study of metformin with N protein of coronavirus and with mTOR (mammalian target of rapamycin), LARP1 (La Ribonucleoprotein 1, Translational Regulator), and FKBP7 (FKBP Prolyl Isomerase 7)), followed by a protein-protein interaction study between LARP1-Nsp7, and FKBP7-Nsp7. The strength of these complexes was further checked in the presence of metformin through Patch Dock. The strength of protein-protein interaction is evaluated in terms of patch dock score.
The molecular docking analysis reveals the interaction of metformin with the LARP1 and FKPB7. The binding energies of the selected targets with MET (metformin), its amino acid residues, the active site involved in the interactions, binding energy, the inhibition constant, H-bond atoms involved, and the H-bond distance are mentioned in Table 1. Negative binding energy shows a relatively strong affinity of LARP1 and FKBP7 with MET. The images of the docked complexes of LARP1 and FKBP7 with MET are shown in Fig. 3.

Pictorial representation of molecular docking analysis. a Binding orientation in the docked complex of MET with FKPB7. b Binding orientation in the docked complex of MET with LARP1. The compound (MET (metformin)) is represented in green. Images were generated using Discovery Studio Visualizer
Further, the patch dock score (PDS) estimates the score of interaction between two protein molecules. PDS was 16,770 when FKPB7 was interacting with the co-factor complex of NSP7 and the C-terminal domain of NSP8 from SARS CoV-2 (PDB ID: 6WIQ), whereas the score decreased to 16,152 in the presence of metformin.
Further, the PDS was 12,070 when LARP1 was interacting with nsp7-nsp8 complex of SARS-CoV-2 (PDB ID: 6YHU), which decreased to 10,818 in the presence of metformin. This decrease in score shows that the interaction between host protein and viral replicative machinery may weaken in the presence of metformin. However, results obtained from in silico studies would need to be validated experimentally.
mTOR inhibitors as sensitizers of viral infections: a double edge sword
The use of mTOR inhibitors is known to immunosuppress individuals, which is a known risk factor for respiratory virus infection and is the reason why patients on mTOR inhibitors, like rapamycin, are considered a part of the highly susceptible, fragile human population that needs to be insulated from SARS-CoV-2 transmission in the community. A study by Shi et al. [68] reported that treatment with mTOR inhibitors such as rapamycin promotes the downregulation of IFN-induced transmembrane (IFITM) proteins. The IFITM proteins, mainly IFITM3, inhibit the virus-cell fusion and helps in preventing viral infection. However, rapamycin increases cellular susceptibility to multiple virus infections (IFITM-sensitive viruses like Influenza A virus) by triggering the degradation of broad-spectrum antiviral proteins, including IFITM. Hence, mTOR inhibitors may enhance the entry of the virus into the cells due to suppressed immune system and promote viral infection making patients taking these drugs susceptible to viral infection, including SARS-CoV-2. Another study by the same author [69] revealed that rapamycin analogs trigger microautophagy by activating TFEB (Transcription Factor EB), resulting in endolysosomal remodeling and turnover of transmembrane proteins like IFITM. As a result, cells are more susceptible to SARS-CoV-2 infection in tissue culture, and hamsters/mice are more vulnerable to SARS-CoV-2 infection in vivo. Hence, patients using mTOR inhibitors may become more susceptible to SARS-CoV-2 infection due to lysosome-mediated suppression of intrinsic immunity (Fig. 4).

Possible mechanism of enhanced SARS-CoV-2 infection through mTOR inhibition. mTOR inhibitors can hinder mTOR mediated TFEB phosphorylation resulting in nuclear translocation of TFEB. Nuclear TFEB activates the genes promoting lysosomal activities including microautophagy, an autophagy related pathway. Microautophagy induces degradation of membrane proteins such as IFITM2 and IFITM3 which in turn promotes the entry of SARS-CoV-2 into the cells by facilitating the fusion between viral and cell membrane
Conclusion
SARS-CoV-2 has emerged as a global health threat. Although vaccines against SARS-CoV-2 have been designed, there is still no approved oral drug against SARS-CoV-2. The growth and progression of SARS-CoV-2 in a host cell may be dependent on the activation of the mTOR pathway. Our article concludes that mTOR inhibitors may inhibit the replication of SARS-CoV-2 through autophagy induction and moderation of the PI3K-AKT-mTOR pathway; thus, bringing a therapeutic impact in COVID-19 patients. Overall, it is essential to find a suitable drug-based therapy against SARS-CoV-2. In this regard, the therapeutic effect of mTOR inhibitors, including metformin should be further studied to design better treatment regimens against SARS-CoV-2. However, mTOR inhibitors can also suppress the immune system, thus, close examination of these inhibitors on respiratory virus acquisition and disease is required.
