
Abstract
Zika virus is a mosquito-borne Flavivirus originally isolated from humans in 1952. Following its re-emergence in Brazil in 2015, an increase in the number of babies born with microcephaly to infected mothers was observed. Microcephaly is a neurodevelopmental disorder, characterised phenotypically by a smaller than average head size, and is usually developed in utero. The 2015 outbreak in the Americas led to the World Health Organisation declaring Zika a Public Health Emergency of International Concern. Since then, much research into the effects of Zika has been carried out. Studies have investigated the structure of the virus, its effects on and evasion of the immune response, cellular entry including target receptors, its transmission from infected mother to foetus and its cellular targets. This review discusses current knowledge and novel research into these areas, in hope of developing a further understanding of how exposure of pregnant women to the Zika virus can lead to impaired brain development of their foetus. Although no longer considered an epidemic in the Americas, the mechanism by which Zika acts is still not comprehensively and wholly understood, and this understanding will be crucial in developing effective vaccines and treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microcephaly
Microcephaly is a diagnosis given to neonates born with significantly smaller heads than expected. The accepted criteria for a diagnosis of microcephaly is a head circumference of three standard deviations or more below the mean for sex and gestational age of the healthy reference population [1]. Head circumference is used as a measurement of brain size, as microcephaly bears a strong correlation with mental retardation. Although the brain is smaller, it is architecturally normal, with reduced brain volume being due to the reduced size of the cerebral cortex [2].
Microcephaly can be primary or secondary; primary microcephaly is present at birth, usually a developmental anomaly due to disturbed neurogenesis or death of neuronal progenitors [3], whereas secondary microcephaly develops after birth, indicating a progressive neurodegenerative disorder. Primary microcephaly can have many non-genetic and genetic causes. For example, various congenital infections such as Toxoplasma gondii, Zika Virus (ZIKV), Cytomegalovirus, Herpes Simplex Virus and Rubella Virus show strong causal evidence to suggest that they result in primary microcephaly and mental retardation [3]. Other factors such as excess alcohol consumption during pregnancy and Rubenstein Taybi syndrome can also result in primary microcephaly [2]. These causes must be excluded before a diagnosis of autosomal recessive primary microcephaly (MCPH) can be reached.
Autosomal recessive primary microcephaly
In order to understand how ZIKV causes microcephaly in infected foetuses, and its viral targets, we will first investigate genes and molecules known to be associated with this condition. Genetic forms of primary microcephaly (collectively known as MicroCephaly Primary Hereditary (MCPH) are rare, genetically heterogeneous disorders which are inherited recessively and cause a large reduction in brain growth in utero [4].
Autosomal recessive primary microcephaly (MCPH) is the result of deficient neurogenesis within the neurogenic epithelium. It remains unclear as to why mutations in these genes only affect the brain, but it is thought to be due to the elongated morphology of neuronal cells and the precise nature in which the last stage of the cytokinesis cleavage furrow has to be executed. The furrow must bisect the cadherin hole (a small apical-membrane fate-determining domain) precisely to ensure that the division producing two daughter cell neural precursors is perfectly symmetrical [5].
Jackson et al. [6] and Roberts et al. [26] reported the initial defining clinical features of MCPH to be:
-
1.
Congenital microcephaly at least four standard deviations below the population mean for age and sex
-
2.
Mental retardation as the only neurological finding. Symptoms such as spasticity, seizures, or progressive cognitive decline rule out MCPH
-
3.
Normal height and weight, appearance, and results on chromosome analysis and brain scan
There have been eighteen genes reported to be associated with MCPH thus far, identified from different populations worldwide (Table 1).
Genes implicated in this condition have effects on varying pathways. The pathways involved include: DNA repair, transcription regulation, cell cycle progression including checkpoint regulation, binding of dynein, centrosome duplication, proliferative capacity of neural progenitor cells and interference with mitotic spindle formation. These pathways may be crucial in understanding ZIKV infection and subsequent microcephaly in foetuses. Recent proteomics studies exploring the ZIKV-host interactome have identified, amongst an extensive array of cellular targets, networks associated with signalling and mitotic pathways [27,28,29,30]. For example, Golubeva et al. identify shared interactions with 14 of the reported MCPH-related genes [30].
