
Abstract
Purpose
In this study, two main research objectives were examined: (1) the cytotoxic and anticancer activities of the aqueous methanol extract from Acacia nilotica flowers on three human cancer cells, namely lung A549, breast MCF-7, and leukemia THP-1 cells, and (2) the genotoxic effects of A. nilotica extract and its influence on DNA damage induced by N-methyl-N-nitrosourea (MNU) in mice.
Methods
Mice were orally treated with A. nilotica extract (200, 500, and 800 mg/kg for 4 days) with or without MNU (80 mg/kg intraperitoneally for 24 h).
Results
In vitro experiments showed that A549 cells were the most sensitive to A. nilotica extract among the tested cell lines. A. nilotica extract inhibited A549 cell proliferation by blocking the cell cycle at the G2/M phase and accumulating apoptotic cells in the sub-G0/G1 phase in A549 cells. In vivo experiments showed that MNU induced positive and negative genotoxicity in bone marrow cells and spermatocytes, respectively. Negative genotoxicity was observed in A. nilotica extract-treated groups only. However, A. nilotica extract (800 mg/kg) remarkably increased comet tail formation in bone marrow cells. Unexpectedly, the absence of antigenotoxicity was observed in three cotreated groups with A. nilotica extract and MNU compared with the MNU-treated group. Astonishingly, cotreatment with MNU and A. nilotica extract at a dose above 200 mg/kg remarkably increased micronucleus and comet tail formation in bone marrow cells compared with the MNU-treated group.
Conclusions
A. nilotica extract possessed anticancer activity with relative genotoxic effects at high doses.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
N-methyl-N-nitrosourea (MNU) is the oldest nitroso compound distributed in air, foods, tobacco smoke, plastics, cutting fluids, gasoline, agrochemicals, dyes, cosmetics, and pharmaceutical products. MNU exogenously occurs in air emissions from sludge incinerators and is endogenously constructed in the stomach and bowel by the nitrosation of methyl urea with nitrate [1]. MNU, a cell-disrupting agent, was initially used as an antineoplastic agent to treat L1210 leukemia cells implanted in mice [2]. MNU is used for carcinogenic, mutagenic, teratogenic, and immunosuppressant purposes. MNU may induce malignant tumors in several tissues based on animal species, strain, age, dosage, and route of administration. MNU, a potent direct-action mutagen, reacts with DNA directly through the transfer of the alkyl group to nitrogen and oxygen atoms in the nitrogen bases and phosphate group, causing a wide range of DNA adducts. Among them, N7 and N3 alkylpurines, O6-alkylguanine, and O4-alkylthymine adducts produce wrong nucleotide incorporation during DNA replication and consequently produce errors in RNA transcription [1].
Medicinal plants are considered the primary source of several valuable pharmaceutical drugs and have been used globally as an alternative and complementary medicine. The search for medicinal plants that can modify or counteract the unwanted side effects of mutagenic agents is in high demand [3]. Acacia, a large genus of the family Fabaceae (Leguminosae), comprises approximately 1350 species and is cultivated in tropical and subtropical regions in Africa, Asia, Australia, and the Caribbean. Acacia nilotica, known as kikar, black babul, and gum Arabic tree, has considerable economic importance represented by gum production, fuel-wood and charcoal production, and livestock feed production [4, 5].
All parts of A. nilotica, including leaves, bark, root, seed, fruit, flower, gum, and immature pods, have been used to treat various disorders, including cough, diarrhea, cold, fever, congestion, dysentery, hemorrhoid, gallbladder disorder, sclerosis, tuberculosis, smallpox, leprosy, and menstrual problems [6]. Phytochemically, A. nilotica plant is a rich source of phenolics, flavonoids, anthocyanins, saponins, carbohydrates, and proteins [5, 6]. Acacias are characterized by their small and fragrant flowers, which are rich in volatile terpenoids. Acacia flowers’ aroma is used for the preparation of aromatic products and essential oils, and its fragrance is used in cosmetics and perfumes. The infusion of Acacia flowers is used in the preparation of digestive, sedative, analgesic and antirheumatic tea [7]. Many literature reports have almost exclusively focused on the phytochemical, toxicological and biological screening of the leaves, seeds, pod, root, and bark of Acacia species [8,9,10,11,12,13]. However, the chemopreventive and genotoxicological effects of A. nilotica flowers are limited. Therefore, this study was conducted to examine the aqueous methanol extract from A. nilotica flowers, based on the following research points: (1) cytotoxicity using sulforhodamine blue (SRB) and 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2 H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) assays for adherent (lung A549 and breast MCF-7); and suspension (leukemia THP-1) cancer cells, respectively; (2) anticancer effect using cell cycle analysis via flow cytometry; and (3) genotoxicity and its influence on MNU-induced genotoxicity using chromosomal aberration (CAs), micronucleus (MN), and comet assays in mice.
Materials and methods
Plant materials, extraction process and isolation
A. nilotica flowers were collected from Upper Egypt in March 2018. The plant specimen was botanically identified and stored in the herbarium at the Department of Botany, Faculty of Science, Cairo University, under voucher number A356. The flowers were defatted with CHCl3 (3 x 1 L) and extracted with methanol (CH3OH)/ H2O (7:3; 5 × 3 L) at room temperature. The combined extracts were filtered, evaporated under reduced pressure and lyophilized (60 g). The dried extract was loaded on a polyamide 6S column chromatography (80 × 3 cm). The column was eluted with H2O, and H2O-ethanol mixtures of decreasing polarity and 10 fractions (1 L, each) were collected. The main phenolic fractions obtained were combined into four fractions after chromatographic analysis. Fraction A (2.2 g) was fractionated via column chromatography on Sephadex LH-20 with aqueous ethanol (0%- 70%) for elution to give compounds 1 and 3. Fraction B (2.5 g) was subjected to column chromatography on cellulose and n-BuOH saturated with H2O as an eluent to yield two main subfractions. Each was separately fractionated on a Sephadex LH-20 to yield pure samples 6 and 5. Under the same procedures, fractions C (2.8 g) and D (2.4 g) gave chromatographically pure samples 4, 2 and 7.
