
Abstract
Apelin, an endogenous ligand for the G protein-coupled receptor APJ, is extensively expressed in various systems, especially the nervous system. This article reviews the role of apelin/APJ system in neurological diseases. In detail, apelin/APJ system can relieve acute brain injury including subarachnoid hemorrhage, traumatic brain injury, and ischemic stroke. Also, apelin/APJ system has therapeutic effects on chronic neurodegenerative disease models, involving the regulation of neurotrophic factors, neuroendocrine, oxidative stress, neuroinflammation, neuronal apoptosis, and autophagy. In addition, through different routes of administration, apelin/APJ system has a biphasic effect on depression, epilepsy, and pain. However, apelin/APJ system exacerbates the proliferation and invasion of glioblastoma. Thus, apelin/APJ system is expected to be a therapeutic target for the treatment of nervous system diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
APJ, a G protein-coupled receptor identified from the human gene by O’Dowd in 1993, was then called an orphan G protein-coupled receptor, consisting of 380 amino acids with 40% to 50% homology to the hydrophobic region of the angiotensin type I receptor [1]. In 1998, Tatemoto et al. extracted and purified the endogenous ligand of APJ from bovine gastric secretions, naming it apelin [2]. Genomics further revealed that the human apelin gene is localized in the q25 to 26.1 region of the X chromosome and consists mainly of 2 introns and 3 exons. The natural apelin peptide is processed from the C-terminal portion of a precursor protein consisting of 77 amino acid residues. The post-translational processing of the precursor protein results in a mature apelin active peptide consisting of 12, 13, 15, 16, 17, 19, 28, 31, and 36 amino acid residues [2,3,4]. Among these apelin isoforms, apelin-13 has the strongest biological potency. The metabolism of apelin can be regulated by ACE-2, and Neprilysin (a metalloprotease) degrades apelin by truncating the RPRL (Arg2-Leu-5) region of apelin. [5, 6]. In addition to apelin, Elabela/Toddler has recently been proved to be another endogenous ligand for APJ [7].
To date, increasing evidence has found that the apelin/APJ system is widely present in diverse tissues, such as heart, liver, kidney, adipose tissue, lung, brain, and so on [8, 9]. Besides, apelin/APJ system acts as a stress receptor in the body and is extensively and sensitively involved in various diseases. Apelin promotes iron citrate-induced mitochondrial ROS and total ion production to induce myocardial hypertrophy via NCOA4-mediated ferritinophagy [10]. Apelin significantly reduces the uptake and oxidation of fatty acids in the myocardium of type 2 diabetic rats [11]. Recent studies have shown that apelin ameliorates acute lung injury and cardiovascular/coagulopathy complications associated with elevated Ang-II/Ang (1–7) ratios in COVID-19 [12]. Indeed, it is found that tumor necrosis factor-α, lipopolysaccharide, hypoxia, and insulin all modulate the activation of the apelin/APJ system under different physiological or pathological conditions [8, 13,14,15]. Altogether, apelin/APJ system engages in the regulation of pathophysiological processes with pleiotropic actions in the various organs.
Actually, the role of apelin/APJ system in the nervous system is of particular interest to us. Apelin-immunoreactive neuronal cell bodies are distributed throughout the extent of the hypothalamic arcuate nucleus [16], which implies a potential role in neuroendocrine. Apelin mRNA and protein have strong signal expression in learning and memory related areas such as caudate nucleus, corpus callosum, and hippocampus gray matter [17]. Wang et al. have analyzed and identified 352 differentially expressed miRNAs and their target genes that play an instrumental role in neuroprotection of apelin-13 [18]. Besides, microinjection of apelin-13 into the nucleus tractus solitarius results in a significant reduction in apnea or phrenic nerve discharge amplitude. In contrast, apelin-13 injection into the rostral ventrolateral medulla resulted in an increase in phrenic nerve discharge amplitude [19]. In rats with experimental autoimmune neuritis, the infiltration of inflammatory cells and demyelination in the apelin-13 group decreased significantly [20]. More importantly, apelin relieves the nerve damage caused by the abuse of many drugs, such as methamphetamine, manifesting as reduced oxidative stress, apoptosis, and autophagy [21].Consequently, the regulation of the apelin/APJ axis in the nervous system is widespread and effective. Herein, the modulatory role of the apelin/APJ system in neurological disorders is reviewed, and its new therapeutic strategies for neurological disorders require exploration.
Apelin/APJ system alleviates cerebrovascular diseases
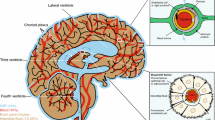
Cerebrovascular diseases are mostly caused by brain injuries, and drug applications are mainly focused on recovery after surgery. The palliative effect of the apelin/APJ system in brain injuries such as subarachnoid hemorrhage, ischemic stroke has been widely reported (Fig. 1). Therefore, apelin/APJ system may be a potential target for cerebrovascular diseases therapy.

Apelin/APJ system alleviates cerebrovascular diseases. The mechanisms of apelin/APJ system alleviates SAH include the following: Apelin/APJ system inhibits Bcl-2-mediated apoptosis through PI3K/Akt agonism of GLP-1R. Apelin/APJ system inhibits TXNIP/NLRP3-mediated caspase-1, IL-1β and TNF-α, and MPO release through AMPK. The apelin/APJ system inhibits endoplasmic reticulum stress through the ATF6/CHOP pathways. The mechanisms of apelin/APJ system alleviates ischemic stroke include the following: Apelin/APJ system inhibits oxidative stress via PLC/IP3/CAMKK/AMPK/GSK-3β/Nrf-2 pathways. Apelin/APJ system inhibits AQP4-mediated edema via PI3K/Akt. Apelin/APJ system inhibits MCP-1 and MIP-1α-mediated inflammation. SAH subarachnoid hemorrhage, Bcl-2 B-cell lymphoma 2, PI3K phosphatidylinositol-3-kinase, Akt protein kinase B, TXNIP thioredoxin-interacting protein, NLRP3 NOD-like receptor protein 3, IL-1β interleukin-1β, TNF-α tumor necrosis factor-α, MPO myeloperoxidase, AMPK AMP-activated kinase, ATF6 activating transcription factor 6, CHOP C/EBP homologous protein, PLC phospholipase C, IP3 inositol triphosphate, CAMKK calmodulin dependent protein kinase kinase, GSK-3 β glycogen synthase kinase-3β, Nrf-2 nuclear erythroid 2-related factor 2, AQP4 aquaporin 4, MCP-1 monocyte chemoattractant protein-1, MIP-1α macrophage inflammatory protein 1α
Subarachnoid hemorrhage
Subarachnoid hemorrhage (SAH) is one of the most serious causes of brain injury. In the early stages of SAH, perfused blood diffuses directly through the subarachnoid space and is not fully absorbed by the cerebral cortex, resulting in increased cerebral blood pressure, as well as the production of toxic cellular debris, inflammatory factors, and reactive oxygen species(ROS), which consequently inhibits normal protein folding and activates endoplasmic reticulum stress, causing irreversible damage to neuronal cells [22].
