
Abstract
Background
Uric acid (UA) transporters mediate the uptake and outflow of UA, and are greatly involved in the control of UA concentrations. Glucose transporter 9 (GLUT9), one of the UA transporters, has been confirmed to be expressed in human umbilical vein endothelial cells (HUVECs). This study aimed to characterize GLUT9’s effect on intracellular UA accumulation in HUVECs in a high-UA environment and to explore the mechanism of cellular dysfunction.
Methods and results
HUVECs were treated with UA to establish a model of cellular dysfunction. Then, UA uptake, GLUT9 expression and endothelial nitric oxide synthase (eNOS) and reactive oxygen species (ROS) amounts were measured. UA uptake was concentration- and time-dependent, and UA treatment significantly reduced nitric oxide (NO) levels and eNOS activity. UA also upregulated pro-inflammatory molecules and GLUT9, and increased intracellular ROS amounts in HUVECs. GLUT9 knockdown reduced UA uptake and ROS content, but antioxidant treatment did not reduce GLUT9 expression. To assess the function of JAK2/STAT3 signaling, HUVECs were treated with UA, and the phosphorylation levels of JAK2, STAT3, IL-6 and SOCS3 were increased by a high concentration of UA. In addition, GLUT9 knockdown reduced the phosphorylation of JAK2/STAT3 intermediates and increased p-eNOS amounts.
Conclusions
GLUT9 mediated the effects of high UA levels on HUVECs by increasing the cellular uptake of UA, activating JAK2/STAT3 signaling, and reduced the production of active eNOS and NO in HUVECs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Uric acid (UA) constitutes the end-product of primates’ metabolic reactions transforming purine nucleosides. Although at normal concentrations, UA plays antioxidative roles in humans, epidemiological evidence indicates abnormally high UA concentrations are associated with cardiovascular abnormalities, such as hypertension, coronary heart disease, and atherosclerosis [1,2,3,4]. However, the underpinning pathophysiological mechanisms require further elucidation. Endothelial dysfunction is the initial defect leading to cardiovascular disease [5]. Recently, animal experiments have demonstrated UA reduces endothelial nitric oxide synthase (eNOS) activity as well as nitric oxide (NO) amounts by inducing mitochondrial calcium overload, endoplasmic reticulum (ER) stress and xanthine oxidase activation in human umbilical vein endothelial cells (HUVECs), which results in apoptosis and endothelial dysfunction [6,7,8].
Serum UA concentration mainly depends on the dynamic balance between UA production and its excretion. UA transporters, including URAT1, GLUT9 and breast cancer resistance protein (BCRP), mediate UA uptake and outflow, greatly regulating UA concentrations in the body [9, 10]. It was previously demonstrated UA uptake level also affects the degree of inflammation [3, 11]. Reactive oxygen species (ROS) concentrations are increased by UA, which can reduce the expression of BCRP in HUVECs and decrease UA efflux, leading to endothelial dysfunction [12]. GLUT9 expression in HUVECs has also been confirmed by previous studies and our preliminary experiments. Therefore, we hypothesized that the balance between intracellular and extracellular UA amounts is regulated by uric acid transport, which should also involve the role of GLUT9. In hyperuricemia, UA transporters increase its intracellular accumulation, thereby promoting cell dysfunction. To test this hypothesis, the effects of high UA concentration on UA’s intracellular accumulation and GLUT9 levels in HUVECs were examined, exploring the mechanism by which UA induces cell damage.
Methods
Cell culture
HUVECs provided by the China Center for Type Culture Collection (CCTCC, China) underwent culture in F12-K basal medium supplemented with 10% FBS, ECGS and 0.1 mg/ml heparin sodium at 37 °C under 5% CO2. UA (Sigma, USA) was dissolved in serum-free F12-K medium, incubated at 37 °C with constant shaking, and filtered using 0.22-μm syringe filters (Millipore, USA) [11]. The UA concentration did not decrease when the urate-medium was stored at 4 °C within 72 h. Therefore, only the urate-medium prepared within 72 h was used. A polarizing microscope (Olympus, Japan) detected no UA crystals during cell treatments.