Cell lines and treatment
Lung adenocarcinoma (A549), breast adenocarcinoma (MCF-7), and acute monocytic leukemia (THP-1) were purchased from the American Type Culture Collection (Manassas, VA, USA). Adherent cancer cell lines (A549 and MCF-7) were grown in a modified eagles medium. The suspension cancer cell line (THP-1) was cultured into an RPMI-1640 medium. Cells were maintained in an appropriate medium supplemented with streptomycin (100 mg/mL), penicillin (100 units/mL), and heat-inactivated fetal bovine serum (10%) in a 5% CO2 atmosphere at 37 °C. Cultured media and serum were obtained from GIBCO™ (Grand Island, NY, USA).
SRB cytotoxicity assay
This assay measures the cellular protein content in the viable cells as described previously [14]. Briefly, adherent cells were harvested through trypsinization and cell suspension (5×103 cells/well, 100 µL) was seeded in 96-well plates for 24 h. Aliquots of a 100 µL medium containing different concentrations of plant extract (10, 20, 50, 75, and 100 µg/mL) were added to each well for 72 h. The medium was aspirated and trichloroacetic acid (10%, 150 µL) was added to each well and left for 1 hour at 4°C to fix the cells attached to the bottom of the wells. The supernatant was removed and the microplates were rinsed with distilled water five times and kept for air-drying. The SRB solution (70 µL, 0.4% w/v in 1% acetic acid) were added to each well, in a dark place at room temperature for 30 min. The microplates were rinsed thrice with acetic acid (1%) and allowed to air-dry overnight. The adsorbed SRB was dissolved by adding 10 mM Tris buffer (pH 10.5, 150 µL) to each well and the plate was gently stirred for 10 min on a shaker platform. The absorbance of the wells was measured using a microplate reader at a wavelength of 540 nm.
WST-1 cytotoxicity assay
This assay is based on the cleavage of the slightly red tetrazolium salt WST-1 to form a dark red formazan dye by cellular mitochondrial dehydrogenases in living cells [15]. Briefly, THP-1 cells were harvested through centrifugation and cell suspension (3 × 103 cells/ well, 50 µL) was seeded into a 96-well plate. After 24 h, cells were treated with an aliquot of 50 µL medium containing different concentrations of plant extract (10, 20, 50, 75, and 100 µg/mL) for 48 h. The WST-1 reagent (10 µL) was added to each well and cells were incubated for an additional hour at 37°C. The optical density (OD) of each well was measured at 450 nm using a microplate reader. The percentage of cell viability was calculated according to the following equation: Cell viability (%) = [(OD of treated cells – OD of blank)/ (OD of control – OD of blank) × 100]. Cytotoxic activity is expressed as IC50 (µg/mL), which is the half-maximal inhibitory concentration of the plant extract required to kill 50% of cancer cells and is used to determine drug effectiveness.
Cell cycle analysis
Cell cycle distribution was performed via flow cytometry as described previously [14]. Briefly, A549 cells (2×106/ 3 mL/six-well plate) were treated with three concentrations of A. nioltica (½ IC50, IC50, and double IC50) for 72 h. Treated cells with dimethylsulfoxide and doxorubicin (0.5µM) were used as negative and positive controls, respectively. Cells were harvested and centrifuged at 2000 rpm for 5 min. The supernatant was discarded, and cell pellets were washed twice with phosphate buffer solution (PBS, 2 mL). Cells were fixed overnight in chilled 70% ethanol in a refrigerator at 4°C. The fixed cells were washed and resuspended in PBS (1 mL) containing RNase digestate (400 µg/mL) for 1 h at 37ºC. Subsequently, cells were stained with propidium iodide (10 µg/mL) for 30 min in the dark. Finally, the cells were analyzed immediately for DNA content via flow cytometry using an FL2 (λex/λem = 535/617 nm) signal detector (ACEA Novocyte™ flowcytometer, ACEA Biosciences Inc., San Diego, CA, USA). Cell cycle histograms were analyzed using ACEA NovoExpress™ software (ACEA Biosciences).
Experimental animals
Mature Swiss male mice of 8–12 weeks old and weighing ~ 25 g were obtained from the animal house colony of the National Research Centre (NRC; Dokki, Cairo, Egypt). Mice were housed in plastic boxes in an air-conditioned room with a temperature of 23 ± 1 °C and relative humidity of 50 ± 20% in a 12-hour light/dark cycle. Mice were provided with a standard balanced pelleted chow diet and chlorinated tap water ad libitum. All experimental procedures were performed following the Guiding Principles for the Care and Use of Laboratory Animals approved by the NRC.
Experimental design
Ninety mice were equally allocated into nine groups of 10 mice each as follows:
Group 1
Negative control group was given distilled water.
Group 2
Positive control group was intraperitoneally (i.p) injected with MNU (80 mg/kg, dissolved in distilled water) for 24 h. This dose was selected based on genotoxic studies of MNU in mice [16].
Groups 3–6: Mice were given orally three doses of A. nilotica extract (200, 500, and 800 mg/kg; solubilized in distilled water) alone for four consecutive days.
Groups 7–9: Mice were treated with A. nilotica extract (200, 500, and 800 mg/kg) for 4 consecutive days followed by an i.p injection with MNU (80 mg/kg) after 1 h from the last dose of plant extract.
Mice were killed by cervical dislocation 24 h after the end of the treatment. For chromosomal analysis, bone marrow and testis were collected from half of the animals in each group. For MN and comet assays, bone marrow cells were collected from another half of the animals in each group.