Apelin activates AMPK inhibition to reduce endoplasmic reticulum stress-mediated oxidative stress and neuroinflammation. The expression of endogenous apelin-13 and APJ in microglia and neurons rose within 24 h after SAH, while contributing to a significant increase and peak of p-AMPK [23]. AMPK reduces neutrophil infiltration after stroke and the neurotoxicity of glutamate in the hippocampus caused by the elevation of TXNIP (Thioredoxin-interacting protein)/NLRP3 (NOD-like receptor protein 3) inflammatory bodies under endoplasmic reticulum overstress [24]. However, after the application of exogenous apelin-13, the level of AMPK continued to increase and significantly decreased the level of NLRP3 and TXNIP, alleviating the nerve injury under oxidative stress and inflammation, which is reflected in the decrease of Bip, cleaved caspase-1, IL-1β and TNF-α, MPO, and ROS [23]. On the other hand, excessive endoplasmic reticulum stress leads to an unfolded protein response (UPR). Activating transcription factor 6 (ATF6), a crucial transmembrane protein that conducts endoplasmic reticulum pressure [25], increased significantly in the cytoplasm and nucleus of neurons after SAH, as did downstream target molecule C/EBP homologous protein (CHOP), and then played a role in promoting apoptosis in the brain [26]. However, this phenomenon is reversed by the application of apelin-13 in the lateral ventricle [27].
In addition to inflammation and endoplasmic reticulum stress, neuronal damage is another risk factor for early brain injury after SAH, which is associated with neuronal apoptosis. Emerging evidence indicates that glucagon-like peptide 1 receptor (GLP-1R) acts as an anti-apoptotic target and attenuates neuronal apoptosis through the PI3K/Akt pathway [28, 29]. The level of GLP-1R increases significantly after SAH and exogenous apelin-13 further up-regulates the expression of GLP-1R in ipsilateral cortical neurons after SAH, and increases the expression of apoptosis-related protein Bcl-2 through PI3K/Akt signal pathways, thus relieving the symptoms of neuronal apoptosis and complicated brain edema. The APJ inhibitor ML221 counteracted these neuroprotective gains [30, 31].
In conclusion, the apelin/APJ system alleviates SAH symptoms by inhibiting endoplasmic reticulum stress and neuronal apoptosis. Further studies should focus on whether apelin upregulation is significantly sufficient to affect ER stress in response to SAH.
Ischemic stroke
Ischemic stroke accounts for about 87% of all stroke patients [32]. Akin to hemorrhagic stroke, oxidative stress and post-ischemic inflammation are considered to be the key pathogenic mechanisms of brain damage caused by ischemic stroke [33].
Duan et al. have concretely elaborated on the process by which apelin-13 protects the brain from ischemic injury [34]. Administration of apelin-13 activates phospholipase C (PLC) following coupling with Gα proteins, and inositol triphosphate (IP3) opens calcium channels after receiving this signal, which promotes the signal transduction of cytoplasmic Ca2+, and calmodulin dependent protein kinase kinase (CaMKK) is activated after binding to calcium [35]. Immediately after, AMPK is activated and phosphorylated by CaMKK in the absence of energy. Glycogen synthase kinase-3β (GSK-3β), as a serine/threonine protein kinase involved in oxidative stress, is then phosphorylated, and mediates the binding of Nrf2, which has been dissociated from its molecular chaperone keap1 and translocated to the nucleus in response to oxidative stress, to antioxidant response elements (ARs), then upregulates heme oxygenase-1 (HO-1) and NADPH quinone oxidoreductase (NQO1), so as to play a neuroprotective role. In summary, apelin-13 protects against oxidative damage through AMPK/GSK-3β/Nrf2 pathways. In addition to the above observation, various subtypes of apelin have been found to protect neurons by inhibiting apoptosis through different pathways, apelin-36 activates PI3K/Akt signaling pathways and drops the levels of cleaved caspase-3 and Bax, ultimately reduces infarct size [36]. Analogously, in cerebral ischemic injury, apelin-13 inhibits apoptosis through the PI3K/Akt and ERK pathways, and suppresses excessive autophagy through PI3K/Akt/mTOR pathways [37,38,39]. For the members of the apoptosis-related MAPKs family such as JNK and P38MAPK, apelin-12 inhibits them thus offering protection to neurons [40].
In terms of brain edema symptoms, aquaporin 4 (AQP4) is a key downstream protein of the apelin/APJ system. AQP4 produces a biphasic effect on the relief of brain edema, which is characterized by promoting the formation of cytotoxic edema but promoting the clearance of angiogenic edema [41]. The main type in the early stage of cerebral ischemia is cytotoxic edema [42]. Consistent with observations by Chu et al. [43], apelin-13 upregulates AQP4 through ERK and PI3K/Akt pathways, and thus relieves brain edema, protects endothelial function and guards the blood–brain barrier in the short term. However, the role of AQP-4 in relatively complex brain edema needs to be further clarified. In addition, apelin-13 protects the blood–brain barrier from oxidative stress damage by inhibiting ocludin, claudin-5, endothelin B receptors (ETBR) and matrix metalloproteinases (MMPs), thereby alleviating edema [44].
Apelin-13 saves brain nerves damaged by ischaemia. Endangered nerves injured by ischemia will synthesize and release chemokines, and induce the release of pro-inflammatory mediators. Apelin-13 inhibits inflammation by decreasing the chemokines monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein 1 (MIP-1) and increasing the anti-apoptotic cytokine IL-10 [45]. What's more, in cerebral hemorrhage, MMPs mediate the imbalance of collagenase activity and brain edema. Apelin-13 inhibits the expression of MMP-9 and activates apoptosis-related proteins to rescue nerve cells [46]. However, the expression of vascular endothelial growth factor (VEGF) and MMP-9 increases in animals with focal ischemia 14 days after treatment with apelin-13 [45]. It is suggested that apelin-13 is a potential target to promote angiogenesis and local cerebral blood flow restoration after ischemic stroke.
In conclusion, apelin/APJ system regulates ischemic stroke through various transcription factors, including AQP4, MMPs, MAPKs, AMPK/GSK-3β/Nrf2 signaling, PI3K/Akt signaling, MIP-1α/MCP-1. Further experiments are needed to disentangle the exact mechanism of apelin/APJ in ischaemic stroke, with a view to targeting apelin as a potential clinical indicator in cerebrovascular disease.
Apelin/APJ system inhibits traumatic brain injury
Under severe traumatic brain injury (TBI), endogenous apelin-13 is depleted, and reduced serum apelin-13 concentration is negatively correlated with the disease and independently related to the mortality within 30 days [47]. The course of TBI can be divided into primary injury and secondary injury, and the changes of biomarkers and drug treatment are generally concentrated in the secondary injury stage.
Indeed, apelin-13 alleviated typical critical conditions such as cerebral edema, elevated intracranial pressure, because application of apelin-13 may inhibit cellular autophagy in the cortex and hippocampus in vivo and promote restoration of mitochondrial function and protein aggregation [48]. In addition, due to the destruction of the blood–brain barrier, the brain's water content reaches its peak within 24–48 h of TBI. The AQP4 protein present in astrocytes, which regulates water transport, also increased at the same time, and the expression of AQP4 is down-regulated by apelin-13, as well as the brain edema is significantly relieved [49]. In addition, apelin-13 may eliminate brain edema by preventing NMDA receptor cytotoxicity, inhibiting TNF-α synthesis, reducing ROS production, and blocking programmed cell death [50]. Another notable finding is that endogenous apelin inhibits VEGF, histamine, or inflammation-induced increases in vascular permeability [51], which may counteract the development of hyperpermeability-induced edema. Thus, autophagy may be a key target of the apelin/APJ system to inhibit secondary impairment of TBI. In addition, inhibition of brain edema may be an important phenotype of apelin/APJ system gain, and its molecular mechanism may involve AQP4, inflammatory factors, and angiogenic factors. The exact mechanism still needs to be further explored.
Apelin/APJ system attenuates neurodegenerative diseases
There is growing evidence for a role of the apelin/APJ system in Parkinson's disease and Alzheimer's disease (Table 1). The involvement of the apelin/APJ system in Huntington's disease, and Amyotrophic lateral sclerosis (ALS)has also been reported. Given the high prevalence and overlap of neurodegenerative diseases, apelin/APJ system may be a promising target for neurodegenerative diseases.