Assessment of the cytotoxicity of uric acid
HUVECs were cultured in 96-well plates and administered diverse amounts of UA (0, 5, 10, 15 and 20 mg/dl) at ~ 80% confluence. Cell Counting Kit-8 (Dojindo, Japan) was utilized to assess cell viability as directed by the manufacturer.
Measurement of uric acid uptake
UA uptake assays were performed in serum-free F12-K medium containing various amounts of UA (5, 10, 15 and 20 mg/dl). A Uric Acid Assay Kit (Sigma) was then utilized for the measurement of UA amounts in the cell culture supernatant after incubation for 24 h and 48 h, as directed by the manufacturer. Next, cell lysis was performed after cell counting with a hemocytometer. UA uptake was quantified by normalizing UA concentration to the cell count.
NO release assessment
NO release was measured in the cell culture supernatant using a Total Nitric Oxide Assay Kit (Beyotime Biotech, China), as directed by the manufacturer. This was achieved by measuring nitrite and nitrate amounts with the Griess reagent. Absorbance was measured on a microplate reader at 520 nm after color development.
Measurement of eNOS
The concentration of eNOS in the cell culture supernatant was assessed with a Human eNOS ELISA kit (Beyotime Biotech), as directed by the manufacturer. Briefly, eNOS in the supernatant was bound by an anti-eNOS antibody in an ELISA plate, and then horseradish peroxidase-labeled avidin and the developing agent were added in sequence. Absorbance at 520 nm was used to calculate eNOS concentration.
Measurement of intracellular ROS concentration
Intracellular ROS concentration was assessed with DCFH-DA (Sigma). The cells underwent seeding in 35-mm plates at 4 × 104 cells/ml, stimulated with UA. The culture medium was replaced with DCFH-DA diluted in serum-free medium to 10 µM, followed by a 20-min incubation. After three washes with serum-free medium, fluorescence was immediately assessed on a Leica fluorescence microscope (Leica, Germany), with excitation and emission at 488 nm and 525 nm, respectively.
Quantitative real-time PCR
RNA extraction utilized Transzol reagent (TransGen Biotech, China), and RNA purity was determined by absorbance measurements at 260 and 280 nm. Reverse transcription was carried out with a Reverse Transcriptase kit (TransGen Biotech), and transcript abundance was quantitated on a 7500 Real-Time PCR System (Applied Biosystems, USA) using the SYBR Green detection system (TransGen Biotech).
Target gene expression was normalized to Gapdh mRNA amounts. The 2−ΔΔCt method was utilized for data analysis. The primers used are listed in Table S1.
Western blot
Lysates were prepared from HUVECs in the RIPA buffer supplemented with protease and phosphatase inhibitors (Solarbio, China). The BCA kit (Thermo Fisher Scientific) was utilized for protein quantitation. Equal amounts of total protein underwent separation by sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE), followed by electro-transfer unto PVDF membranes (Millipore, USA). Upon blocking with 5% skim milk (2 h at ambient), successive incubations were carried out with primary (overnight at 4 °C) and secondary (50 min at ambient) antibodies. After washing, detection of immunoreactive bands utilized the enhanced chemiluminescence reagent (Thermo Fisher Scientific). ImageJ was used for imaging and quantitation. Target protein expression was normalized to that of GAPDH. The utilized antibodies, provided by Abcam (UK), Proteintech (USA) or Cell Signaling (USA), were directed against GLUT9 (1:1000), eNOS (1:1000), phosphorylated (p)-eNOS (1:1000), total (t)-JAK2 (1:1500), p-JAK2 (1:1000), t-STAT3 (1:1500), p-STAT3 (1:1000), IL-6 (1:2000), SOCS3 (1:2000) and GAPDH (1:20,000).