CAs assay
Mitotic and meiotic CAs were prepared from bone marrow and testis of the same animal, respectively, as described previously in detail [17]. The slides were stained with 10% Giemsa solution in PBS. One hundred cells in mitotic and meiotic metaphases (diakinesis metaphase I spermatocytes) were examined per mouse under a light microscope at 1600× magnification for the presence of CAs.
MN assay
The bone marrow MN assay was conducted as described previously in detail [17]. The slides were stained using the May-Grünwald–Giemsa staining protocol and examined under a light microscope at ×1600 magnification. Two thousand polychromatic erythrocytes (PCE, immature erythrocytes) were analyzed per mouse for the presence of MN.
Single cell gel electrophoresis (Comet assay)
The comet assay was conducted in mouse bone marrow under alkaline conditions (pH > 13) as described previously in details [17]. The slides were stained with ethidium bromide and immediately examined at ×400 magnification using an upright fluorescent microscope equipped with a digital camera and green light excitation filter. Two hundred nucleoid were analyzed per animal using comet score™ version 2.0.0.0 (TriTek Corp., Sumerduck, VA, USA). The percentage of DNA in the comet tail (tail DNA) is considered the most reliable parameter to quantify DNA damage.
Data analysis
Data were analyzed using SPSS version 20 (Statistical Package of Social Science, Armonk, NY, UAS; IBM Corp). Data were checked for normality and the homogeneity of the variance using the Kolmogorov–Smirnov’s and Levene’s tests, respectively. The differences among groups with normal distribution were analyzed via one-way analysis of variance (ANOVA) followed by Tukey honestly significant difference (HSD) test. Results were regarded as significant when P < 0.05.
Results
Identification of isolated polyphenol compounds
Seven polyphenol compounds were isolated from the aqueous methanol extract from A. nilotica flowers. By comparing their chromatographic and spectroscopic data with those reported in the literature [18, 19], these compounds were identified as follows:
Gallic acid (1): 1 H-nuclear magnetic resonance (NMR) δ (ppm): 6.98 (s, H-2 and H-6).13 C-NMR δ (ppm): 120.60 (C-1), 108.80 (C-2), 145.50 (C-3), 138.10 (C-4), 145.50 (C-5), 108.80 (C-6), 167.70 (C-7).
Catechin (2): 1 H-NMR δ (ppm): 6.72 (d, J = 1.4 Hz, H-2’), 6.69 (d, J = 8.1 Hz, H-5’), 6.59 (dd, J = 8.1 and J = 1.4 Hz, H-6’), 5.88 (d, J = 2.1 Hz, H-6), 5.70 (d, J = 2.1 Hz, H-8), 4.49 (d, J = 7.34 Hz, H-2), 3.63 (m, H-3), 2.65 (eq., dd, J = 15.87 and J = 5.31, H-4) or 2.35 (ax., dd, J = 15.94 and J = 7.86 Hz, H-4). 13 C-NMR δ (ppm): 81.27 (C-2), 66.63 (C-3), 28.09 (C-4), 156.50 (C-5), 95.50 (C-6), 156.76 (C-7), 94.24 (C-8), 155.67 (C-9), 99.45 (C-10), 130.94 (C-1’), 114.81 (C-2’), 145.16 (C-3’), 145.16 (C-4’), 115.46 (C-5’), 118.82 (C-6’).
7-Galloyl catechin (3): 1 H-NMR δ (ppm): 7.27 (s, galloyl), 6.95 (d, J = 1.8 Hz, H-2′), 6.85 (d, J = 7.8 Hz, H-5’), 6.84 (dd, J = 9.9 Hz and 1.8 Hz, H-6’), 6.38 (d, J = 2.1 Hz, H-6), 6.28 (d, J = 2.1 Hz, H-8), 4.71 (d, J = 7.5 Hz, H-2), 3.02 (m, H-3), 2.70 (eq., dd, J = 16.5 and 8.1 Hz, H-4) or 2.65 (ax., dd, J = 16.5 and 8.1 Hz, H-4).13 C-NMR δ (ppm): 82.21 (C-2), 67.23 (C-3), 28.14 (C-4), 156.31 (C-5), 101.47 (C-6), 150.82 (C-7), 101.19 (C-8), 156.00 (C-9), 106.25 (C-10), 131.22 (C-1′), 114.53 (C-2′), 145.15 (C-3′), 145.04 (C-4′), 115.13 (C-5′), 119.38 (C-5′), 120.43 (galloyl C-1), 109.68 (galloyl C-2), 145.59 (galloyl C-3), 138.76 (galloyl C-4), 145.59 (galloyl C-5), 109.68 (galloyl C-6), 164.63 (-COO-).
Quercetin 3-O-rhanmnopyranosyl (1→6) glucopyranoside (4): 1 H-NMR, aglycone, δ (ppm): 7.57 (1 H, d, J = 2.1 Hz, H-2′), 7.54 (1 H, dd, J = 9, 2.1 Hz, H-6′), 6.85 (1 H, d, J = 9 Hz, H-5′), 6.39 (1 H, d, J = 2.1 Hz, H-8), 6.2 (1 H, d, J = 2.1 Hz, H-6). Sugar: 5.35 (1 H, d, J = 7.5 Hz, H-1″), 4.4 (1 H, d, J = 2.0 Hz, H-1′″), 3.16–3.65 (9 H, m, H-2″–H-6″, H-2′″–H-5′″), 1.01 (3 H, d, J = 6.3 Hz, CH3 of rhamnose). 13 C-NMR, aglycone δ (ppm): 158.6 (C-2), 135.6 (C-3), 179.4 (C-4), 163.0 (C-5), 100.1 (C-6), 166.5 (C-7), 95.0 (C-8), 159.3 (C-9), 105.5 (C-10), 123.1 (C-1′), 117.7 (C-2′), 145.9 (C-3′), 149.9 (C-4′), 116.1 (C5′), 123.5 (C-6′). Sugar: glucose at 3-position: δ (ppm): 104.8 (C-1″), 75.7 (C-2″), 78.2 (C-3″), 71.4 (C-4″), 77.2 (C-5″), 68.6 (C-6″). Rhamnose at 3-position as terminal sugar: δ (ppm): 102.4 (C-1′″), 72.1 (C-2′″), 72.2 (C-3′″), 73.9 (C-4′″), 69.7 (C-5′″), 17.9 CH3 of rhamnose.