Alzheimer's disease
Till date, three cerebrospinal fluid biomarkers have been described for the diagnosis of AD, namely β-amyloid (Aβ), total tau (t-tau), and phospho-tau (p-tau) [52]. Toxic free radicals, pro-inflammatory and nutrient factors are also involved in the progression of the disease [53]. Due to the complex pathogenesis of AD, elucidation of crosstalk between apelin and the pathological targets of ADfacilitates the discovery of new targets.
Apelin-13 can improve the AD-like phenotype induced by streptozotocin(STZ) [54], which effect is mainly dependent on the brain-derived neurotrophic factor/tyrosine receptor kinase B (BDNF/TrkB) pathway. Reduced IL-1, TNF-, and elevated Ach are observed in the hippocampus of AD rats via the apelin/BDNF/TrkB pathway, which contributes to the reduction of amyloid plaques, decrease in tau phosphorylation, and recovery of cholinergic neurons [55]. In addition, apelin/APJ system regulates BDNF transcription through PI3K/Akt, ERK, or eNOS signaling pathways [17]. Thus, apelin may regulate the BDNF/TrkB axis through the above transcription factors thereby producing anti-inflammatory and anti-tau phosphorylation effects.
On the other hand, Aβ aggregation interferes with the function of hippocampal neurons and damages the hypothalamic–pituitary–adrenal axis. Glucocorticoids are released in a stressful manner with the abnormality of HPA axis, and FK506 binding protein 5 (FKBP5) is a helper protein of glucocorticoid receptor, which inhibits its sensitivity and translocation [56]. Apelin promotes the nuclear translocation of glucocorticoid receptor (GC) and enhance the sensitivity of insulin receptor by inhibiting FKBP5 [57, 58]. Another point of view suggests that Aβ-induced autophagy stress is involved in the development of AD. For details, autophagy is inhibited and neuronal apoptosis rises after intrahippocampal injection of Aβ25-35, and 2 μg of apelin treatment inhibits the autophagy flux in the early stage of Aβ aggregation through mTOR pathway, alleviates the autophagy load and maintains the turnover of autophagy and apoptosis [59]. In addition, apelin-13 at doses of 1, 2.5 and 5 μg/mL but not 10 μg/mL has inhibited intracellular reactive oxygen species and calcium production in Aβ-treated neuroblastoma cell lines [60]. Excitingly, calcium homeostasis is thought to play an instrumental role in the regulation of Aβ as well as familial Alzheimer's disease proteins such as PSENs [61]. Altogether, apelin counteracts the damage of Aβ aggregation by balancing neuroautophagy and apoptosis, alleviating oxidative stress and restoring HPA axis function. Whether apelin perturbs Aβ effects or even Alzheimer's disease progression by modulating calcium signaling needs to be supported by new evidences.
Thus, apelin/APJ system inhibits inflammation via the BDNF/TrkB axis thereby suppressing p-tau production and protects the HPA axis by inhibiting FKBP5 thereby counteracting Aβ production. Its role in balancing neuronal autophagy and apoptosis in the brain via PI3K/Akt/mTOR pathway cannot be ignored either.
Parkinson's disease
Parkinson's disease, characterized by resting tremors and bradykinesia, is a progressive neurodegenerative disease. Currently, oxidative stress, enhanced autophagy, and apoptosis inhibition are widely involved in the progression of Parkinson's disease. In addition, the restoration of learning and memory functions helps to alleviate the symptoms of Parkinson's disease. To highlight the association between apelin and Parkinson's disease, various studies focus on the potential roles and mechanisms of apelin/APJ system in autophagy, apoptosis, oxidative stress, memory and learning.
After the accumulation of -synuclein, inositol-requiring enzyme 1 (IRE1) is overactivated in drosophila PD models. IRE1 catalyzes the transcription of X-box binding protein 1 (XBP1) mRNA to induce the production of XBP1, which causes autophagy-dependent dopaminergic neuronal death [62]. However, in the nigrostriatal injection of apelin-13 in mice, the IRE1/XBP1/CHOP pathway is significantly inhibited, and autophagy is repaired so itreduces the aggregation of α-synuclein [63]. In addition, under oxidative stress caused by neurotoxins that cannot be removed reasonably by autophagy, reactive oxygen species (ROS) are produced in large quantities. Attractively, apelin-36 has been centrally reported in alleviating oxidative stress and apoptosis. In MPTP-induced Parkinson's disease model mice, apelin-36 restores antioxidant system activity by increasing superoxide dismutase (SOD) and glutathione (GSH) while decreasing malondialdehyde (MDA), and simultaneously inhibits apoptosis signal-regulated kinase 1(ASK1)/JNK/caspase-3 apoptosis signaling pathway [64]. A separate report said apelin-36 mainly inhibits the increase of endoplasmic reticulum stress-related proteins GRP78, CHOP and cleaved caspase-12, then reverses neuroapoptosis [65]. Intuitively, apelin-13 enhances autophagy while reducing misfolded proteins via the AMPK/mTOR/ULK1 pathway [66]. Together, the above results suggest that apelin alleviates neuronal damage caused by oxidative stress, insufficient autophagy, and excessive apoptosis in drug-induced Parkinson's disease models.
Except for the central role of the above axis in PD, apelin is also related to proteins in the brain that are related to the transmission of memory and learning signals. Long-term potentiation (LTP) is the foundation of learning and consolidating memory. Postsynaptic density protein 95(PSD-95) and dopaminergic receptor (DR1) are driven by dopamine and affect LTP and many synaptic signals. apelin restores the expression of PSD-95 and DR1 in the hippocampus and striatum, and improves the early long-term potentiation of the hippocampal CA1 area of Parkinson’s disease rats, thereby inducing and maintaining LTP [67, 68]. Notably, metabotropic glutamate receptors (mGluRs) are new medications used to relieve PD dyskinesia. In the striatum of the substantia nigra characterized by acute dopamine exhaustion, exogenous apelin reduces overexpressed mGluR1 to a normal level [69], and behaviorally improves PD mice's completion of novel object recognition and object location tasks [70].
In conclusion, various factors including autophagy, apoptosis, and oxidative stress are involved in the aggregation of α-synuclein. The apelin/APJ system alleviates PD by normalizing such effects. Additionally, apelin alleviates PD symptoms such as impaired learning memory and slowed movements in animal models. Thus, targeting apelin/APJ system might be a potential therapeutic strategy for PD.
Other neurodegenerative diseases
Huntington's disease, named for the causative gene Huntingtin (HTT), which encodes the Htt protein. The mutated HTT protein not only contributes to an increase in the abnormal function of the protein but also leads to a loss of normal function [71]. Although there is no direct evidence about the crosstalk of the Apelin/APJ system on HTT proteins,Considering the existence of prodromal molecular mechanisms in Huntington's disease, the crosstalk of the apelin/APJ system in Huntington's disease is worth discussing. Mutant HTT protein (mHTT), one of the biomarkers of Huntington's disease, has been reported to be an autophagic substrate and is promoted for clearance by autophagy inducers [72]. Apelin may promote the degradation of HTT proteins by initiating the autophagic pathway. Moreover, ferroptosis, as an emerging target for the treatment of neurodegenerative diseases, affects Huntington's disease by influencing iron metabolism, lipid metabolism, and oxidative stress [73]. Elabela/APJ system promotes ferroptosis through the IL-6/STAT3/GPX4 signaling pathway [74], apelin-13 causes mitochondrial iron overload and ferritinophagy [10], which may lead to ferrous ion concentration-dependent ferroptosis. Apelin/APJ system scavenges HTT and promotes ferroptosis, which may be a mechanism of interest as a future therapeutic target for Huntington's disease.