References
Zhong J, Tang J, Ye C, Dong L (2020) The immunology of COVID-19: is immune modulation an option for treatment? Lancet Rheumatol 2(7):e428–e436. doi: https://doi.org/10.1016/S2665-9913(20)30120-X
Culp WC Jr (2020) Coronavirus disease 2019: in-home isolation room construction. A & A Practice 14(6):e01218. https://doi.org/10.1213/XAA.0000000000001218
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16(1):69. doi: https://doi.org/10.1186/s12985-019-1182-0
Xu J, Shi PY, Li H, Zhou J (2020) Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect Dis 6(5):909–915. https://doi.org/10.1021/acsinfecdis.0c00052
Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses Methods Mol Biol 1282:1–23. https://doi.org/10.1007/978-1-4939-2438-7_1
Hui DS, Azhar I, Madani E, Ntoumi TA, Kock F, Dar R, Ippolito O, Mchugh G, Memish TD, Drosten ZA, Zumla C, Petersen A E (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis 91:264–266. doi: https://doi.org/10.1016/j.ijid.2020.01.009
Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J, Xing F, Liu J, Yip CC, Poon RW, Tsoi HW, Lo SK, Chan KH, Poon VK, Chan WM, Ip JD, Cai JP, Cheng VC, Chen H, Hui CK, Yuen KY (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395(10223):514–523. doi: https://doi.org/10.1016/S0140-6736(20)30154-9
National Institute of Allergy and Infectious Diseases. COVID-19, MERS & SARS | NIH: National Institute of Allergy and Infectious Diseases. [online], Website https://www.niaid.nih.gov/diseases-conditions/covid-19 [Accessed 06-04-2020]
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269. doi: https://doi.org/10.1038/s41586-020-2008-3
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26(4):450–452. doi: https://doi.org/10.1038/s41591-020-0820-9
Song HC, Seo MY, Stadler K, Yoo BJ, Choo QL, Coates SR, Uematsu Y, Harada T, Greer CE, Polo JM, Pileri P, Eickmann M, Rappuoli R, Abrignani S, Houghton M, Han JH (2004) Synthesis and characterization of a native, oligomeric form of recombinant severe acute respiratory syndrome coronavirus spike glycoprotein. J Virol 78(19):10328–10335. doi: https://doi.org/10.1128/JVI.78.19.10328-10335.2004
McBride R, van Zyl M, Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein. Viruses 6(8):2991–3018. doi: https://doi.org/10.3390/v6082991
de Haan CA, Rottier PJ (2005) Molecular interactions in the assembly of coronaviruses. Adv Virus Res 64:165–230. doi: https://doi.org/10.1016/S0065-3527(05)64006-7
Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ (1994) Coronavirus M proteins accumulate in the golgi complex beyond the site of virion budding. J Virol 68(10):6523–6534. doi: https://doi.org/10.1128/JVI.68.10.6523-6534.1994
Siu YL, Teoh KT, Lo J, Chan CM, Kien F, Escriou N, Tsao SW, Nicholls JM, Altmeyer R, Peiris JS, Bruzzone R, Nal B (2008) The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J Virol 82(22):11318–11330. doi: https://doi.org/10.1128/JVI.01052-08
Neuman BW, Kiss G, Kunding AH, Bhella D, Baksh MF, Connelly S, Droese B, Klaus JP, Makino S, Sawicki SG, Siddell SG, Stamou DG, Wilson IA, Kuhn P, Buchmeier MJ (2011) A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol 174(1):11–22. doi: https://doi.org/10.1016/j.jsb.2010.11.021
Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66:193–292. doi: https://doi.org/10.1016/S0065-3527(06)66005-3
Venkatagopalan P, Daskalova SM, Lopez LA, Dolezal KA, Hogue BG (2015) Coronavirus envelope (E) protein remains at the site of assembly. Virology 478:75–85. doi: https://doi.org/10.1016/j.virol.2015.02.005
Curtis KM, Yount B, Baric RS (2002) Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J Virol 76(3):1422–1434. https://doi.org/10.1128/jvi.76.3.1422-1434.2002
Mortola E, Roy P (2004) Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett 576(1–2):174–178. doi: https://doi.org/10.1016/j.febslet.2004.09.009
Narayanan K, Makino S (2001) Characterization of nucleocapsid-M protein interaction in murine coronavirus. Adv Exp Med Biol 494:577–582. doi: https://doi.org/10.1007/978-1-4615-1325-4_85