Quercetin-3-O-α-L-rhamnopyranoside (5): 1 H-NMR, aglycone δ (ppm): 7.33 (1 H, d, J = 2.0 Hz, H-2′), 7.28 (1 H, dd, J = 2.0 and 8.5 Hz, H-6′), 6.9 (1 H, d, J = 8.5 Hz, H-5′), 6.42 (1 H, d, J = 2.0 Hz, H-8), 6.23 (1 H, d, J = 2.0 Hz, H-6). Sugar: δ (ppm): 5.29 (1 H, d, J = 1.41 Hz, H-1″), 3.16–3.94 (4 H, m, H-2″–H-5″),1.03 (3 H, d, J = 6.15 Hz, CH3 of rhamnose). 13 C-NMR, aglycone: δ (ppm): 157.34 (C-2), 134.27 (C-3), 177.79 (C-4), 161.35 (C-5), 98.78 (C-6), 164.34 (C-7), 93.70 (C-8), 156.86 (C-9), 104.16 (C-10), 121.17 (C-1′), 115.52 (C-2′), 145.25 (C-3′), 148.50 (C-4′), 115.72 (C-5′), 120.80 (C-6′). Sugar: δ (ppm): 101.87 (C-1″), 70.43 (C-2″), 70.63 (C-3″), 71.25 (C-4″), 70.11 (C-5″), 17.54 (C-6″).
Quercetin 3-O-β-glucopyranoside (6): 1 H-NMR, aglycone: δ (ppm): 7.67 (1 H, dd, J = 2.12 and 8.62 Hz, H-6′), 7.53 (1 H, d, J = 2.12 Hz, H-2′), 6.82 (1 H, d, J = 8.62 Hz, H-5′), 6.4 (1 H, d, J = 1.8 Hz, H-8), 6.2 (1 H, d, J = 1.8 Hz, H-6). Sugar: δ (ppm): 5.37 (1 H, d, J = 7.63 Hz, H-1″), 3.28–3.65 (5 H, m, H-2″-H-6″). 13 C-NMR, aglycone: δ (ppm): 156.80 (C-2), 133.60 (C-3), 177.50 (C-4), 161.60 (C-5), 98.90 (C-6), 164.60 (C-7), 93.80 (C-8), 156.60 (C-9), 104.00 (C-10), 121.60 (C-1′), 115.80 (C-2′), 145.80 (C-3′), 148.80 (C-4′), 116.20 (C-5′), 122.00 (C-6′). Sugar: δ (ppm): 101.20 (C-1″), 71.60 (C-2″), 74.40 (C-3″), 70.02 (C-4″), 77.70 (C-5″), 61.50 (C-6″).
Quercetin (7): 1 H-NMR δ (ppm): 7.69 (1 H, d, J = 2.1 Hz, H-2′), 7.57 (1 H, dd, J = 2.1 and 8.4 Hz, H-6′), 6.9 (1 H, d, J = 8.4 Hz, H-5′), 6.42 (1 H, d, J = 1.8 Hz, H-8), 6.2 (1 H, d, J = 1.8 Hz, H-6). 13 C-NMR δ (ppm): 147.50 (C-2), 136.44 (C-3), 176.55 (C-4), 161.43 (C-5), 98.88 (C-6), 164.59 (C-7), 94.05 (C-8), 156.83 (C-9), 103.71 (C-10), 122.66 (C-1′), 116.31 (C-2′), 145.76 (C-3′), 148.40 (C-4′), 115.76 (C-5′), 120.68 (C-6′).
Effects ofA. niloticaextract on cell viability.
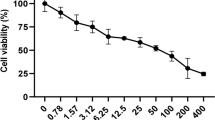
In Table 1, A. nilotica extract displayed very lower cytotoxicity against THP-1 cells at concentrations of > 100 µg/mL. By contrast, A. nilotica extract showed 50% growth inhibition against A549 and MCF-7 cells at concentrations of < 100 µg /mL. Accordingly, A549 cells were more sensitive to A. nilotica extract and better candidates for studying molecular cytotoxicity using cell cycle analysis.
Effects of A.niloticaextract on the cell cycle distribution.
To elucidate the molecular mechanism by which A. nilotica extract caused growth inhibition in A549 cells, cell cycle distribution was analyzed via flow cytometry (Table 2; Fig. 1). Results showed that treatment with the half-IC50 concentration of A. nilotica extract had no significant effect on all cell cycle phases compared with control cells. Treatment with IC50 and double IC50 concentrations of A. nilotica extract induced a significant increase in the sub-G0/G1 apoptotic peak (13.99% and 16.46% vs. 1.34% and 3.12% for negative and positive control cells, respectively). This increase was accompanied by a decrease in cell population in G0/G1 and S phases. Treatment with A. nilotica (IC50 and double IC50) significantly increased the cell population in the G2/M phase (16.29% and 19.91% vs. 9.56% and 46.45% for the negative and positive control cells, respectively).

Flow cytometric DNA histogram of A549 cells incubated with three concentrations of A. nilotica extract (half IC50, IC50, and double IC50) for 72 h. Representative of one of three similar experiments
Acute toxicity study
Throughout the 14 day study period, no death was observed in the three groups treated with A. nilotica extract at 800, 1600, and 2400 mg/kg. Accordingly, LD50 of A. nilotica extract was > 2400 mg/kg in male mice.