ALS is a fatal neurodegenerative disease caused by the loss of motor neurons from the brain and spinal cord. Current hypotheses to explain motor neuron degeneration include glutamate toxicity and oxidative damage [75]. The expression of apelin in the spinal cord of ALS model mice decreases with the paralytic phenotype, which is attributed to the enhanced protective effect of apelin on oxidative stress-induced neuronal death by VEGF [76]. Moreover, apelin alleviates the neurotoxicity of glutamate receptor NMDA, which has been widely reported [77, 78]. These studies illustrate apelin/APJ system is involved in ALS via inhibiting redox injury and saving NMDA receptor. Maybe in some cases, apelin/APJ system could be a key factor in hindering the development of ALS.
Apelin/APJ system is involved in psychosis
Depression
Depression is a psychological disorder with complex origins, and apelin/APJ system currently has crosstalk through neuroendocrine, trophic factors in the brain, and inflammation (Fig. 2).

Apelin/APJ system is involved in depression. Apelin/APJ system inhibits depression by agonizing BDNF/Trk through PI3K/Akt and ERK pathways. Apelin/APJ system protects the HPA axis and thus suppresses depression. LPS promotes NF-κB nuclear translocation and promotes STAT3-mediated rise in the pro-inflammatory factor iNOS and fall in the anti-inflammatory factor Arg-1, thereby promoting inflammation, and apelin reverses this effect. PI3K phosphatidylinositol-3-kinase, Akt protein kinase B, ERK extracellular regulated protein kinases, HPA axis hypothalamic–pituitary–adrenal axis, STAT3 signal transducer and activator of transcription 3, iNOS inducible nitric oxide synthase, Arg-1 Arginase 1
Neuroendocrine is considered as a new target to understand the occurrence of depression. The link between the abnormalities of the hypothalamic–pituitary–adrenal (HPA) axis and depression has been one of the most consistently reported findings in psychiatry [79]. Importantly, the plasma cortisol in patients with depression is excessively secreted, and the circadian rhythm of secretion is changed, which also indicates that patients with depression may have HPA axis dysfunction [80]. Moreover, the serum glucocorticoid level in patients with depression increased significantly, which in turn attacked the hippocampus, which is rich in glucocorticoid receptors and vulnerable to stress [81]. Intriguingly, increased expression of apelin/APJ is found in the hippocampus of rats with forced swimming stress and chronic water-immersion restraint stress [82]. Apelin-13 ameliorates HPA hyperactivity, as evidenced primarily by dexamethasone-mediated negative feedback resistance and increased relative adrenal weight, to attenuate depression-like manifestations in rats exposed to CWIRS [58]. Therefore, the mechanism of apelin/APJ system involvement in depression via neuroendocrine needs to be urgently elucidated.
Recently, the involvement of neurotrophic factors in the development of depression has also been emphasized. BDNF, as a member of the neurotrophic factor family, is a pivotal indicator of antidepressant treatment in clinical and animal models [83]. Apelin-13 activates PI3K/Akt signaling pathways related to the activation of BDNF/TrkB [84]. In detail, the therapeutic effects of apelin-13 on depression and memory impairment in rats in the forced swim test (FST) are blocked by LY294002 (PI3K inhibitor) or PD98059 (ERK1/2 inhibitor) [85]. In a word, apelin/APJ system plays a vital role in the neuroprotection mediated by BDNF/PI3K/Akt signaling pathway.
On the other hand, inflammatory processes also play a significant role in depression. Nowadays, a wealth of evidence demonstrates the role of the apelin/APJ system in the mitigation of stress and inflammation [86, 87]. Zhang et al. [88]. have detailed the protective mechanism of apelin under lipopolysaccharide (LPS)-induced inflammation. LPS induces microglia to transform to the M1 phenotype, phosphorylate and directly activate NF-κB, thereby activating NF-κB nuclear translocation, leading to increase of cytokines IL-1β, IL-6 and TNF-α, the hippocampus will suffer additional damage leading to inflammation attack, synaptic neuron damage and cognitive dysfunction. However, repeated ventricular injections of apelin-13 can reverse LPS/NF-κB-mediated neuroinflammation. Moreover, apelin inhibits LPS-induced polarization of N9 microglia [89]. Carefully, apelin-13 regulates microglial polarization by inhibiting the activation of IL-6/STAT3 pathway induced by LPS. In this process, apelin-13 decreased N9 microglial pro-inflammatory factor iNOS and upregulated anti-inflammatory factors Arg-1 [90]. Interestingly, pSTAT3 is co-expressed with NLRP3-positive cells in dorsal root ganglion neurons, and apelin-13 alleviates acute lung injury caused by LPS-mediated NLRP3 activation [8, 89]. Thus, apelin may further inhibit STAT3-activated NLRP3 to exert anti-inflammatory effects. In short, apelin-13 can reduce glial cell activation and relieve depression-like inflammation in rats by acting via NF-κB and STAT3.
There are complex brain mechanisms involved in the pathogenesis and treatment of depression, with strong links to both neuroendocrine and neuroinflammatory conditions. apelin alleviates the development of depression with its function of supporting neurotrophic factor BDNF production and anti-inflammation. In addition, central administration of apelin is reported to induce depressive behavior via κ-opioid receptors [91], and also intrahypothalamic injection of apelin-13 fails to reverse immobility in FST rats [82]. Therefore, apelin targeting to the hippocampus may be a novel target for the treatment of depression.
Epilepsy
More than 30% of patients do not respond to any of the 20 available antiepileptic drugs [92]. Currently, apelin has been identified as one of the biomarkers for temporal lobe epilepsy. Its expression shows elevated levels in patients with temporal lobe epilepsy than in healthy controls, increases gradually after seizures, and reaches higher levels in the chronic epileptic phase [93]. Severe seizures and repeated brief seizures will induce intensifying deprivation of hippocampal neurons, and rats with seizure-induced hippocampal damage show prolonged episodes of recurrent seizures and more frequent severe epileptic seizures [94]. Excitotoxicity is thought to be a crucial contributor for seizure-induced neuronal injury, in which excessive glutamatergic transmission leads to a massive influx of calcium ions into cells, ultimately results in the death of the neuron. Meanwhile, up-regulation of mGluR1 mRNA and protein occurs in the hippocampus of different animal models of epilepsy, and apelin inhibits their abnormal high expression [95]. On the other hand, the critical role of brain inflammation in epilepsy is highlighted. Expression of IL-1β, TNF-α, and high-mobility group box protein 1 (HMGB1), has elevated during seizure [92, 96]. Such inflammatory factors are mainly secreted by astrocytes. Aperin/APJ signaling disruption exacerbates PTZ-induced seizures, and apelin-13 treatment inhibits astrocyte activation and subsequent neuro-inflammation. [96].
Indeed, the expression of apelin is up-regulated in the epileptic models, but the neurological death is still observed [97]. Maybe endogenous apelin is not enough to maintain a large amount of nerve survival, more experiments are necessitated about the pathophysiologic effects of exogenous apelin.
Apelin/APJ system is related to pain
Pain is a complex psychological activity that is difficult to describe. Concurrently, as one of the most common clinical symptoms, it can be used as a warning of injury to the body and cause a series of defensive protective responses, while some long-term severe pain has become an unbearable torture to the body. So far, apelin/APJ system is mainly related to acute pain, inflammatory pain, and neuropathic pain [98].