Globally, ASPM and WDR62 are the most commonly implicated genes, contributing to over 50% of gene mutations found in MCPH [31]. Most cases of MCPH are caused by point mutations within these genes involved in mitosis. For example, the ASPM gene encodes a spindle pole protein [32], WDR62 associates and interacts with the spindle assembly factor Aurora A to regulate mitotic progression and spindle formation [33], and STIL encodes a pericentriolar and centrosomal protein [14].
MCPH is inherited, and is most common in areas where consanguineous marriage is practiced. The incidence is around 1 in 10,000 in consanguineous populations, but this figure is lower in non-consanguineous populations [34], estimated to be 1–3 per 100,000 births in the Western world [35].
ASPM
The abnormal spindle-like microcephaly-associated (ASPM) gene is found at the MCPH5 locus, and encodes a nuclear mitotic spindle pole associated protein [32]. ASPM is the human orthologue of the abnormal spindle gene (asp) found in Drosophila melanogaster, and is essential in the function of normal mitotic spindles in embryonic neuroblasts [11, 36]. Mouse Aspm is expressed in the primary sites of prenatal cerebral cortical neurogenesis [11]. Asp is essential for the organisation and bundling of microtubules at the spindle poles during cell division, and is necessary for the correct development of the mitotic spindles [37]. During interphase, ASPM is localised to the centrosome. During mitosis, from prophase to telophase, ASPM is localised at the spindle poles.
ASPM has been established to contribute to the evolution of the brain both in humans and in other primates [38,39,40]. ASPM in mice encodes a protein associated with the centrosome [11] and the midbody [32, 41]. It is expressed during neurogenesis primarily in the ventricular zone of the neocortex [11, 26, 42] and is important in determining the position of the spindle poles in apical progenitor cells, allowing them to divide symmetrically during mitosis [42].
WDR62
Mutations in WDR62 (MCPH2) are the second most common cause of MCPH, after ASPM mutations, and it encodes a spindle pole protein [2]. The expression pattern of WDR62 during mitosis was found to be identical to that of ASPM in that WDR62 was concentrated at the spindle poles but was not present at the midbody during cytokinesis [5].
This awareness of identified microcephaly-related genes raises the question as to whether the expression of these MCPH genes is affected by ZIKV infection. Additionally, mutations in these genes are understood to cause mitotic defects, therefore the question arises as to whether this is a mechanism employed by ZIKV in causing microcephaly.
Zika virus
ZIKV is a positive-stranded RNA virus within the Flaviviridae family belonging to the same genus, Flavivirus, as Dengue and West Nile Viruses (DENV and WNV respectively) and is transmitted through Aedes spp. mosquitos. The virus was first isolated in the Zika forest in Entebbe, Uganda, in 1947, from a rhesus monkey. It was then isolated from mosquitos in 1948 and humans in 1952 [43]. Urban transmission of the virus is maintained primarily in an infection cycle between humans and predominantly A. aegypti mosquitoes in tropical/sub-tropical regions; however transmission has also been reported via A. albopictus, which has a wider geographical distribution, including more temperate regions.
Acute clinical symptoms of ZIKV infection in adults are similar to those of other mosquito transmitted arbovirus such as dengue fever and chikungunya, displaying as fever, arthralgia, myalgia, headache and maculopapular rash. Other symptoms, such as neurological manifestations and Guillain–Barré syndrome, were described during ZIKV outbreaks in French Polynesia and Brazil [44,45,46]. Despite their close phylogenetic relationship, ZIKV is the only member of the Flavivirus genus that is classified as a teratogen, or an agent that intervenes with the foetal development process [47].
Prior to 2015, ZIKV had been responsible for outbreaks and epidemics mostly in remote islands, including Yap Island and Micronesia in 2007, when an estimated 75% of the population of Yap Island became infected within a 4 month period [48]. There were also outbreaks in French Polynesia, New Caledonia, the Cook Islands and Easter Island in 2013 and 2014 [49, 50]. The virus was detected in the Americas, most notably in Brazil, in early 2015 [51].