Effects ofA. niloticaextract on MNU-induced CAs.
Tables 3 and 4 summarize the occurrence of CAs induced by treatment with MNU and A. nilotica alone or in combination in mouse bone marrow cells and spermatocytes. Oral supplementation with A. nilotica extract (200, 500, and 800 mg/kg) did not statistical increase CAs in mouse bone marrow (2.20%, 2.80%, and 3.80%, respectively) and spermatocytes (4.20%, 4.40%, and 6.40%, respectively). Single i.p injection of MNU (80 mg/kg) significantly increased CA levels in mouse bone marrow cells (21.00% vs. 3.40% for the control, P < 0.05). However, MNU had no statistical effect on CA levels (4.20% vs. 3.20% for thecontrol, P > 0.05) in mouse spermatocytes. Unexpectedly, when three doses of A. nilotica extract were given before MNU administration, CA levels did not significantly change in mouse bone marrow cells and spermatocytes compared with the MNU-only treated group in mouse spermatocytes.
Effects ofA. niloticaextract on MNU-induced micronucleated PCEs (MNPCEs) and comet tail formation.
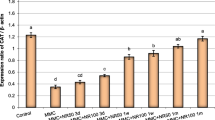
In Table (3), three doses of A. nilotica extract did not cause a significant increase in the frequency of MNPCEs (0.90%, 1.07%, and 1.39%, respectively vs. 0.51% for the control) in bone marrow cells. Low and middle doses of A. nilotica extract had no statistical effect on the percentage of tail DNA (8.21% and 9.00%, respectively) compared with the control group (Supplementary Fig. 1). However, a high dose of A. nilotica extract produced a detectable increase in comet tail formation (11.61% vs. 8.01% for the control).Treatment with MNU considerably increased the MNPCE levels (3.85% vs. 0.51% for the control) and comet tail formation (13.97% vs. 8.01% for the control) in mouse bone marrow cells. Unexpectedly, no inhibitory activity in MNPCEs (2.96%, 5.62%, and 7.40%, respectively) and comet tail formation (14.67%, 15.55%, and 19.21%, respectively) were observed between cotreated groups and the MNU-only-treated group. Surprisingly, cotreatment with middle and high doses of A. nilotica extract statistically increased MNPCEs (5.62% and 7.40%, respectively) compared with the MNU-treated group. Cotreatment with MNU and a high dose of A. nilotica extract statistically increased comet tail formation (19.21% vs. 13.97% for the positive control).
Discussion
Generally, medicinal plants contain a mixture of phytochemical constituents that may stimulate or inhibit DNA damage induced by mutagens. In this sense, this study aimed to evaluate two main research objectives: (1) in vitro anticancer activity of A. nilotica against three human cancer cells namely, A549, MCF-7, and THP-1 cells, and (2) in vivo genotoxic activity of A. nilotica extract and its effect against MNU in mouse tissues.
In vitro experiments showed that A. nilotica extract exerted moderate cytotoxicity toward A549 (IC50 = 59.3 µg/mL) and MCF-7 cells (IC50 = 96.9 µg /mL) cells and weak cytotoxic activity toward THP-1 cells (IC50 > 100 µg/mL). This finding reflected the fact that cytotoxic criteria of crude extract, based on the IC50 value, can be classified into four classes: (1) very active potent extract with IC50 ≤ 20 µg/mL, (2) moderate active extract with IC50 > 20–100 µg/mL, (3) weak active extract with IC50 > 100–1000 µg/mL, and (4) extract without cytotoxicity with IC50 > 1000 µg/mL [20]. The anticancer activity of natural products depends on several factors, such as (1) the concentration and chemical structure of polyphenols and (2) the origin, morphology, and genomes of cancer cells [21,22,23,24]. These observations coincided with previous studies from other parts of A. nilotica and other different Acacia species [25]. For example, various extracts from A. nilotica leaves displayed cytotoxic activity in cervical cancer HeLa cells with IC50 values ranging from 28.9 to > 100 µg/mL [26]. Similarly, aqueous ethanol extract from A. catechu fruits displayed potent to moderate active cytotoxic activities, with IC50 values ranging from 9.7 to 42.8 µg/mL against nine human cancer cell lines, including A549, MCF-7, and THP-1 cells [14]. Moreover, methanol extract from Acacia. hydaspica (twigs and leaves) and its fractions ethyl acetate and n-butanol exhibited moderate active cytotoxicity against two breast cancer cells, namely MDA361 and HCC38 cells with IC50 values ranging from 29.9 to 75.9 µg/mL [8]. Furthermore, ethanol extract from aerial parts of four Acacia species, namely Acacia salicina, Acacia laeta, Acacia hamulosa, and Acacia tortilis, possessed moderate active cytotoxicity against four human cancer cell lines, including breast, liver and kidney [9].
Flow cytometric data showed that A. nilotica extract (IC50 and double IC50) markedly reduced A549 cell viability by inducing apoptosis and arresting the cell cycle at the G2/M phase. A. nilotica polyphenols can bind to DNA and proteins involved in cell cycle progression. Such bindings activate DNA fragmentation, diminish nucleic acids synthesis, and modify the expression of genes implicated in cell cycle arrest at the G2/M phase [27, 28]. A similar pattern of results reported by Sundarraj et al. [29] found that ethanol extract from A. nilotica leaves blocked the cell cycle at the G2/M phase in A549 and MCF-7 cells. Several studies have reported that Acacia species arrested cell cycle at the G2/M phase in leukemia K562 cells [14], breast cancer MDA-MB-231 cells [13], and colon cancer HT-29 cells [12]. Doxorubicin had no pronounced effect on triggering apoptosis in A549 cells compared with A. nilotica extract. However, doxorubicin was more prominent in arresting the cell cycle at the G2/M phase (46.45%) than A. nilotica extract, indicating that apoptosis might not be the core molecular mechanism of cell death in A549 cells treated with 0.5 µM doxorubicin for 72 h. Indeed, various doxorubicin concentrations activate different cytotoxic pathways to induce apoptosis or cell death through mitotic catastrophe [30].