For inflammatory pain, the existing findings indicate that in the experiment of electroacupuncture stimulation to relieve inflammatory pain, the transcription and expression of the apelin/APJ system are significantly improved [99]. Attractively, Lv et al. have concretely elaborated the mechanism of apelin attenuating inflammatory pain [100, 101]. In the lumbar spinal cord of mice with Complete Freund's Adjuvant (CFA)-induced inflammatory pain, apelin is found to reduce the significant increase of GluN2B, one of the subtypes of NMDAR. Fos, as a proto-oncogene involved in the transmission of pain information to the brain, its expression is also decreased by intrathecal injection of apelin. Besides, in the murine formalin-induced paw inflammatory pain model, intravenous and intramuscular injection of (pyr)apelin-13 (10 and 20 mg/kg) reduced licking caused by inflammatory pain, which effect is thought to be caused by the up-regulation of Adenylate cyclase (ADCY) in the prefrontal cortex by apelin/APJ system [102]. Meanwhile, i.v. (pyr)apelin-13 significantly upregulates prodynorphin (PDYN) and κ1 opioid receptor, kappa 1(OPRK1) gene expression in the mouse prefrontal cortex. Conformably, Naloxone, a non-selective opioid receptor antagonist, blocked the antinociceptive effect of (Pyr) apelin-13. Collectively, the anti-pain effect of apelin/APJ system may be achieved by inhibiting the release of excitatory neurotransmitters in spinal cord, reducing Fos in spinal dorsal horn neurons, and upregulating dynorphin and KOR gene expression and protein levels in the mouse prefrontal cortex.
Only a few studies have illustrated the relationship between apelin and pain other than inflammatory pain. In general, apelin-13(0.3 μg/mouse, i.c.v. or i.t) triggers an analgesic effect in an acetic acid-induced visceral pain model [103]. In addition, in vaso-occlusive episodes (VOE) of sickle cell disease (SCD), molecules that contract and dilate blood vessels are assumed to play a key role in severe pain. Smith et al. have demonstrated that apelin and vasoconstrictor endothelin-1 (ET-1) are potent targets to relieve the pain of VOE in children with SCD [104]. In a tail immersion test, intraventricular injection of apelin-13 suppresses pain over time and the μ1 opioid receptor is involved [105]. Whereas, there are reports show that peripheral application of apelin to increase pain sensitivity [106, 107], and apelin-mediated synovial vascular endothelial growth factor causes painful knee osteoarthritis [108]. Perhaps because of the difference in the site of action, more experiments are needed to elucidate the specific mechanisms of apelin for pain.
Apelin/APJ system promotes glioblastoma
Apelin is a marker of increased angiogenesis in many tumor types including glioblastoma (GBM) [109]. Apelin levels in glioblastoma are reduced by bevacizumab, one of the traditional therapeutic agents for GBM [110]. Furthermore, APJ is expressed in putative stem cell niche in GBM tumor tissue, high Elabela expression is associated with poor survival in glioblastoma patients, and Elabela expression is also associated with glioma grade [111]. Thus, the apelin system is a significant influential factor in GBM.
Nowadays, inhibition of vascular growth signaling to target tumor endothelial cells is a new concept in oncology therapy. Although VEGFA inhibition failed to prolong overall patient survival in clinical trials of GBM treatment [112], blocking VEGFA/VEGFR2 signaling in glioblastoma accelerates the invasion of APJ+ glioblastoma cells. Furthermore, co-administration of F13A and DC101, a blocking antibody to VEGFR2, synergistically attenuates the vascularization of glioblastoma and reduces the pro-invasive side effects associated with VEGFA/VEGFR2 inhibition [110]. Notably, the GBM vascular system in the brain of Apelin KO mice was greatly reduced, the survival rate of tumor-bearing mice was improved, and in situ injection of apelin-13 specifically restored tumor angiogenesis [113]. Thus, apelin, but not VEGF, specifically controls GBM angiogenesis in the brain.
The heterogeneity of glioblastoma is thought to be intimately related to glioblastoma stem-like cells (GSCs). GSC interacts with the tumor microenvironment and resides in a hypoxic niche near the endothelial vasculature. In a model in which brain endothelial cells are co-cultured with patient-derived GSCs, exogenous and endothelial secreted apelin are shown to be one of the signals that maintain GSC [114]. Elizabeth et al. have found that apelin is released by endothelial cells in a hypoxic microenvironment and upregulated GSK-3β levels in tumor cells, and that the APJ inhibitor MM54 reduced GSC self-renewal and enhanced sensitivity to Temozolomide [115]. Trillet et al.[116] depict a novel perspective on the involvement of the apelin/APJ system in GSC self-renewal, the arbitration of APJ expression on the GSC plasma membrane surface by the glycoprotein GP130 through ELMO domain containing 1(ELMOD1)-mediated GSC vesicle transport of ADP ribosylation factor (ARF), and GP130 modulates apelin binding to the receptor via an endocytosis/cycling mechanism. On the contrary, when GP130 is missing, apelin/GSK3β-mediated GSC complementation of tumors is abolished.
Together, apelin/APJ system intervenes in the complex GBM heterogeneity both through its pro-angiogenic capacity and through its involvement in the sorting of GSC (Fig. 3). As research advances, targeting apelin/APJ system may be a promising GBM therapy.

Apelin/APJ system promotes glioblastoma invasion. Apelin/APJ system promotes RGC aggregation for GBM growth via GSK-3β, the effect is promoted by hypoxia. GP130 regulates ARF to control apelin binding to APJ. GBM glioblastoma, RGC glioblastoma stem-like cell, GP130 glycoprotein 130, GSK-3β glycogen synthase kinase 3β
Clinical application of apelin/APJ system in nervous system
To date, a large number of reports have emerged on the use of apelin as an indicator of neurological disorders in clinical settings (Table 2). In a series of cerebrovascular diseases, apelin has also demonstrated neuroprotective capabilities similar to those of biochemical experiments. Serum apelin-13 concentrations in patients with ischemic stroke are strongly associated with disability and mortality [47]. Patients with high apelin-13 levels had a lower incidence of stroke over one year [117]. Apelin is used as an independent protective factor and predictor of hemorrhagic transformation after intravenous thrombolytic therapy [118]. In clinical cases of psychiatric disorders, plasma apelin levels are significantly higher in the first episode psychosis group than in the chronic schizophrenia and healthy groups [119]. Meanwhile, serum apelin-13 indicates total oxidation levels and may be used to screen for progression of AD development [120]. Notably, clinical testing for apelin appears to have different implications in patients with preeclampsia. Serum apelin-13 and apelin-36 concentrations are significantly lower in patients with preeclampsia compared to healthy pregnant women [121,122,123], and in placental samples, apelin concentration was proportional to the disease [124]. Another investigation has shown low intraplacental apelin mRNA and protein levels but elevated circulating apelin in patients with pre-eclampsia. The reason for this conflicting report may be attributed to the fact that previous studies on apelin serum concentrations measured total apelin rather than its active fragment. There are also many reports on the use of apelin for the diagnosis of diabetic neuropathy. Apelin negatively correlates with diabetic peripheral nerve conduction velocity [125,126,127], which is thought to be related to apelin's function regarding pro-endothelial dysfunction, angiogenesis, and inflammation. In conclusion, apelin has been used clinically to diagnose brain injury, preeclampsia, psychosis, and diabetic neuropathy and to predict the onset of AD by indicating total oxidative status. As clinical samples increase and a deeper understanding of the biological function of apelin is gained, the significance of apelin as an accurate and efficient clinical indicator will be elucidated, ultimately bridging the gap between theory and clinical application.
Conclusion
In summary, the above results prove the extensive and unique role of apelin/APJ system in neurological diseases. Apelin is highly expressed in special parts of the brain, such as the hypothalamus and hippocampus, and its levels increase when pathological changes occur in the brain area. More importantly, apelin/APJ system alleviates the secondary damage of acute brain injuries such as SAH, TBI, and ischemic stroke to the brain area, and inhibits the generation of pathological indicators in chronic neurodegenerative diseases such as AD, PD, ALS, and HTT. In addition, targeting apelin/APJ system in the hippocampus suppressed the depressive phenotype and relieved the severity of inflammatory pain. Furthermore, apelin/APJ promotes the invasion and mutation of GBM. In view of the intricate role of apelin/APJ in the nervous system, rationally design experiments to distinguish the effects of apelin/APJ system at different levels, different parts, and different experimental models on the nervous system in order to design it as a reasonable target for neurological diseases point.