The ZIKV genome is a single-stranded, positive-sense RNA molecule of ~ 10.8 kilobases (kb) in length (Fig. 1). The genome contains a 5′ untranslated region (5′UTR) of ~ 100 nucleotides with a a type-I 5′ cap, a single open reading frame of ~ 10 kb, and a 3′UTR of ~ 420 nucleotides [52]. Both the 5′ and 3′UTRs contain a number of RNA structure elements and primary sequence motifs, essential for stabilising alternative linear and cyclised genome conformations and initiation of negative-strand replication (reviewed for example in [53]). The open reading frame encodes a single polyprotein, which is co- and post-translationally cleaved into the capsid (C), the precursor membrane (prM) and the envelope protein (E), as well as seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) involved in replication, assembly, and antagonising the innate immune response of the host [54].

The Zika virus genome consists of 10.8 kb, coding for three structural proteins—capsid (C), precursor of membrane (prM) and envelope (E)—and seven non-structural proteins—NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5
The general life cycle of a virus is shown in Fig. 2. In brief, ZIKV enters the host-cell via receptor-mediated endocytosis [55], through candidate receptors AXL and Tyro3 [56,57,58,59]. Following endosome acidification and subsequent conformational changes to the E protein, ZIKV RNA is released into the host cell and is translated as a polyprotein—that is subsequently cleaved by viral and host-cell proteases, into the structural and non-structural proteins [54]. The non-structural proteins are essential for formation of the virus replication complex and subsequent ZIKV genome replication and translation. For examples, among other functions, non-structural protein 5 (NS5) has both viral RNA dependent RNA polymerase and methyltransferase activity, responsible for both replication and capping of the ZIKV genome respectively. The capsid protein packages genomic viral transcripts and along with the other structural proteins assemble on the surface of the ER as immature non-infectious virions. Subsequent maturation, during transportation through trans-Golgi apparatus, involves cleavage of prM by the host protease furin into the pr peptide and M protein before release of infectious virions from the cell by exocytosis [60, 61].

ZIKV replication cycle. Virus enters the cell through receptor-mediated endocytosis, RNA/protein synthesis is carried out, leading to genome replication and then assembly of the infectious virus particles, and finally the virus is released from the cell and goes on to infect a new cell
How does Zika cause microcephaly?
Primary microcephaly may be caused by depletion of the founder population of radial glia (neural stem cells found in the developing foetal brain), caused either by mass cell death or premature differentiation [62]. There have previously been cases reported of neurodevelopmental brain malformations, including microcephaly, associated with infections from various other viruses such as cytomegalovirus (CMV), rubella virus, West Nile Virus (WNV), Human Immunodeficiency Virus (HIV), herpes simplex virus and chikungunya [63,64,65,66,67,68]. These mechanisms are thought to bear similarities to those observed with ZIKV and this will be discussed further in this report.
Initial infection
ZIKV is primarily spread via Aedes spp. mosquitoes, predominantly A. aegypti but increasingly A. albopictus (Fig. 3). The virus is transmitted through the bite of an infected mosquito, but may also be transmitted sexually or through blood transfusions [69]. Within an urban transmission cycle, mosquitoes are commonly infected following a blood meal from a ZIKV viremic individual, however vertical transmission via infected A. aegypti eggs has also been observed [70].

Source some images from https://www.cdc.gov/ncbddd/birthdefects/images/microcephaly-comparison-500px.jpg
The ‘story of infection’. Simple representation of the events leading up to the development of foetal microcephaly.
It is suggested that only 20% of ZIKV infections result in a symptomatic phenotype [71]. However, when symptoms do develop following infection, they tend to be mild and self-limiting, and include fever, maculopapular rash, joint-pain, headache and conjunctivitis or red eyes [71]. These symptoms are non-specific and are similar to infections caused by other arboviruses such as dengue or chikungunya viruses, consequently diagnosis of ZIKV infection upon clinical presentation alone is almost impossible. More severe neurological symptoms such as Guillain–Barré syndrome have also been described but are comparatively rare [45, 69, 72].