In vivo experiments displayed that treatment with MNU significantly increased the occurrence of CAs, MNPCEs, and comet tail formation in mouse bone marrow cells. These findings referred that MUN can methylate DNA at the O6-position of guanine, causing GC→AT mutations, DNA double-strand breaks, interstrand cross-links, nonrepaired N-methylpurines, and abasic sites [31,32,33,34]. Similarly, MNU produces CAs, MNPCE, and comet tail formation in rodent bone marrow cells, liver, glandular stomach, and peripheral blood lymphocytes of rodents [16, 35,36,37,38]. Surprisingly, the absence of genotoxicity was observed in mouse spermatocytes treated with MNU (80 mg/kg) under experimental conditions. This observation was not been mentioned in earlier studies using CA assay in mouse spermatocytes. However, treatment with MNU (75 and 150 mg/kg) produced a positive response in the rat spermatocyte unscheduled DNA synthesis assay [39].
In this study, four days of repeated oral administration of A. nilotica extract did not markedly induce the levels of CAs, MNPCEs, and comet tail formation, suggesting its absence of clastogenicity under experimental conditions. However, a high dose of A. nilotica extract (800 mg/kg) significantly induced comet tail formation in mouse bone marrow cells. These findings suggested that the comet assay is more sensitive in identifying DNA damage than classical cytogenetic assays. The comet assay measures a variety of DNA damages: DNA strand breaks, DNA-adducts, purinic/apyrimidinic sites or lacking nitrogen base in the DNA region, DNA–protein cross-links, incomplete excision repair sites, and apoptotic nuclei in the cells [40, 41]. These observations revealed that chemical constituents presented in A. nilotica extract at high doses showed prooxidant and genotoxic activities in cells. These findings explained that plant-derived antioxidant polyphenols act as prooxidative and antioxidative compounds under certain circumstances in certain tissues, depending on the concentration, metal-reducing potential, chelating behavior, pH, and solubility characteristics [42]. For example, gallic acid, ellagic acid, protocatechuic acid, syringic acid, vanillic acid, caffeic acid, coumaric acid, chlorogenic acid, ferulic acid, myricetin, quercetin, rutin, kaempferol, (+)-catechin, (−)-epicatechin, delphinidin, and malvidin possess dual antioxidant and prooxidant properties [42].Similar to these findings, repeated administration of an aqueous extract from A. nilotica roots (500 mg/kg) for 28 days caused hepatotoxicity in rats on biochemical liver function enzymes [10]. Likewise, Mattana et al. [43] reported that hot aqueous and ethanol extracts from A. aroma leaves (20 mg/mL) exhibit negative and moderate genotoxicity in human blood lymphocytes using the comet assay, respectively.
Contrary to expectations, this study exhibited no antigenotoxicity of A. nilotica extract against MNU in mouse bone marrow and spermatocytes. These observations implied that the chemical constituents of A. nilotica extract did not prevent or reduce the formation of DNA adducts generated by MNU. Unexpectedly, administration of A. nilotica at doses of > 200 mg/kg potentiated the DNA- damaging effect induced by the direct-action alkylating agent MNU in MN and comet assays. These findings can be explained by the fact that many flavonoids intercalate into DNA and inhibit DNA topoisomerases (I and II) by exerting their mutagenic effects [44, 45]. This observation directly agreed with previous literature, which showed that some antioxidant compounds with biphasic nature might exert mutagenic and antimutagenic effects depending on the mutagenic agent and test system used [46, 47]. For example, the absence of antimutagenicity of the ginger extract was observed against DNA damage (comet assay) in blood leukocytes of a mouse model of MNU-induced bladder carcinogenesis [48]. Furthermore, Arora et al. [49] examined the antimutagenicity of three extracts from A. nilotica bark against direct- and indirect-acting mutagens in the Salmonella typhimurium TA 100 strain. The authors found that water and chloroform extracts have no antimutagenicity against direct-acting mutagen (sodium azide), whereas acetone extract was more effective antimutagen against indirect-action mutagen (2-aminofluorene). Furthermore, the antimutagenic activity of Acacia species against indirect-action mutagens has been reported in earlier studies. For example, aqueous extracts from the gum, flower, and leaf of A. nilotica at doses of 800 mg/kg decreased the frequency of MNPCE and CAs in mouse bone marrow cells induced by indirect mutagen dimethylbenz(a)anthracene [50]. In vitro studies showed that different extracts from A. salicina leaves reduced comet tail formation induced by H2O2 in human chronic myelogenous leukemia K562 cells [11].
Given the above data, a possible explanation for the discrepancy between this study and those of earlier investigations might be due to several factors: (1) animal species, gender, age, and tissue response; (2) Acacia species and their geographical origin, extraction process, degree of polymerization, and interaction of chemical constituents with each other in the extract; (3) polyphenol concentration in the extract, route of the administration, and duration of treatment; (4) type of mutagen and its mode of action (direct or indirect); and (5) sensitivity or reliability of the genetic marker utilized to detect DNA damage.
Conclusions
In vitro experiments showed that A. nilotica extract decreased A549 cell viability by inducing apoptosis and blocking cell cycle progression at the G2/M phase. In vivo experiments showed no inhibitory activity in the three cotreated groups with A. nilotica and MNU compared with the MNU-only-treated group. Surprisingly, A. nilotica administration at a high dose potentiated the DNA-damaging effect induced by the direct-action alkylating agent MNU in MN and comet assays. Overall, the dosage use of A. nilotica extract may be adjusted for long-term safety.