Prospects
Notably, apelin/APJ system promotes the growth of GBM while generally alleviating brain edema, epilepsy, and other brain tumor symptoms as reviewed previously. Considering the unclear pathogenesis and complex microenvironment of GBM. We speculate that singular targeting of cellular pathways in glioblastoma would lead to redundant compensatory mechanisms, inadequate coverage of foci-related targets, or poor tolerability and safety. Multiple pathways and precise targeting may be potential avenues to combat GBM. In our opinion, specifically targeting APJ-positive cerebrovascular tumors and precisely depleting apelin is a promising pathway. This strategy will specifically prevent sprouting angiogenesis by not targeting the established cerebral vasculature, while avoiding increased GBM cell invasion and potentially reducing tumor hypoxia and edema formation. Interestingly, an apelin-based synNotch receptor has been designed to recognize the surface marker APJ in the neovascular endothelium [128]. The synNotch circuit appears to be a promising precision therapeutic strategy for targeting tumor microenvironmental xenobiotic vessels. In addition, many fluorescent probes that can precisely target tumors and lose small molecule signals have emerged [129]. There is an urgent need to develop a probe that targets GBM tracing apelin concentration changes in order to facilitate high-fidelity reproduction of apelin fluctuations and thus facilitate accurate surgical navigation. By taking advantage of the biological effects of the apelin/APJ system, combined with the precision targeting advantages of technologies such as immune delivery and molecular probes, treatments that combine safety and efficacy for complex tumors may open up in the future.
References
O’Dowd BF et al (1993) A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 136(1–2):355–360
Tatemoto K et al (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 251(2):471–476
Cano Martinez LJ et al (2019) Serum concentrations of apelin-17 isoform vary in accordance to blood pressure categories in individuals with obesity class 3. Clin Exp Hypertens 41(2):168–173
Castan-Laurell I et al (2011) Apelin, diabetes, and obesity. Endocrine 40(1):1–9
McKinnie SM et al (2016) The metalloprotease neprilysin degrades and inactivates apelin peptides. ChemBioChem 17(16):1495–1498
Kalea AZ, Batlle D (2010) Apelin and ACE2 in cardiovascular disease. Curr Opin Investig Drugs 11(3):273–282
Pauli A et al (2014) Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science 343(6172):1248636
Zhang H et al (2018) Apelin-13 administration protects against LPS-induced acute lung injury by inhibiting NF-kappaB pathway and NLRP3 inflammasome activation. Cell Physiol Biochem 49(5):1918–1932
Yan J et al (2020) Apelin/APJ system: an emerging therapeutic target for respiratory diseases. Cell Mol Life Sci 77(15):2919–2930
Tang M et al (2019) Ferritinophagy activation and sideroflexin1-dependent mitochondria iron overload is involved in apelin-13-induced cardiomyocytes hypertrophy. Free Radic Biol Med 134:445–457
Feng J et al (2019) The effect of apelin-13 on pancreatic islet beta cell mass and myocardial fatty acid and glucose metabolism of experimental type 2 diabetic rats. Peptides 114:1–7
Saeedi Saravi SS, Beer JH (2020) Apelin-potential therapy for COVID-19? J Mol Cell Cardiol 145:84–87
Chu J et al (2013) Apelin ameliorates TNF-alpha-induced reduction of glycogen synthesis in the hepatocytes through G protein-coupled receptor APJ. PLoS ONE 8(2):e57231
He L et al (2015) Apelin/APJ signaling in hypoxia-related diseases. Clin Chim Acta 451(Pt B):191–198
Reaux-Le Goazigo A et al (2004) Dehydration-induced cross-regulation of apelin and vasopressin immunoreactivity levels in magnocellular hypothalamic neurons. Endocrinology 145(9):4392–4400
Reaux-Le Goazigo A et al (2011) Apelin and the proopiomelanocortin system: a new regulatory pathway of hypothalamic alpha-MSH release. Am J Physiol Endocrinol Metab 301(5):E955–E966
O’Carroll AM et al (2013) The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol 219(1):R13-35
Wang CM et al (2018) High-throughput sequencing analysis of differentially expressed miRNAs and target genes in ischemia/reperfusion injury and apelin-13 neuroprotection. Neural Regen Res 13(2):265–271
Seyedabadi M, Goodchild AK, Pilowsky PM (2002) Site-specific effects of apelin-13 in the rat medulla oblongata on arterial pressure and respiration. Auton Neurosci 101(1–2):32–38
Wei YF et al (2019) Effects of APELIN-13 on the expression of IL-6, TNF-alpha, and IFN-gamma in rats with experimental autoimmune neuritis. J Biol Regul Homeost Agents 33(5):1369–1376
Foroughi K et al (2019) Apelin-13 protects PC12 cells against methamphetamine-induced oxidative stress, autophagy and apoptosis. Neurochem Res 44(9):2103–2112
Yan F et al (2017) Pharmacological Inhibition of PERK attenuates early brain injury after subarachnoid hemorrhage in rats through the activation of Akt. Mol Neurobiol 54(3):1808–1817
Xu W et al (2019) Apelin-13/APJ system attenuates early brain injury via suppression of endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation and oxidative stress in a AMPK-dependent manner after subarachnoid hemorrhage in rats. J Neuroinflamm 16(1):247
Li Y et al (2015) Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol Appl Pharmacol 286(1):53–63
Hillary RF, FitzGerald U (2018) A lifetime of stress: ATF6 in development and homeostasis. J Biomed Sci 25(1):48
Tungkum W et al (2017) Melatonin suppresses methamphetamine-triggered endoplasmic reticulum stress in C6 cells glioma cell lines. J Toxicol Sci 42(1):63–71
Xu W et al (2018) Apelin-13 alleviates early brain injury after subarachnoid hemorrhage via suppression of endoplasmic reticulum stress-mediated apoptosis and blood-brain barrier disruption: possible involvement of ATF6/CHOP pathway. Neuroscience 388:284–296
Xie Z et al (2018) Exendin-4 attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early brain injury after subarachnoid hemorrhage in rats. Neuropharmacology 128:142–151
Zhu H et al (2016) The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci Rep 6:26859
Liu Y et al (2019) Apelin-13 attenuates early brain injury following subarachnoid hemorrhage via suppressing neuronal apoptosis through the GLP-1R/PI3K/Akt signaling. Biochem Biophys Res Commun 513(1):105–111
Shen X et al (2022) Apelin-13 attenuates early brain injury through inhibiting inflammation and apoptosis in rats after experimental subarachnoid hemorrhage. Mol Biol Rep 49(3):2107–2118
Benjamin EJ et al (2019) Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 139(10):e56–e528
Lakhan SE, Kirchgessner A, Hofer M (2009) Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 7:97
Duan J et al (2019) Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling. J Neuroinflamm 16(1):24
Hurtado de Llera A et al (2014) The calcium/CaMKKalpha/beta and the cAMP/PKA pathways are essential upstream regulators of AMPK activity in boar spermatozoa. Biol Reprod 90(2):29
Gu Q et al (2013) Apelin-36, a potent peptide, protects against ischemic brain injury by activating the PI3K/Akt pathway. Neurochem Int 63(6):535–540
Yang Y et al (2014) Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci Lett 568:44–49