Infection with ZIKV results in downregulation of various genes that are involved in cell cycle-related pathways, e.g. mitosis and cell cycle phase genes [73]. One of these cell cycle-related genes, CENPF [74], encodes a kinetochore protein. This protein is essential for the proper alignment of fully formed chromosomes during mitosis, as well as recruiting additional proteins that, collectively, play a part in spindle-pole formation. Primary microcephaly phenotypes have previously been shown to be affected by downregulation of genes involved in various stages of the cell cycle including centrosome maturation and spindle-pole formation [34]. This may suggest that ZIKV could interact with genes previously implicated in MCPH.
In addition to affecting genes involved in the cell cycle, ZIKV infection has been shown to significantly downregulate expression of genes associated with neural stem cells, neuronal cell types, and oligodendrocytes. As well as this downregulation, infection has been shown to upregulate certain genes, for example those associated with astrocyte differentiation as well as antiviral genes such as Toll-Like Receptor 3 (TLR3), Viperin and CCL2 [75]. This data suggests that, during infection, ZIKV targets several neural cell types, while also inducing differentiation of astrocytes and promoting TLR3 in order to initiate cellular disorder.
Evasion of immune response
Following virus infection, human cells initiate the production of small chemical messenger molecules known as cytokines. Critical cytokines are the initial response to viral infection, the first being interferons. The type I interferons α and β are produced by all nucleated cells in the body, particularly plasmacytoid dendritic cells. They inhibit viral replication by inducing an antiviral state through the expression of interferon-stimulated genes (ISGs) [76]. Examples of ISGs include double stranded (ds) RNA activated protein kinase (PKR), whose activation results in the inhibition of protein synthesis and therefore of new virus particles, and Oligoadenylate synthetase, whose activation by dsRNA results in the degradation of viral RNA. IFNs act as the initial response to infection, leading to the activation of natural killer cells and MHC Class I antigen presentation.
As with many viruses, Flaviruses have evolved various strategies in order to escape the immune response exhibited by the host cells, often involving the non-structural proteins encoded in their genomes. These mechanisms include inhibiting IFN, complement system, natural killer cells, and T and B cell responses [55]. For example, infection with dengue virus has been shown to inhibit expression of IFNα, even following stimulation by strong inducers of type I IFN [77].
ZIKV infection has been shown to be able to be restricted by several ISGs [78]. Studies have found that ZIKV NS5 protein inhibits type I IFN signalling by binding to and degrading STAT2, an essential transcription factor involved in type I IFN-mediated signalling [79, 80]. Although the NS5 protein appears to play a role in targeting type I IFN signalling, it is unknown if this interaction is cell-type dependent. More research needs to be done to establish the effect of the virus on STAT expression, phosphorylation and degradation in cell types that are known to be involved in the pathogenesis of the virus. Further to signalling regulation activity by the NS5 protein, the NS1 and NS4B proteins have been shown to inhibit RIG-I-like receptor signalling, supressing type I IFN induction [81].
These findings suggest that the non-structural proteins encoded by the ZIKV genome are used to evade the host immune system by inhibiting pathways that lead to the induction of type I IFN and ISGs. Clearly, the virus has evolved several mechanisms allowing the interference of type I IFN signalling, a crucial step permitting infection of the host mother, and hence infection of the foetus that leads to microcephaly.
As observed in other viruses, it is likely that ZIKV non-structural proteins are responsible for similar roles. For instance, the NS1 protein of dengue virus is thought to inhibit the innate antiviral immune response by activating TLR4 and interfering with TLR signalling [82, 83]. The NS1 protein of WNV has likewise been shown to inhibit TLR3-induced antiviral effects [84]. Furthermore, in a more distantly related member of the Flaviviridae family, Hepatitis C Virus (HCV) NS2 protein, a transmembrane polypeptide, has been implicated in playing a role in the inhibition of apoptosis [85], as well as in cell growth and interference with cell cycle regulation, all roles that may be beneficial for replication of the virus in host cells [86].
Likewise, NS2A, NS2B, NS4A, NS4B and NS5 proteins of dengue virus, NS3/4A, NS4B and NS5A proteins of HCV, and the NS4B protein of WNV have all been shown or suggested to be involved in immune evasion [87,88,89,90,91,92,93,94,95].
Cellular entry
Following transmission to the human host, ZIKV is likely to enter target cells through cellular receptors as with other Flaviviruses. However, this has not yet been studied in detail and the specific mechanism for ZIKV entry is not yet clear.