Corse E, Machamer CE (2001) Infectious bronchitis virus envelope protein targeting: implications for virus assembly. Adv Exp Med Biol 494:571–576. doi: https://doi.org/10.1007/978-1-4615-1325-4_84
Seyran M, Pizzol D, Adadi P, El-Aziz TMA, Hassan SS, Soares A, Kandimalla R, Lundstrom K, Tambuwala M, Aljabali AAA, Lal A, Azad GK, Choudhury PP, Uversky VN, Sherchan SP, Uhal BD, Rezaei N, Brufsky AM (2021) Questions concerning the proximal origin of SARS-CoV-2. J Med Virol 93(3):1204–1206. doi: https://doi.org/10.1002/jmv.26478
Surjit M, Lal SK (2008) The SARS-CoV nucleocapsid protein: a protein with multifarious activities. Infect Genet Evol 8(4):397–405. doi: https://doi.org/10.1016/j.meegid.2007.07.004
Setti L, Passarini F, De Gennaro G, Barbieri P, Perrone MG, Borelli M, Palmisani J, Di Gilio A, Piscitelli P, Miani A (2020) Airborne transmission route of COVID-19: why 2 meters/6 feet of inter-personal distance could not be enough. Int J Environ Res Public Health 17(8):2932. https://doi.org/10.3390/ijerph17082932
Antibody News: Novus Biologicals (n.d.). [online], Website https://www.novusbio.com/antibody-news [Accessed 24-02-2021]
Shapira M, Kakiashvili E, Rosenberg T, Hershko DD (2006) The mTOR inhibitor rapamycin down-regulates the expression of the ubiquitin ligase subunit Skp2 in breast cancer cells. Breast Cancer Res 8(4):R46. doi: https://doi.org/10.1186/bcr1533
Fingar DC (2015) Rag ubiquitination recruits a GATOR1: attenuation of amino acid-induced mTORC1 signaling. Mol Cell 58(5):713–715. https://doi.org/10.1016/j.molcel.2015.05.029
Brooks GF, Carroll KC, Butel JS, Morse SA (2007) Jawetz Melnick & Adelbergs Medical Microbiology, 24th edn. McGraw-Hill, New York
Hui DS, Azhar EI, Kim YJ, Memish ZA, Oh MD, Zumla A (2018) Middle East respiratory syndrome coronavirus: risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect Dis 18(8):e217–e227. doi: https://doi.org/10.1016/S1473-3099(18)30127-0
Le Sage V, Cinti A, Amorim R, Mouland AJ (2016) Adapting the stress response: viral subversion of the mTOR Signaling Pathway. Viruses 8(6):152. doi: https://doi.org/10.3390/v8060152
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221(1):3–12. https://doi.org/10.1002/path.2697
Fruman DA, Meyers RE, Cantley LC (1998) Phosphoinositide kinases. Annu Rev Biochem 67:481–507. doi: https://doi.org/10.1146/annurev.biochem.67.1.481
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307(5712):1098–1101. doi: https://doi.org/10.1126/science.1106148
Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520(7548):563–566. doi: https://doi.org/10.1038/nature14147
Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE (1997) Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 11(11):1464–1478. doi: https://doi.org/10.1101/gad.11.11.1464
Frescas D, Pagano M (2008) Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 8(6):438–449. doi: https://doi.org/10.1038/nrc2396
Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM (2001) A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 413(6853):323–327. doi: https://doi.org/10.1038/35095083
Xu D, Shan B, Sun H, Xiao J, Zhu K, Xie X, Li X, Liang W, Lu X, Qian L, Yuan J (2016) USP14 regulates autophagy by suppressing K63 ubiquitination of beclin 1. Genes Dev 30(15):1718–1730. doi: https://doi.org/10.1101/gad.285122.116
Geng Q, Liu J, Gong Z, Chen S, Chen S, Li X, Lu Y, Zhu X, Lin HK, Xu D (2017) Phosphorylation by mTORC1 stablizes Skp2 and regulates its oncogenic function in gastric cancer. Mol Cancer 16(1):83. doi: https://doi.org/10.1186/s12943-017-0649-0
Jin G, Lee SW, Zhang X, Cai Z, Gao Y, Chou PC, Rezaeian AH, Han F, Wang CY, Yao JC, Gong Z, Chan CH, Huang CY, Tsai FJ, Tsai CH, Tu SH, Wu CH, Sarbassov D, Ho YS, Lin HK (2015) Skp2-mediated raga ubiquitination elicits a negative feedback to prevent amino-acid-dependent mTORC1 hyperactivation by recruiting GATOR1. Mol Cell 58(6):989–1000. https://doi.org/10.1016/j.molcel.2015.05.010