Shao ZQ et al (2021) Apelin-13 inhibits apoptosis and excessive autophagy in cerebral ischemia/reperfusion injury. Neural Regen Res 16(6):1044–1051
Yan XG et al (2015) Lateral intracerebroventricular injection of Apelin-13 inhibits apoptosis after cerebral ischemia/reperfusion injury. Neural Regen Res 10(5):766–771
Liu DR, Hu W, Chen GZ (2018) Apelin-12 exerts neuroprotective effect against ischemia-reperfusion injury by inhibiting JNK and P38MAPK signaling pathway in mouse. Eur Rev Med Pharmacol Sci 22(12):3888–3895
Verkman AS et al (2017) The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets 21(12):1161–1170
Hirt L et al (2017) Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J Cereb Blood Flow Metab 37(1):277–290
Chu H et al (2017) Apelin-13 protects against ischemic blood-brain barrier damage through the effects of aquaporin-4. Cerebrovasc Dis 44(1–2):10–25
Gholamzadeh R et al (2021) Apelin-13 attenuates injury following ischemic stroke by targeting matrix metalloproteinases (MMP), endothelin- B receptor, occludin/claudin-5 and oxidative stress. J Chem Neuroanat 118:102015
Chen D et al (2015) Intranasal delivery of apelin-13 is neuroprotective and promotes angiogenesis after ischemic stroke in mice. ASN Neuro 7(5):175909141560511
Bao H et al (2016) The neuroprotective effect of apelin-13 in a mouse model of intracerebral hemorrhage. Neurosci Lett 628:219–224
Zhuang Y et al (2021) Serum apelin-13 and risk of death following severe traumatic brain injury. Clin Chim Acta 516:64–68
Bao HJ et al (2015) Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem Res 40(1):89–97
Bao HJ et al (2016) Apelin-13 as a novel target for intervention in secondary injury after traumatic brain injury. Neural Regen Res 11(7):1128–1133
Khaksari M et al (2012) Apelin-13 protects the brain against ischemic reperfusion injury and cerebral edema in a transient model of focal cerebral ischemia. J Mol Neurosci 48(1):201–208
Morgan AR et al (2019) Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement 15(6):776–787
Masoumi J et al (2018) Apelin, a promising target for Alzheimer disease prevention and treatment. Neuropeptides 70:76–86
Nasseri B, Zareian P, Alizade H (2020) Apelin attenuates streptozotocin-induced learning and memory impairment by modulating necroptosis signaling pathway. Int Immunopharmacol 84:106546
Luo H et al (2019) Apelin-13 suppresses neuroinflammation against cognitive deficit in a streptozotocin-induced rat model of Alzheimer’s disease through activation of BDNF-TrkB signaling pathway. Front Pharmacol 10:395
Touma C et al (2011) FK506 binding protein 5 shapes stress responsiveness: modulation of neuroendocrine reactivity and coping behavior. Biol Psychiatry 70(10):928–936
Aminyavari S et al (2019) Anxiolytic impact of Apelin-13 in a rat model of Alzheimer’s disease: involvement of glucocorticoid receptor and FKBP5. Peptides 118:170102
Dai TT et al (2018) Apelin-13 upregulates BDNF against chronic stress-induced depression-like phenotypes by ameliorating HPA axis and hippocampal glucocorticoid receptor dysfunctions. Neuroscience 390:151–159
Aminyavari S et al (2019) Protective role of Apelin-13 on amyloid beta25-35-induced memory deficit; involvement of autophagy and apoptosis process. Prog Neuropsychopharmacol Biol Psychiatry 89:322–334
Samandari-Bahraseman MR, Elyasi L (2021) Apelin-13 protects human neuroblastoma SH-SY5Y cells against amyloid-beta induced neurotoxicity: Involvement of anti oxidant and anti apoptotic properties. J Basic Clin Physiol Pharmacol 33:599
Alzheimer’s Association Calcium Hypothesis W (2017) Calcium Hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 13(2):178–18217
Yan C et al (2019) IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis 10(11):800
Zhu J et al (2019) Apelin-13 protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through inhibiting endoplasmic reticulum stress and promoting autophagy. Brain Res 1715:203–212
Zhu J et al (2020) Apelin-36 mediates neuroprotective effects by regulating oxidative stress, autophagy and apoptosis in MPTP-induced Parkinson’s disease model mice. Brain Res 1726:146493
Zhu J et al (2019) Apelin-36 mitigates MPTP/MPP(+)-induced neurotoxicity: Involvement of alpha-synuclein and endoplasmic reticulum stress. Brain Res 1721:146334
Chen P et al (2020) Apelin-13 protects dopaminergic neurons against rotenone-induced neurotoxicity through the AMPK/mTOR/ULK-1 mediated autophagy activation. Int J Mol Sci 21(21):8376
Esmaeili-Mahani S et al (2021) Apelin-13 prevents hippocampal synaptic plasticity impairment in Parkinsonism rats. J Chem Neuroanat 111:101884
Gazmeh S et al (2022) Apelin-13 protects against memory impairment and neuronal loss, induced by Scopolamine in male rats. Metab Brain Dis 37(3):701–709
Haghparast E et al (2019) Apelin-13 attenuates motor impairments and prevents the changes in synaptic plasticity-related molecules in the striatum of Parkinsonism rats. Peptides 117:170091
Haghparast E et al (2018) Apelin-13 ameliorates cognitive impairments in 6-hydroxydopamine-induced substantia nigra lesion in rats. Neuropeptides 68:28–35
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10(1):83–98
Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11(9):1107–1117
Qiu Y et al (2020) The application of ferroptosis in diseases. Pharmacol Res 159:104919
Zhang Z et al (2022) Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic Biol Med 181:130–142
Oskarsson B, Gendron TF, Staff NP (2018) Amyotrophic lateral sclerosis: an update for 2018. Mayo Clin Proc 93(11):1617–1628
Kasai A et al (2011) Apelin deficiency accelerates the progression of amyotrophic lateral sclerosis. PLoS ONE 6(8):e23968
Zeng XJ et al (2010) Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp Cell Res 316(11):1773–1783
Cook DR et al (2011) NMDA receptor modulation by the neuropeptide apelin: implications for excitotoxic injury. J Neurochem 118(6):1113–1123
Galts CPC et al (2019) Depression in neurodegenerative diseases: common mechanisms and current treatment options. Neurosci Biobehav Rev 102:56–84
Jia Y et al (2019) Increased serum levels of cortisol and inflammatory cytokines in people with depression. J Nerv Ment Dis 207(4):271–276
Hryhorczuk C, Sharma S, Fulton SE (2013) Metabolic disturbances connecting obesity and depression. Front Neurosci 7:177
Xiao ZY et al (2018) The hippocampus is a critical site mediating antidepressant-like activity of Apelin-13 in rats. Neuroscience 375:1–9
Caviedes A et al (2017) BDNF/NF-kappaB signaling in the neurobiology of depression. Curr Pharm Des 23(21):3154–3163
Saral S et al (2021) Apelin-13 activates the hippocampal BDNF/TrkB signaling pathway and suppresses neuroinflammation in male rats with cisplatin-induced cognitive dysfunction. Behav Brain Res 408:113290
Li E et al (2016) Apelin-13 exerts antidepressant-like and recognition memory improving activities in stressed rats. Eur Neuropsychopharmacol 26(3):420–430
Faridvand Y et al (2019) Dihydroxyvitamin D3 activates Apelin/APJ system and inhibits the production of adhesion molecules and inflammatory mediators in LPS-activated RAW264.7 cells. Pharmacol Rep 71(5):811–817
Xia F et al (2021) Apelin-13 protects the lungs from ischemia-reperfusion injury by attenuating inflammatory and oxidative stress. Hum Exp Toxicol 40(4):685–694