Immunofluorescence (IF)
HUVECs underwent fixation with 4% formalin in PBS and permeabilization with 0.3% Triton X-100 for 10 min. Upon blocking with 3% BSA for 30 min, incubation was carried out with mouse anti-p-eNOS (1:500) and rabbit anti-GLUT9 (1:500) antibodies, respectively, overnight at 4 °C in a humidified environment. After washing, the cells were covered with secondary antibodies and incubated at ambient for 50 min. DAPI counterstaining was performed, and cells were finally examined under a Leica fluorescence microscope (Leica).
Effects of GLUT9 siRNA transfection into HUVECs
Small interfering (si)RNAs targeting SLC2A9 (GLUT9), shown in Table S2, were designed and produced by GenePharma (China). A 20-μM siRNA stock solution was prepared as directed by the manufacturer. Then, the transfection complex was prepared by mixing Opti-MEM (Thermo Fisher Scientific, 200 µl), siRNA stock solution (5 µl) and siRNA-Mate transfection reagent (GenePharma, 10 µl). HUVECs were transfected using these transfection complexes with siRNA concentration of 50 nM in a 6-well plate at 37 °C for 24 h. Next, UA was supplemented to appropriate wells, and cells were subsequently collected to measure transfection efficiency by RT-PCR and immunoblot. N-acetylcysteine (NAC, 10 mM; Beyotime), an antioxidant, was supplemented to some wells for determining the impact of oxidative stress on GLUT9 expression.
Statistical analysis
SPSS 22.0 (SPSS, USA) was utilized for data analysis. Data are mean ± SD. Group pairs and multiple groups were compared by Student’s t-test and one-way ANOVA for normally distributed data. Otherwise, a non-parametric test was applied. P < 0.05 was deemed to reflect statistical significance.
Results
Effects of UA on survival and UA uptake in HUVECs
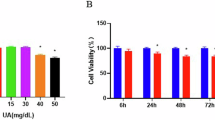
UA at 5, 10 and 15 mg/dl had no clear effect on cell survival, but decreased survival was detected at a UA concentration of 20 mg/dl (Fig. 1A). The uptake of UA increased concentration- and time-dependently; thus, high UA concentrations were associated with elevated uptake by endothelial cells (Fig. 1B).

UA’s effects on uric acid uptake, NO and eNOS amounts and inflammation in HUVECs. A Cell survival was measured using CCK-8 following incubation with 5, 10, 15, or 20 mg/dl UA. B Effect of treatment with the indicated concentrations of urate on UA uptake by HUVECs. *P < 0.05 UA groups versus Con (24 h); #P < 0.05 UA groups versus Con (48 h). C NO production, measured with Griess reagent. D–F eNOS concentration in the cell culture supernatant, and relative protein and mRNA expression, measured using an eNOS assay kit, real-time PCR and western blot analysis, respectively. G–I MCP-1, ICAM-1 and VCAM-1 mRNA amounts. *P < 0.05 UA group versus Con. Con control group, UA5 uric acid 5 mg/dl, UA10 uric acid 10 mg/dl, UA15 uric acid 15 mg/dl, UA20 uric acid 20 mg/dl, HUVEC human umbilical vein endothelial cell, NO nitric oxide, eNOS endothelial nitric oxide synthase. Data are mean ± SD (n = 3)
Uric acid causes concentration-dependent dysfunction and inflammation in HUVECs
As shown in Fig. 1C, D, NO concentration and eNOS activity in the cell culture supernatant were significantly reduced by 15 mg/dl and 20 mg/dl UA. The gene and protein expression levels of eNOS were similar in all treatment groups, but eNOS phosphorylation was significantly lower in UA-treated cells (Fig. 1E, F), indicating impaired endothelial cell function. In addition, qRT-PCR analysis of proinflammatory factors (MCP-1, ICAM-1 and VCAM-1) showed that UA also increased their expression dose-dependently in HUVECs (Fig. 1G–I).
The effect of GLUT9 on UA uptake and oxidative stress in HUVECs
The expression of GLUT9 was measured by qRT-PCR and immunoblot. The results revealed GLUT9 mRNA and protein amounts were markedly increased after administration of 10, 15 and 20 mg/dl UA, respectively, compared with control cells (Fig. 2A–C). In addition, these concentrations of UA increased intracellular ROS content (Fig. 2D).