Li Y, Tsang CK, Wang S, Li XX, Yang Y, Fu L, Huang W, Li M, Wang HY, Zheng XF (2016) MAF1 suppresses AKT-mTOR signaling and liver cancer through activation of PTEN transcription. Hepatology 63(6):1928–1942. doi: https://doi.org/10.1002/hep.28507
Miyazaki Y, Matsubara S, Ding Q, Tsukasa K, Yoshimitsu M, Kosai K, Takao S (2016) Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol Cancer 15(1):49. doi: https://doi.org/10.1186/s12943-016-0534-2
Thomas JD, Zhang YJ, Wei YH, Cho JH, Morris LE, Wang HY, Zheng XF (2014) Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 26(5):754–769. doi: https://doi.org/10.1016/j.ccell.2014.09.008
Gassen NC, Niemeyer D, Muth D, Corman VM, Martinelli S, Gassen A, Hafner K, Papies J, Mösbauer K, Zellner A, Zannas AS, Herrmann A, Holsboer F, Brack-Werner R, Boshart M, Müller-Myhsok B, Drosten C, Müller MA, Rein T (2019) SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat Commun 10(1):5770. doi: https://doi.org/10.1038/s41467-019-13659-4
Wang Z, Fukushima H, Inuzuka H, Wan L, Liu P, Gao D, Sarkar FH, Wei W (2012) Skp2 is a promising therapeutic target in breast cancer. Front Oncol 1(57):57. doi: https://doi.org/10.3389/fonc.2011.00057
Gassen NC, Hartmann J, Zschocke J, Stepan J, Hafner K, Zellner A, Kirmeier T, Kollmannsberger L, Wagner KV, Dedic N, Balsevich G, Deussing JM, Kloiber S, Lucae S, Holsboer F, Eder M, Uhr M, Ising M, Schmidt MV, Rein T (2014) Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: evidence in cells, mice, and humans. PLoS Med 11(11):e1001755. doi: https://doi.org/10.1371/journal.pmed.1001755
Cong Y, Verlhac P, Reggiori F (2017) The interaction between nidovirales and autophagy components. Viruses 9(7):182. https://doi.org/10.3390/v9070182
Kindrachuk J, Ork B, Hart BJ, Mazur S, Holbrook MR, Frieman MB, Traynor D, Johnson RF, Dyall J, Kuhn JH, Olinger GG, Hensley LE, Jahrling PB (2015) Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother 59(2):1088–1099. doi: https://doi.org/10.1128/AAC.03659-14
Lundin A, Dijkman R, Bergström T, Kann N, Adamiak B, Hannoun C, Kindler E, Jónsdóttir HR, Muth D, Kint J, Forlenza M, Müller MA, Drosten C, Thiel V, Trybala E (2014) Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the middle East respiratory syndrome virus. PLoS Pathog 10(5):e1004166. doi: https://doi.org/10.1371/journal.ppat.1004166
O’Shea C, Klupsch K, Choi S, Bagus B, Soria C, Shen J, McCormick F, Stokoe D (2005) Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J 24(6):1211–1221. doi: https://doi.org/10.1038/sj.emboj.7600597
Moody CA, Scott RS, Amirghahari N, Nathan CO, Young LS, Dawson CW, Sixbey JW (2005) Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J Virol 79(9):5499–5506. doi: https://doi.org/10.1128/JVI.79.9.5499-5506.2005
Pringle ES, Robinson CA, McCormick C (2019) Kaposi’s sarcoma-associated herpesvirus lytic replication interferes with mTORC1 regulation of autophagy and viral protein synthesis. J Virol 93(21):e00854–e00819. https://doi.org/10.1128/JVI.00854-19
Beatman E, Oyer R, Shives KD, Hedman K, Brault AC, Tyler KL, Beckham JD (2012) West Nile virus growth is independent of autophagy activation. Virology 433(1):262–272. https://doi.org/10.1016/j.virol.2012.08.016
Shives KD, Beatman EL, Chamanian M, O’Brien C, Hobson-Peters J, Beckham JD (2014) West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J Virol 88(16):9458–9471. doi: https://doi.org/10.1128/JVI.01323-14
Urbanowski MD, Hobman TC (2013) The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. J Virol 87(2):872–881. doi: https://doi.org/10.1128/JVI.02030-12
Lee CJ, Liao CL, Lin YL (2005) Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J Virol 79(13):8388–8399. doi: https://doi.org/10.1128/JVI.79.13.8388-8399.2005
McNulty S, Flint M, Nichol ST, Spiropoulou CF (2013) Host mTORC1 signaling regulates andes virus replication. J Virol 87(2):912–922. doi: https://doi.org/10.1128/JVI.02415-12