Flaviviruses are known to enter host cells through the process of endocytosis following the interaction of the E glycoprotein with host cell surface receptors and attachment factors [77, 96, 97] (Fig. 4). Differential expression of these molecules determines the cellular tropism of the virus [98].

Mechanism of viral entry, and subsequent actions within the cell, including proposed functions of non-structural proteins ultimately resulting in microcephaly
It is believed that Flaviviruses use at least two types of molecule in the processes of receptor recognition and attachment: attachment factors and primary receptors. Attachment factors are thought to concentrate/recruit virus particles to the surface of the cell, allowing faster entry, and primary receptors are used to bind virions, directing them to the endocytic pathway. Specifically, C-type (calcium dependant) lectin receptors, phosphatidylserine receptors, TIM (T-cell immunoglobulin and mucin domain) and TYRO3, AXL and MER (TAM) receptors have all been indicated to be involved in cellular entry of Flaviviruses [58].
Several studies have suggested glycosaminoglycans (GAGs), such as heparan-sulphate proteoglycans or syndecans, as being initial host-cell attachment factors for the Flaviviruses [99,100,101,102,103,104]. GAGs are long, unbranched, sulphated polysaccharides with a repeating disaccharide unit. They are found linked to core proteins which are attached to cellular surfaces (proteoglycans) and are primarily exposed on the surface of cells in all tissues [105].
Due to their position on the cell surface, GAGs are an ideal candidate, easily accessible for viral attachment. The electrostatic interaction caused by the difference in charge between the positive residues of the E glycoprotein and the negative sulphate groups exposed on the polysaccharide appears to be the primary interaction allowing the attachment of the virus particle [99]. In this instance, the main role of GAGs is as attachment factors allowing the concentration of the Flavivirus particles at the surface of target cells, facilitating their interaction with primary entry receptors such as, in the case of ZIKV, the candidate receptor AXL [99,100,101,102,103]. It is likely that there are various cellular attachment factors and receptors that allow the concentration and entry of these viruses into cells, and that these receptors can function independently of each other, meaning that even by blocking one receptor the virus may still manage to enter the host cells via an alternative mechanism.
In their study on the biology of the ZIKV, Hamel et al. [98] showed that virus entry is mediated by several cellular factors such as DC-SIGN, AXL, Tyro3 and TIM-1. It seemed that some of these factors may not contribute to the infection itself, but rather that they co-operate with other factors/receptors to cause an additive effect. For example, TIM-1 made little contribution to the entry of the virus, but its expression increased the efficacy of viral entry mediated by the AXL receptor. This suggests that TIM-1 may act as an attachment factor, binding viral particles and transferring them to the AXL receptor, allowing internalisation of the virus. In this case, the TIM-1 receptor would concentrate virions on the cell surface, hence easing their interaction with the AXL receptor and facilitating viral infection. However, the exact role of the TIM and TAM receptors in the infection of ZIKV is not well understood and requires additional research.
AXL belongs to the Tyro AXL MER (TAM) family of receptors. The TAM family are a group of tyrosine kinase receptors, known to be involved in the processes of clearing apoptotic cells and regulating the innate immune response [106]. AXL mRNA has been found to be present in human neural progenitor cells (NPCs), the cells which differentiate to produce the neurons and glia of the nervous system during embryonic development, as well as other brain cells such as radial glial cells, microglial cells and astrocytes [57]. Poorly susceptible cells have been shown to demonstrate sensitivity to ZIKV infection following the expression of various known Flavivirus entry factors, including AXL [98]. The same study also found that, by blocking or silencing the AXL receptor, in vitro infectivity is reduced by up to 90% in cultured fibroblasts and alveolar epithelial cells. These results suggest that the AXL receptor is a main contributor in the cellular entry and infection process of ZIKV.
It has been suggested that ZIKV targets in vitro-derived progenitor cells as opposed to immature neurons [73]. This has been supported by Nowakowski et al. [57], who reported that the AXL receptor is highly expressed in radial glia, with 78/96 radial glia displaying expression greater than 6 log2 normalised read counts. These results are in contrast with the limited expression shown by TYRO3 (5/96 radial glia). Radial glia are the neural stem cells of the foetal cerebral cortex in humans. A large abundance of the AXL receptor in these cells may provide a suggestion as to why these cells appear to be particularly susceptible to infection by ZIKV. The disruption of radial glia may explain how ZIKV causes microcephaly—by interrupting the growth of the cerebral cortex in the foetus.