Zhang ZX et al (2019) Apelin attenuates depressive-like behavior and neuroinflammation in rats co-treated with chronic stress and lipopolysaccharide. Neuropeptides 77:101959
Zhou S et al (2019) Apelin-13 regulates LPS-induced N9 microglia polarization involving STAT3 signaling pathway. Neuropeptides 76:101938
Zhou S et al (2020) Microglia polarization of hippocampus is involved in the mechanism of Apelin-13 ameliorating chronic water immersion restraint stress-induced depression-like behavior in rats. Neuropeptides 81:102006
Lv SY et al (2012) Centrally administered apelin-13 induces depression-like behavior in mice. Brain Res Bull 88(6):574–580
Paudel YN et al (2019) High mobility group box 1 (HMGB1) as a novel frontier in epileptogenesis: from pathogenesis to therapeutic approaches. J Neurochem 151(5):542–557
Elhady M et al (2019) Oxidative stress contribution to attention deficit hyperactivity disorder in children with epilepsy. Appl Neuropsychol Child 8(4):347–354
Dingledine R, Varvel NH, Dudek FE (2014) When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol 813:109–122
Dong H et al (2020) microRNA-182 negatively influences the neuroprotective effect of apelin against neuronal injury in epilepsy. Neuropsychiatr Dis Treat 16:327–338
Kalantaripour TP et al (2016) Anticonvulsant and neuroprotective effects of apelin-13 on pentylenetetrazole-induced seizures in male rats. Biomed Pharmacother 84:258–263
Zhang X et al (2011) Up-regulation of apelin in brain tissue of patients with epilepsy and an epileptic rat model. Peptides 32(9):1793–1799
Lv SY, Chen WD, Wang YD (2020) The apelin/APJ system in psychosis and neuropathy. Front Pharmacol 11:320
Wang K et al (2016) The effects of electroacupuncture on the apelin/APJ system in the spinal cord of rats with inflammatory pain. Anesth Analg 123(6):1603–1610
Lv S et al (2020) Intrathecally administered apelin-13 alleviated complete Freund’s adjuvant-induced inflammatory pain in mice. Front Pharmacol 11:1335
Lv S et al (2020) Intravenous administration of pyroglutamyl apelin-13 alleviates murine inflammatory pain via the kappa opioid receptor. Front Neurosci 14:929
Burghi V et al (2019) Participation of galphai-adenylate cyclase and ERK1/2 in mas receptor signaling pathways. Front Pharmacol 10:146
Lv SY et al (2012) Supraspinal antinociceptive effect of apelin-13 in a mouse visceral pain model. Peptides 37(1):165–170
Smith TP et al (2015) Modulation of pain in pediatric sickle cell disease: understanding the balance between endothelin mediated vasoconstriction and apelin mediated vasodilation. Blood Cells Mol Dis 54(2):155–159
Xu N et al (2009) Supraspinal administration of apelin-13 induces antinociception via the opioid receptor in mice. Peptides 30(6):1153–1157
Canpolat S et al (2016) Effects of apelin-13 in mice model of experimental pain and peripheral nociceptive signaling in rat sensory neurons. J Recept Signal Transduct Res 36(3):243–247
Xiong Q et al (2017) Effect of the spinal apelinAPJ system on the pathogenesis of chronic constriction injuryinduced neuropathic pain in rats. Mol Med Rep 16(2):1223–1231
Takano S et al (2018) Vascular endothelial growth factor expression and their action in the synovial membranes of patients with painful knee osteoarthritis. BMC Musculoskelet Disord 19(1):204
Masiero M et al (2013) A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 24(2):229–241
Mastrella G et al (2019) Targeting APLN/APLNR improves antiangiogenic efficiency and blunts proinvasive side effects of VEGFA/VEGFR2 blockade in glioblastoma. Cancer Res 79(9):2298–2313
Ganguly D et al (2019) APELA expression in glioma, and its association with patient survival and tumor grade. Pharmaceuticals (Basel) 12(1):45
Chinot OL (2014) Cilengitide in glioblastoma: when did it fail? Lancet Oncol 15(10):1044–1045
Frisch A et al (2020) Apelin controls angiogenesis-dependent glioblastoma growth. Int J Mol Sci 21(11):4179
Harford-Wright E, Gavard J (2018) Apelin, the devil inside brain tumors. J Exp Neurosci 12:1179069518759680
Harford-Wright E et al (2017) Pharmacological targeting of apelin impairs glioblastoma growth. Brain 140(11):2939–2954
Trillet K et al (2021) The glycoprotein GP130 governs the surface presentation of the G protein-coupled receptor APLNR. J Cell Biol 220(9):e202004114
Wang X et al (2020) The association between serum apelin-13 and the prognosis of acute ischemic stroke. Transl Stroke Res 11(4):700–707
Zhu B et al (2021) Predictive value of apelin and vaspin on hemorrhagic transformation in patients with acute ischemic stroke after intravenous thrombolysis and analysis of related factors. Evid Based Complement Alternat Med 2021:5020622
Sahpolat M, Ari M, Kokacya MH (2020) Plasma apelin, visfatin and resistin levels in patients with first episode psychosis and chronic schizophrenia. Clin Psychopharmacol Neurosci 18(1):109–115
Yildiz Z et al (2021) Serum apelin-13 levels and total oxidant/antioxidant status of patients with Alzheimer’s disease. Aging Med (Milton) 4(3):201–205
Gurlek B et al (2020) Evaluation of serum apelin-13 and apelin-36 concentrations in preeclamptic pregnancies. J Obstet Gynaecol Res 46(1):58–65
Mazloomi S et al (2021) Association of glutamate cystein ligase (GCL) activity Peroxiredoxin 4 (prxR4) and apelin levels in women with preeclampsia. Pregnancy Hypertens 23:163–168
Deniz R et al (2019) Evaluation of elabela, apelin and nitric oxide findings in maternal blood of normal pregnant women, pregnant women with pre-eclampsia, severe pre-eclampsia and umbilical arteries and venules of newborns. J Obstet Gynaecol 39(7):907–912
Yamaleyeva LM et al (2015) Downregulation of apelin in the human placental chorionic villi from preeclamptic pregnancies. Am J Physiol Endocrinol Metab 309(10):E852–E860
Xu H et al (2021) Clinical significance of apelin in the treatment of type 2 diabetic peripheral neuropathy. Medicine (Baltimore) 100(17):e25710
Hosny SS et al (2019) Relation between plasma Apelin level and peripheral neuropathy in Type 2 diabetic patients. Diabetes Metab Syndr 13(1):626–629
Bilir B et al (2016) Association of apelin, endoglin and endocan with diabetic peripheral neuropathy in type 2 diabetic patients. Eur Rev Med Pharmacol Sci 20(5):892–898
Wang Z et al (2020) Using apelin-based synthetic Notch receptors to detect angiogenesis and treat solid tumors. Nat Commun 11(1):2163
Wang Y, Chen L (2021) Smart fluorescent probe strategy for precision targeting hypoxic tumor. J Med Chem 64(6):2967–2970
Kitagawa Y et al (2021) A novel topical fluorescent probe for detection of glioblastoma. Clin Cancer Res 27(14):3936–3947
Acknowledgements
The study was supported by grants from the National Natural Science Foundation of China (81973326), Hunan Provincial Natural Science Foundation (2019JJ40255)
Funding
Funding was provided by National Natural Science Foundation of China (81973326) and Natural Science Foundation of Hunan Province (2019JJ40255)
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, A., Zhao, Q., Chen, L. et al. Apelin/APJ system: an emerging therapeutic target for neurological diseases. Mol Biol Rep 50, 1639–1653 (2023). https://doi.org/10.1007/s11033-022-08075-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-08075-9