Data Availability
All data generated or analysed during this study are included in this published article.
References
Kobayashi J (2018) Effect of diet and gut environment on the gastrointestinal formation of N-nitroso compounds: A review. Nitric Oxide 73:66–73
Faustino-Rocha AI, Ferreira R, Oliveira PA, Gama A, Ginja M (2015) N-Methyl-N-nitrosourea as a mammary carcinogenic agent. Tumour Biol 36:9095–9117
Słoczyńska K, Powroźnik B, Pękala E, Waszkielewicz AM (2014) Antimutagenic compounds and their possible mechanisms of action. J Appl Genet 55:273–285
Abdullah M, Farghaly M, Youssef I (2018) Effect of feeding Acacia nilotica pods to sheep on nutrient digestibility, nitrogen balance, ruminal protozoa and rumen enzymes activity. J Anim Physiol Anim Nutr 102:662–669
Rather LJ, Mohammad F (2015) Acacia nilotica (L.): A review of its traditional uses, phytochemistry, and pharmacology. Sustain Chem Pharm 2:12–30
Ali A, Akhtar N, Khan BA, Khan MS et al (2012) Acacia nilotica: a plant of multipurpose medicinal uses. J Med Plant Res 6:1492–1496
Isla MI, Ezquer ME, Leal M, Moreno MA et al (2021) Flower beverages of native medicinal plants from Argentina (Acacia caven, Geoffroea decorticans and Larrea divaricata) as antioxidant and anti-inflammatory. J Ethnopharmacol 5:281:114490
Afsar T, Razak S, Khan MR, Mawash S et al (2016) Evaluation of antioxidant, anti-hemolytic and anticancer activity of various solvent extracts of Acacia hydaspica R. Parker aerial parts. BMC Complement Altern Med 16:258
Alajmi MF, Alam P, Alqasoumi SI, Ali Siddiqui N et al (2017) Comparative anticancer and antimicrobial activity of aerial parts of Acacia salicina, Acacia laeta, Acacia hamulosa and Acacia tortilis grown in Saudi Arabia. Saudi Pharma J 25:1248–1252
Alli LA, Adesokan AA, Salawu OA, Akanji MA (2015) Toxicological studies of aqueous extract of Acacia nilotica root. Interdiscip Toxicol 8:48–54
Bouhlel I, Kilani S, Skandrani I, Ben Amar R et al (2008) Acacia salicina extracts protect against DNA damage and mutagenesis in bacteria and human lymphoblast cell K562 cultures. Nutr Res 28:190–197
Chiaino E, Micucci M, Durante M, Budriesi R et al (2020) Apoptotic-induced effects of Acacia catechu Willd. extract in human colon cancer cells. Int J Mol Sci 21:2102
Zingue S, Michel T, Cisilotto J, Tueche AB et al (2018) The hydro-ethanolic extract of Acacia seyal (Mimosaceae) stem barks induced death in an ER-negative breast cancer cell line by the intrinsic pathway of apoptosis and inhibited cell migration. J Ethnopharmacol 223:41–50
Diab KA, Guru SK, Bhushan S, Saxena AK (2015) In vitro anticancer activities of Anogeissus latifolia, Terminalia bellerica, Acacia catechu and Moringa oleiferna Indian plants. Asian Pac. J Cancer Prev. 16:6423–6428
Berridge MV, Herst PM, Tan AS (2005) Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev 11:127–152
Diab KA, Aboul-Ela EI (2012) In vivo comparative studies on antigenotoxicity of date palm (Phoenix Dactylifera L.) Pits extract against DNA damage induced by N-Nitroso-N-methylurea in mice. Toxicol Int. 19:279–286
Diab KA, Fahmy MA, Hassan ZM, Hassan EM et al (2018) Genotoxicity of carbon tetrachloride and the protective role of essential oil of Salvia officinalis L. in mice using chromosomal aberration, micronuclei formation, and comet assay. Environ Sci Pollut Res 25:1621–1636
Markham KR (1982) Techniques of flavonoid identification. Academic press
Malan E, Pienaar DH (1987) (+)-catechin-galloyl esters from the bark of Acacia gerrardii. Phytochemistry 26:2049–2051
Atjanasuppat K, Wongkham W, Meepowpan P, Kittakoop P et al (2009) In vitro screening for anthelmintic and antitumour activity of ethnomedicinal plants from Thailand. J Ethnopharmacol 123:475–482
Abdal Dayem A, Choi HY, Yang G-M, Kim K et al (2016) The Anti-Cancer Effect of Polyphenols against Breast Cancer and Cancer Stem Cells: Molecular Mechanisms. Nutrients 8:581
Sak K (2014) Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn Rev 8:122–146
Romagnolo DF, Selmin OI (2012) Flavonoids and cancer prevention: a review of the evidence. J Nutr Gerontol Geriatr 31:206–238
Ravishankar D, Rajora AK, Greco F, Osborn HM (2013) Flavonoids as prospective compounds for anti-cancer therapy. Int J Biochem Cell Biol 45:2821–2831
Manríquez-Torres JdJ, Hernández-Lepe MA, Chávez-Méndez JR, González-Reyes S et al (2020) Isolation and cytotoxic activity of phyllocladanes from the roots of Acacia schaffneri (Leguminosae). Molecules 25:3944
Kalaivani T, Rajasekaran C, Suthindhiran K, Mathew L (2011) Free radical scavenging, cytotoxic and hemolytic activities from leaves of Acacia nilotica (L.) Wild. ex. Delile subsp. indica (Benth.) Brenan. Evid.-based Complement. Altern. Med. 2011:274741
Das A, Majumder D, Saha C (2017) Correlation of binding efficacies of DNA to flavonoids and their induced cellular damage. J Photochem Photobiol B 170:256–262
Atrahimovich D, Avni D, Khatib S (2021) Flavonoids-macromolecules interactions in human diseases with focus on alzheimer, atherosclerosis and Cancer. Antioxidants10.