Wang CH, Chung FT, Lin SM, Huang SY, Chou CL, Lee KY, Lin TY, Kuo HP (2014) Adjuvant treatment with a mammalian target of rapamycin inhibitor, sirolimus, and steroids improves outcomes in patients with severe H1N1 pneumonia and acute respiratory failure. Crit Care Med 42(2):313–321. doi: https://doi.org/10.1097/CCM.0b013e3182a2727d
Fang Y, Zhang H, Xu Y, Xie J, Pang P, Ji W (2020) CT manifestations of two cases of 2019 Novel Coronavirus (2019-nCoV) pneumonia. Radiology 295(1):208–209. https://doi.org/10.1148/radiol.2020200280
Lewis EL, Harbour DA, Beringer JE, Grinsted J (1992) Differential in vitro inhibition of feline enteric coronavirus and feline infectious peritonitis virus by actinomycin D. J Gen Virol 73(Pt 12):3285–3288. doi: https://doi.org/10.1099/0022-1317-73-12-3285
Zhou Y, Hou Y, Shen J, Huang Y, Martin W (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery 6:14. doi: https://doi.org/10.1038/s41421-020-0153-3
Chen Y, Gu F, Guan JL (2018) Metformin might inhibit virus through increasing insulin sensitivity. Chin Med J (Engl) 131(3):376–377. doi: https://doi.org/10.4103/0366-6999.223856
El-Arabey AA, Abdalla M (2020) Metformin and COVID-19: a novel deal of an old drug. J Med Virol 92(11):2293–2294. doi: https://doi.org/10.1002/jmv.25958
Mortensen E, Anzueto A (2018) Association of metformin and mortality for patients with diabetes who are hospitalized with pneumonia. Eur Respir J 52:PA2639. https://doi.org/10.1183/13993003.congress-2018.pa2639
Sharma S, Ray A, Sadasivam B (2020) Metformin in COVID-19: a possible role beyond diabetes. Diabetes Res Clin Pract 164:108183. doi: https://doi.org/10.1016/j.diabres.2020.108183
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’Meara MJ, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Naing ZZC, Zhou Y, Peng S, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Shen W, Shi Y, Zhang Z, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Ramachandran R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Lin Y, Wankowicz SA, Bohn M, Trenker R, Young JM, Cavero D, Hiatt J, Roth T, Rathore U, Subramanian A, Noack J, Hubert M, Roesch F, Vallet T, Meyer B, White KM, Miorin L, Agard D, Emerman M, Ruggero D, García-Sastre A, Jura N, von Zastrow M, Taunton J, Schwartz O, Vignuzzi M, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor S, Fraser JS, Gross J, Sali A, Kortemme T, Beltrao P, Shokat K, Shoichet BK, Krogan NJ (2020) A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. bioRxiv [Preprint]. https://doi.org/10.1101/2020.03.22.002386
Shi G, Ozog S, Torbett BE, Compton AA (2018) mTOR inhibitors lower an intrinsic barrier to virus infection mediated by IFITM3. Proc Natl Acad Sci USA 115(43):E10069–E10078. doi: https://doi.org/10.1073/pnas.1811892115
Shi G, Chiramel AI, Li T, Lai KK, Kenney AD, Zani A, Eddy AC, Majdoul S, Zhang L, Dempsey T, Beare PA, Kar S, Yewdell JW, Best SM, Yount JS, Compton AA (2022) Rapalogs downmodulate intrinsic immunity and promote cell entry of SARS-CoV-2. J Clin Invest. https://doi.org/10.1172/JCI160766
Acknowledgements
The authors are thankful to Hon’ble Vice-Chancellor, Integral University, Lucknow for providing infrastructural support and Dean Office, R&D, Integral University for providing manuscript communication number (IU/R&D/2021-MCN0001268).
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
TK: Conceptualization, Writing- original draft; AH: Conceptualization, Writing - original draft, Writing - review & editing; JeF: Writing - review & editing; SAF: Writing - review & editing, AFK: Writing - review & editing; SSM: Conceptualization, Supervision, Writing - original draft, Writing - review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khalid, T., Hasan, A., Fatima, J.e. et al. Therapeutic role of mTOR inhibitors in control of SARS-CoV-2 viral replication. Mol Biol Rep 50, 2701–2711 (2023). https://doi.org/10.1007/s11033-022-08188-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-08188-1