The role of GLUT9 in urate uptake by and oxidative stress in HUVECs. A–C Relative GLUT9 mRNA and protein expression in cells administered different UA amounts. D Impact of UA at various concentrations on intracellular reactive oxygen species (ROS) content, assessed using the specific probe DCFH-DA (scale bars 200 μm). *P < 0.05 UA group versus Con. Data are mean ± SE (n = 3)
To confirm that GLUT9 affects UA uptake by HUVECs, GLUT9 siRNA was transfected into these cells. This caused marked reductions in GLUT9 mRNA and protein amounts, by 78.90% and 77.47%, respectively, indicating a high transfection efficiency (Fig. 3A–C). Knockdown of GLUT9 also significantly reduced UA uptake (P < 0.05) (Fig. 3D). Next, to determine the role of GLUT9 in oxidative stress, cells were treated with siRNA-GLUT9 and/or the antioxidant NAC. siRNA-GLUT9 transfection reduced ROS content in cells (Fig. 3E). While NAC inhibited ROS production, it did not inhibit GLUT9 expression (Fig. 3F, G).

Effects of GLUT9 knockdown on urate uptake by and oxidative stress in HUVECs. A–C The mRNA and protein amounts of GLUT9 after GLUT9 siRNA transfection. D Effect of GLUT9 knockdown on urate uptake by HUVECs. E Effect of GLUT9 knockdown and N-acetylcysteine (NAC) treatment on intracellular ROS content (scale bars 200 μm). F and G Effect of NAC on GLUT9 protein expression in HUVECs. *P < 0.05 versus Con. #P < 0.05 versus UA. Data are mean ± SE (n = 3)
Uric acid causes oxidative stress in HUVECs by inducing JAK2/STAT3 signaling
JAK2/STAT3 signaling activation is known to worsen cellular oxidative stress. Therefore, we aimed to determine whether UA increases intracellular ROS content by inducing JAK2/STAT3 signaling, subsequently leading to endothelial cell dysfunction and inflammation. The results showed that UA treatment had no effects on JAK2 and STAT3 mRNA amounts, but SOCS3 was significantly upregulated by UA at concentrations of 10, 15 and 20 mg/dl (Fig. 4A–C). Furthermore, phosphorylated JAK2 and STAT3 levels were dose-dependently elevated after UA treatment; IL-6 and SOCS3 protein amounts were increased by UA at concentrations of 15 and 20 mg/dl (Fig. 4D–E). However, knockdown of GLUT9 reduced phosphorylated JAK2 and STAT3 amounts (Fig. 5A) and enhanced eNOS phosphorylation in HUVECs (Fig. 5B).

Uric acid triggers HUVEC dysfunction by inducing JAK2/STAT3 signaling. A–C Relative mRNA levels of JAK2/STAT3 signaling intermediates. D–E Relative protein amounts of JAK2/STAT3 signaling intermediates. p-JAK2/JAK2, p-STAT3/STAT3, SOCS3 and IL-6 were examined. *P < 0.05 vs. Con (p-JAK2/JAK2), #P < 0.05 vs. Con (p-STAT3/STAT3), $P < 0.05 vs. Con (SOCS3), &P < 0.05 vs. Con (IL-6). Data are mean ± SE (n = 3)

Effect of GLUT9 knockdown on the JAK2/STAT3 pathway and eNOS expression. A Relative protein amounts of JAK2/STAT3 signaling intermediates. *P < 0.05 vs. Con (p-JAK2/JAK2), #P < 0.05 vs. Con (p-STAT3/STAT3), $P < 0.05 vs. Con (SOCS3), &P < 0.05 vs. Con (IL-6). Data are mean ± SE (n = 3). B Immunofluorescence for GLUT9 (green) and eNOS (red) in HUVECs (scale bars 200 μm). (Color figure online)
Discussion
Multiple mechanisms explaining UA’s role in endothelial dysfunction and cardiovascular diseases have been suggested. It was previously demonstrated UA increases calcium amounts in the mitochondria, which leads to greater production of mitochondrial superoxide, mitochondrial dysfunction, ROS generation [13, 14], lower eNOS expression, activation of the p38/ERK1/2 pathway, and endoplasmic reticulum stress in, and therefore damage to, endothelial cells [3, 15]. However, it remains unclear whether extracellular urate itself or intracellular accumulation of urate causes endothelial dysfunction. Here, HUVECs were treated with high UA concentrations to mimic hyperuricemia, and the roles played by the UA transporter GLUT9 in the deleterious effects of UA on HUVECs were evaluated, attempting to explore the molecular mechanisms involved.