In addition to its role as a viral entry factor, AXL signalling has been shown to suppress the innate immune response [107]. The receptor’s kinase domain has been seen to enhance infectivity of dengue virus following cell entry [108]. ZIKV may similarly activate AXL signalling upon binding to the AXL receptor, suppressing the innate immune response and therefore enhancing the ability of the virus to establish a persisting infection in the host and prevent viral clearance [109]. It has been suggested that the interaction between the virus and the AXL receptor on human astrocytes and the subsequent subversion of the host immune response is an important contributing factor leading to pathogenesis [56].
However, other studies indicate that the AXL receptor may not be the main entry receptor used by this virus. A study by Hastings et al. [110] concluded that TAM receptors were not required for cell entry and viral infection in mice. This study performed comparative infection studies in wild type and in TAM receptor knockout mice. The study found that TAM receptors were not required for the entry of ZIKV through trans-placental, vaginal, subcutaneous, or intracranial routes of infection. They also suggested that cellular targets in the brain and placenta are similar, regardless of whether TAM receptors are present or not. This indicates that, in mice, ZIKV infection is not dependant on the presence of TAM receptors. However, it is worth noting that these results may suggest that there are several viral entry receptors facilitating infection of cells by ZIKV. As discussed previously, C-lectins, phosphatidylserine receptors and TIM-1 receptors could all be implicated in virus cellular entry, which may be why the virus is still able to infect target cells in the absence of TAM (AXL) receptors.
Initial cellular targets
Given the fact that ZIKV is transferred to humans mainly through the blood-feeding process of mosquitos, it is understandable to think that initial target cells for the virus are likely to be localised to the skin layers, i.e. the dermis and the epidermis, where the virus is deposited. Skin fibroblasts have previously been recognised as being highly permissive targets for arboviruses [111]. ZIKV has been shown to infect numerous cell types, including cells of the dermis and epidermis, dendritic cells, placental macrophages, and neural progenitor cells [57, 73, 98, 112].
In a study by Hamel et al. [98], active ZIKV replication was demonstrated in infected skin fibroblasts, found in the dermis layer. Infection of these cells resulted in the presence of high RNA copy numbers and an increase in the number of ZIKV particles produced over time. This study also found that, upon ZIKV infection, keratinocytes, cells found in the epidermis, showed the presence of cytoplasmic vacuolation and pyknotic nuclei in the stratum granulosum—indicators of cells which are undergoing apoptosis. In addition to dermal fibroblasts and epidermal keratinocytes, it was also shown that dendritic cells, another cell type present in skin, are permissive to ZIKV infection. Induction of cell death in these skin cells could be a mechanism which allows the virus to divert the initial antiviral immune response, allowing easier entry of the virus into the host and, in the context of this report, ultimately leading to entry into the foetal brain. Interestingly, replication of ZIKV in human skin fibroblasts was shown to be enhanced by cellular autophagy.
Some studies have reported that the early cellular targets of the virus appear to be astrocytes [75, 113]. Astrocytes are some of the first responders to viral infections. They are characteristically star-shaped glial cells which are found in the brain and spinal cord, an important component of the central nervous system. Astrocytes perform an assortment of tasks, including axon guidance, synaptic support and control of the blood brain barrier. They have roles in maintaining synapse homeostasis, regulating neuronal signalling, determining the outcome of endogenous neural precursors, and protecting neurons from the damage of oxidation [114].
The significance of astrocytes being an early target for the virus may be due to their interaction with the vascular system. This would allow the virus to reach the target organ more rapidly. This was demonstrated in a study by Kozak et al. [115] during which the virus was detected in several brain regions as early as 4 days following infection. This study found that miRNAs and mRNAs are dysregulated in astrocytes infected with the virus. Globally, miRNAs were downregulated. However, the small number of miRNAs that were upregulated appeared to be correlated with host pathways associated with infection of the virus. This would suggest that the regulation of astrocyte miRNA is involved in allowing the virus to infect the human host, and may be responsible for the subversion of the host immune response.