Sundarraj S, Thangam R, Sreevani V, Kaveri K, Gunasekaran P, Achiraman S et al (2012) γ-Sitosterol from Acacia nilotica L. induces G2/M cell cycle arrest and apoptosis through c-Myc suppression in MCF-7 and A549 cells. J Ethnopharmacol 141:803–809
Eom Y-W, Kim MA, Park SS, Goo MJ et al (2005) Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 24:4765–4777
Loechler EL, Green CL, Essigmann JM (1984) In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci USA 81:6271–6275
Hofnung M (1985) [Molecular mechanisms of the adaptive response to alkylating agents]. Biochimie. 67:Ix-xiv
Swann PF (1990) Why do O6-alkylguanine and O4-alkylthymine miscode? The relationship between the structure of DNA containing O6-alkylguanine and O4-alkylthymine and the mutagenic properties of these bases. Mutat Res 233:81–94
Spratt TE, Levy DE (1997) Structure of the hydrogen bonding complex of O6-methylguanine with cytosine and thymine during DNA replication. Nucleic Acids Res 25:3354–3361
Sohn SJ, Huh IH, Au WW, Heo MY (1998) Antigenotoxicity of galangin against N-methyl-N-nitrosourea. Mutat Res 402:231–236
Okada E, Fujiishi Y, Yasutake N, Ohyama W (2008) Detection of micronucleated cells and gene expression changes in glandular stomach of mice treated with stomach-targeted carcinogens. Mutat Res 657:39–42
Vrzoc M, Petras ML (1997) Comparison of alkaline single cell gel (Comet) and peripheral blood micronucleus assays in detecting DNA damage caused by direct and indirect acting mutagens. Mutat Res 381:31–40
Kitamoto S, Matsuyama R, Uematsu Y, Ogata K et al (2015) Optimal dose selection of N-methyl-N-nitrosourea for the rat comet assay to evaluate DNA damage in organs with different susceptibility to cytotoxicity.Mutat Res Genet Toxicol Environ Mutagen.786–788:129 – 36
Bentley KS, Working PK (1988) Activity of germ-cell mutagens and nonmutagens in the rat spermatocyte UDS assay. Mutat Res 203:135–142
Araldi RP, de Melo TC, Mendes TB, de Sá Júnior PL et al (2015) Using the comet and micronucleus assays for genotoxicity studies: A review. Biomed Pharmacother 72:74–82
Kawaguchi S, Nakamura T, Yamamoto A, Honda G, Sasaki YF (2010) Is the comet assay a sensitive procedure for detecting genotoxicity? J Nucleic Acids 2010:541050
Fukumoto LR, Mazza G (2000) Assessing antioxidant and prooxidant activities of phenolic compounds. J Agric Food Chem 48:3597–3604
Mattana CM, Cangiano MA, Alcaráz LE, Sosa A et al (2014) Evaluation of cytotoxicity and genotoxicity of Acacia aroma leaf extracts.Sci. World J:380850
Müller L, Schütte LRF, Bücksteeg D, Alfke J et al (2021) Topoisomerase poisoning by the flavonoid nevadensin triggers DNA damage and apoptosis in human colon carcinoma HT29 cells. Arch Toxicol 95:3787–3802
Bandele OJ, Osheroff N (2007) Bioflavonoids as poisons of human topoisomerase IIα and IIβ. Biochemistry 46:6097–6108
Zeiger E (2003) Illusions of safety: antimutagens can be mutagens, and anticarcinogens can be carcinogens. Mutat Res 543:191–194
Acésio NO, Carrijo GS, Batista TH, Damasceno JL et al (2017) Assessment of the antioxidant, cytotoxic, and genotoxic potential of the Annona muricata leaves and their influence on genomic stability. J Toxicol Environ Health A 80:1290–1300
Bidinotto LT, Spinardi-Barbisan AL, Rocha NS, Salvadori DM, Barbisan LF (2006) Effects of ginger (Zingiber officinale Roscoe) on DNA damage and development of urothelial tumors in a mouse bladder carcinogenesis model. Environ Mol Mutagen 47:624–630
Arora S, Kaur K, Kaur S (2003) Indian medicinal plants as a reservoir of protective phytochemicals. Teratog Carcinog Mutagen Suppl 1:295–300
Meena PD, Kaushik P, Shukla S, Soni AK et al (2006) Anticancer and antimutagenic properties of Acacia nilotica (Linn.) on 7,12-dimethylbenz(a)anthracene-induced skin papillomagenesis in Swiss albino mice. Asian Pac J Cancer Prev 7:627–632
Acknowledgements
We thank the Microbial Biotechnology Department, NRC, for allowing us to use the fluorescence microscopy facility. We thank Mohammed Ismail (laboratory technician) for his skilled technical assistance.
Funding
There was no funding.
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
KAD: conceptualization, experimental works, statistical analysis, interpretation the results, writing-original draft, and writing-review and editing. MAF: conceptualization and design of the article, experimental work, supervision, literature search, and critical revision of the manuscript. EMH and SAE: collection of the plant sample, preparation of extract, and isolation and identification of polyphenol compounds presented in the extract. All authors read and approved the final version of manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal handling and experiment procedures were approved by the ethical committee National Research Centre (Dokki, Cairo, Egypt).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Diab, K.A., Fahmy, M.A., Hassan, E.M. et al. Evaluation of the cytotoxic, anticancer, and genotoxic activities of Acacia nilotica flowers and their effects on N-methyl-N-nitrosourea-induced genotoxicity in mice. Mol Biol Rep 49, 8439–8448 (2022). https://doi.org/10.1007/s11033-022-07662-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07662-0