Hyperuricemia was associated with lower plasma membrane expression of the UA efflux transporter BCRP, which leads to decreased UA efflux and intracellular UA accumulation [12]. In addition, elevated UA concentrations resulted in greater UA uptake via UA transporter-1 and greater production of cell adhesion factors [16]. As shown above, survival of HUVECs was decreased by UA only at a concentration of 20 mg/dl at the 24-h time point. Therefore, it seemed that UA did not have an acute effect on endothelial cell survival. We also determined that the uptake of UA increased dose- and time-dependently. Additionally, the UA transporter GLUT9 was upregulated by UA concentration-dependently, and GLUT9 knockdown reduced UA uptake. These data suggest that UA at a relatively high extracellular concentration upregulates GLUT9 to increase its uptake into endothelial cells.
NO release by endothelial cells plays a critical role in vascular homeostasis [17, 18]. In this study, UA had deleterious effects on endothelial cells: NO content, and eNOS activity and phosphorylation in HUVECs decreased with increasing UA concentrations. Additionally, MCP-1, ICAM-1 and VCAM-1 mRNA were increased, which implies increased endothelial cell inflammation. Previous reports demonstrated UA inhibits NO production by suppressing the phosphorylation of eNOS [7, 19]. However, UA is also considered an effective antioxidant that scavenges singlet oxygen and free radicals [20, 21]. As demonstrated above, 5 mg/dl UA had no significant effects on endothelial function and inflammatory response, but when the concentration was increased to 10 mg/dl, the inflammatory response increased significantly.
To explore the mechanism underpinning this effect, we examined GLUT9’s role. GLUT9’s effect on oxidative stress was determined by treating HUVECs with siRNA-GLUT9 and the antioxidant NAC, respectively. The results showed that siRNA and NAC treatment reduced intracellular ROS content, and GLUT9 expression was not inhibited when oxidative stress was relieved by NAC administration. Therefore, we can speculate that overexpression of GLUT9 would increase the accumulation of UA in cells and stimulate oxidative stress, which has never been addressed previously.
Inflammation is critical in initiating and sustaining vascular injury. Previous studies have shown that UA causes inflammation in endothelial cells, which manifests as upregulation and enhanced release of pro-inflammatory molecules, including IL-6, THF-α, IL-1β and MCP-1 [22]. However, the specific mechanism by which UA causes inflammation in vascular endothelial cells remains unclear. High UA amounts trigger inflammation and endothelial damage by activating NF-κB/ERK signaling [23]. In addition, UA crystals activate NLRP3 or interact with receptors on the cell membrane, including toll-like receptors 2/4, to mediate the inflammatory response [24,25,26]. Activation of JAK2/STAT3 signaling is known to cause oxidative stress [27, 28]. As shown above, JAK2/STAT3 was upregulated by UA in a concentration-dependent manner in HUVECs, as well as the pro-inflammatory cytokine IL-6. Knockdown of GLUT9 inhibited JAK2/STAT3 signaling, reduced the secretion of pro-inflammatory factors and upregulated eNOS in endothelial cells. These data indicate a very important role for GLUT9 in UA-dependent endothelial cell dysfunction.
Conclusions
High UA levels induce endothelial cell dysfunction, which is characterized by decreased eNOS activity and NO production. These effects require GLUT9, which mediates the accumulation of UA in cells, which in turn activates the JAK2/STAT3 pathway, inducing inflammation and oxidative stress.