Vertical transmission: crossing the placental barrier
Zika virus RNA has been detected in placental and neural tissue in infected humans, as well as in amniotic fluid and brain tissues from new-borns who had been diagnosed with foetal microcephaly [116,117,118,119]. These findings suggest that the mechanism by which ZIKV infects the foetus is by disrupting and passing through the placental barrier during vertical viral transmission. Subsequently, various reports have established that ZIKV has the ability to cross the human foetal-placental barrier in order to infect the developing brain and nervous system [109, 118, 119].
Few viruses are known to cross the placental barrier. One such virus is cytomegalovirus (CMV), a dsDNA virus belonging to the Herpesvirida family. Although unrelated to ZIKV, infection with CMV causes similar neurodevelopmental brain abnormalities [67, 120]. The neuroinvasiveness of CMV is mediated by various entry factors, such as EGFR and integrins, which are particularly highly expressed in radial glia and neural stem/progenitor cells [121]. Higher expression of these entry proteins, particularly of β1 integrin in this case, determines the initial susceptible cell population in the brain. It is therefore plausible to theorise that the Zika virus infects cells with high expression of the entry receptor AXL, such as neural progenitor cells.
Rubella is another virus that is able to cross the placental barrier, with infection of the placenta occurring during maternal viraemia. This infection results in areas of necrosis that are focally distributed within the epithelium of chorionic villae, as well as in the endothelial cells of its capillaries [96].
However, this behaviour has not previously been associated with Flaviviruses. For example, congenital acquisition of West Nile Virus has previously been investigated, but most infants were born at term and without complications that could be attributed to WNV infection, suggesting that the virus was unable to cross the placental barrier [65, 122]. Furthermore, there are an estimated 390 million cases of Dengue infection per year [123], but the virus has not been shown to be able to cross the placental barrier and there have been no reported cases of associated congenital microcephaly.
There have been various proposed mechanisms by which ZIKV can enter the foetal compartment in order to gain access to foetal target cells. Firstly, it was proposed that the virus would be carried by viraemia from the maternal blood stream into the maternal blood space between the developing placenta and the decidua, or the uterine lining which forms the maternal part of the placenta [124].
Primary placental cells, such as amniotic epithelial cells that proliferate in the amniotic membranes, were shown to be susceptible to infection with ZIKV. The virus was seen to grow well in these cells, particularly in those from mid-gestation compared to those from late-gestation [124]. This suggests that infection of a pregnant woman during the mid-gestational phase, while the foetus is growing, would allow the transmission of the virus to the foetus through the close contact of the parietal decidua with the anmio-chorionic membranes. The maternal blood may infect the trophoblasts in these membranes, allowing the virus to cross into the foetal compartment and hence directly infect the growing foetus. However, it has been suggested that primary human placental trophoblast cells from full-term placentas are in fact refractory to infection with ZIKV due to Type III Interferon production [125]. Therefore, this route would require further investigation including control of gestational age to investigate whether these cells are always resistant to infection or if resistance is developed with gestational age of the foetus. This hypothesis is supported in a study by Bayer et al. [125] which suggested that the less differentiated trophoblasts present in the first trimester of pregnancy, along with extra-villous trophoblasts, are more permissive to infection when compared to villous trophoblasts present during late stages of pregnancy.
Many studies have been conducted investigating the cells infected by ZIKV during its crossing of the placental barrier. It has been shown that ZIKV has the ability to infect human placental macrophages, also known as Hofbauer cells [112, 117, 125]. Hofbauer cells are a heterogeneous, foetal population of macrophages residing in the chorionic villus, a functional unit of the placenta. These cells are associated with a wide range of functions that are crucial in a successful pregnancy, such as immune regulation, placental morphogenesis, transfer of ions and serum proteins across the maternal-foetal barrier, and control of stromal water content. Imbalances in the homeostasis of Hofbauer cells are associated with placental pathologies that involve infections, inflammation, and inadequate development of the placenta [126].