Data availability
The data are available from the corresponding author upon reasonable contact.
References
Kuwabara M, Hisatome I, Niwa K et al (2018) Uric acid is a strong risk marker for developing hypertension from prehypertension: a 5-year Japanese cohort study. Hypertension 71(1):78–86. https://doi.org/10.1161/HYPERTENSIONAHA.117.10370
Mutluay R, Deger SM, Bahadir E, Durmaz AO, Citil R, Sindel S (2012) Uric acid is an important predictor for hypertensive early atherosclerosis. Adv Ther 29(3):276–286. https://doi.org/10.1007/s12325-012-0006-z
Prasad M, Matteson EL, Herrmann J et al (2017) Uric acid is associated with inflammation, coronary microvascular dysfunction, and adverse outcomes in postmenopausal women. Hypertension 69(2):236–242. https://doi.org/10.1161/HYPERTENSIONAHA.116.08436
Gaffo AL, Calhoun DA, Rahn EJ et al (2021) Effect of serum urate lowering with allopurinol on blood pressure in young adults: a randomized, controlled, crossover trial. Arthritis Rheumatol 73(8):1514–1522. https://doi.org/10.1002/art.41749
Yu W, Cheng JD (2020) Uric acid and cardiovascular disease: an update from molecular mechanism to clinical perspective. Front Pharmacol 11:582680. https://doi.org/10.3389/fphar.2020.582680
Park JH, Jin YM, Hwang S et al (2013) Uric acid attenuates nitric oxide production by decreasing the interaction between endothelial nitric oxide synthase and calmodulin in human umbilical vein endothelial cells: a mechanism for uric acid-induced cardiovascular disease development. Nitric Oxide 32:36–42. https://doi.org/10.1016/j.niox.2013.04.003
Li P, Zhang L, Zhang M, Zhou C, Lin N (2016) Uric acid enhances PKC-dependent eNOS phosphorylation and mediates cellular ER stress: a mechanism for uric acid-induced endothelial dysfunction. Int J Mol Med 37(4):989–997. https://doi.org/10.3892/ijmm.2016.2491
Itano S, Kadoya H, Satoh M et al (2020) Non-purine selective xanthine oxidase inhibitor ameliorates glomerular endothelial injury in InsAkita diabetic mice. Am J Physiol Ren Physiol 319(5):765–772. https://doi.org/10.1152/ajprenal.00236.2020
Andrade Sierra J, Flores Fonseca MM (2018) Renal handling of uric acid. Contrib Nephrol 192:1–7. https://doi.org/10.1159/000484271
Mandal AK, Mount DB (2015) The molecular physiology of uric acid homeostasis. Annu Rev Physiol 77:323–345. https://doi.org/10.1146/annurev-physiol-021113-170343
Kanellis J, Watanabe S, Li JH et al (2003) Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 41(6):1287–1293. https://doi.org/10.1161/01.HYP.0000072820.07472.3B
Komori H, Yamada K, Tamai I (2018) Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim Biophys Acta Biomembr 1860(5):973–980. https://doi.org/10.1016/j.bbamem.2018.01.006
Hong Q, Qi K, Feng Z et al (2012) Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 51(5):402–410. https://doi.org/10.1016/j.ceca.2012.01.003
Wiik BP, Larstorp AC, Høieggen A et al (2010) Serum uric acid is associated with new-onset diabetes in hypertensive patients with left ventricular hypertrophy: the LIFE Study. Am J Hypertens 23(8):845–851. https://doi.org/10.1038/ajh.2010.89
Yan M, Chen K, He L, Li S, Huang D, Li J (2018) Uric acid induces cardiomyocyte apoptosis via activation of calpain-1 and endoplasmic reticulum stress. Cell Physiol Biochem 45(5):2122–2135. https://doi.org/10.1159/000488048
Liang WY, Zhu XY, Zhang JW, Feng XR, Wang YC, Liu ML (2015) Uric acid promotes chemokine and adhesion molecule production in vascular endothelium via nuclear factor-kappa B signaling. Nutr Metab Cardiovasc Dis 25(2):187–194. https://doi.org/10.1016/j.numecd.2014.08.006
Huang PL (2009) eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab 20(6):295–302. https://doi.org/10.1016/j.tem.2009.03.005
Moncada S, Higgs EA (2006) The discovery of nitric oxide and its role in vascular biology. Br J Pharmacol 147(Suppl 1):S193-201. https://doi.org/10.1038/sj.bjp.0706458
Mishima M, Hamada T, Maharani N et al (2016) Effects of uric acid on the NO production of HUVECs and its restoration by urate lowering agents. Drug Res (Stuttg) 66(5):270–274. https://doi.org/10.1055/s-0035-1569405
Glantzounis GK, Tsimoyiannis EC, Kappas AM, Galaris DA (2005) Uric acid and oxidative stress. Curr Pharm Des 11(32):4145–4151. https://doi.org/10.2174/138161205774913255
Ya BL, Liu Q, Li HF et al (2018) Uric acid protects against focal cerebral ischemia/reperfusion-induced oxidative stress via activating Nrf2 and regulating neurotrophic factor expression. Oxid Med Cell Longev 2018:6069150. https://doi.org/10.1155/2018/6069150
Jiao H, Zhang Y, Yan Z et al (2013) Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signaling and sepsis-induced lung inflammation. J Immunol 191(12):6191–6199. https://doi.org/10.4049/jimmunol.1300873
Janda E, Visalli V, Colica C et al (2011) The protective effect of tianeptine on Gp120-induced apoptosis in astroglial cells: role of GS and NOS, and NF-κB suppression. Br J Pharmacol 164(6):1590–1599. https://doi.org/10.1111/j.1476-5381.2010.01172.x
Milanesi S, Verzola D, Cappadona F et al (2019) Uric acid and angiotensin II additively promote inflammation and oxidative stress in human proximal tubule cells by activation of toll-like receptor 4. J Cell Physiol 234(7):10868–10876. https://doi.org/10.1002/jcp.27929
Zhang J, Diao B, Lin X et al (2019) TLR2 and TLR4 mediate an activation of adipose tissue renin–angiotensin system induced by uric acid. Biochimie 162:125–133. https://doi.org/10.1016/j.biochi.2019.04.013
Allaeys I, Marceau F, Poubelle PE (2013) NLRP3 promotes autophagy of urate crystals phagocytized by human osteoblasts. Arthritis Res Ther 15(6):R176. https://doi.org/10.1186/ar4365
Zhang DM, Li YC, Xu D, Ding XQ, Kong LD (2012) Protection of curcumin against fructose-induced hyperuricaemia and renal endothelial dysfunction involves NO-mediated JAK-STAT signalling in rats. Food Chem 134(4):2184–2193. https://doi.org/10.1186/ar4365
Eid RA, Alkhateeb MA, Eleawa S et al (2018) Cardioprotective effect of ghrelin against myocardial infarction-induced left ventricular injury via inhibition of SOCS3 and activation of JAK2/STAT3 signaling. Basic Res Cardiol 113(2):13. https://doi.org/10.1007/s00395-018-0671-4
Funding
The current study was funded by the Natural Science Foundation of Hebei Province (Grant Number H2018307071) and Hebei Province Medical Science Research Organization (Grant Number 20200731).
Author information
Authors and Affiliations
Contributions
QN carried out experiments, data analysis and manuscript writing. ML, ZZ and XZ performed experiments and prepared the figures. GS and CW designed and supervised the study. All authors read the manuscript and approved its publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Informed consent
The current report contains no human or animal studies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nie, Q., Liu, M., Zhang, Z. et al. The effects of hyperuricemia on endothelial cells are mediated via GLUT9 and the JAK2/STAT3 pathway. Mol Biol Rep 48, 8023–8032 (2021). https://doi.org/10.1007/s11033-021-06840-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06840-w