Bhatnager et al. [117] indicated that Hofbauer cells, which can reach foetal blood vessels, may be responsible for the dissemination of ZIKV into the foetal brain, particularly in early pregnancy as this is when Hofbauer cells are most abundant in the placenta. Bayer et al. [125] demonstrated that ZIKV infects and primarily replicates within Hofbauer cells in vitro. Interestingly, Dengue virus has also been shown to target macrophages to aid in its dissemination [127]. These findings suggest that Hofbauer cells may be significant targets for ZIKV, facilitating transfer or dissemination into the foetal brain, as demonstrated in Fig. 5.

ZIKV crosses the placental barrier, infecting Hofbauer cells which then allow the virus to transfer to the foetus through foetal blood vessels, particularly in early pregnancy
In addition to the infection of these placental macrophages, it has been shown that ZIKV is able to replicate in anchoring villi and basal decidua from first-trimester placentas. This would allow prolonged viraemia during the earlier stages of development, and would lead to trans-placental transmission [124].
Infection of foetal cells
Nowakowski et al. [57] proposed that, after breaching the foetal-placental barrier, the virus reaches the brain of the developing foetus by haematogenous spread, or via the cerebrospinal fluid. Once at the target site, it is hypothesised that radial glia cells are the first target cell population with the highest AXL expression. Zika would then enter these cells, either through their contact with blood vessels, or via their direct contact with the cerebrospinal fluid through their apical end-feet.
Neural progenitor cells and radial glia have been widely accepted in recent literature as being the major cellular target of ZIKV in the developing foetal brain. NPCs are similar to stem cells which differentiate to produce the neurons and glia of the nervous system, critical during brain development of foetuses.
Radial glia span the width of the cortex in the developing central nervous system, serving as primary progenitor cells that are capable of differentiating into neurons, astrocytes, and oligodendrocytes [128]. As the virus targets these cortex spanning cells, this may explain why infected foetuses display the phenotype known as microcephaly, whereby the brain is smaller than usual (i.e. three or more standard deviations below the mean).
Research has suggested that ZIKV can replicate in both mouse astroglial cells and in neurons [116]. The viral RNA has been detected in human placental and neural tissue [118, 119]. It has also been suggested that the virus is able to infect human neural progenitors as well as mature neurons, and increase expression of apoptotic markers. Infection has additionally been shown to impair growth of human neural stem cells and brain organoids [73, 129].
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase, a component of two protein complexes that are functionally distinct from each other, known as mTORC1 and mTORC2. Each complex responds to varied environmental stimuli and has different regulatory properties and cellular activities [130].
The Akt-mTOR signalling pathway plays a crucial role in regulating various processes in NPCs, including cellular development, proliferation, survival and inhibition of autophagy [131]. This pathway has been shown to be crucial in establishing proper neuronal architecture, and in regulating neural specification and function [132]. A study by Liang et al. [133] suggested that the NS4A and NS4B proteins encoded in the ZIKV genome cooperate in order to inhibit the activation of Akt, thus inhibiting downstream signalling via this pathway in NPCs. Preliminary studies have indicated that the NS4A and NS4B protein complex is able to inhibit PI3K (phosphatidylinositide 3 kinase), the upstream activator of Akt, thereby terminating the Akt-mTOR signalling cascade [134]; however, this phenomenon is not well understood and requires further investigation. Likewise, a correlation between Dengue virus (DENV) infection and decreased activity of mTORC1 has been found [135]. This was inversely correlated to lipophagy induction, which was in turn said to promote viral replication. These studies suggest that Flaviviruses are able to disrupt the mTOR pathway in infected cells, with the result (i.e. inhibition of neurogenesis or induction of autophagy or lipophagy) depending on the target cell type of the individual virus.
In contrast, a related Flavivirus, West Nile Virus (WNV), has been seen to upregulate the mTOR pathway through a PI3K-dependent mechanism. Shives et al. [136] demonstrated that infection of serum-starved mammalian cells with WNV leads to activation of mTOR Complex 1, which is involved in governing diverse anabolic and catabolic processes including protein synthesis and autophagy. Activating this complex allows the support of viral growth and protein expression within the host cell. This mechanism was proven to be crucial for viral replication, evidenced by a reduction in WNV growth following pharmacological inhibition of PI3K. Shives et al. concluded that TORC1 supports protein synthesis of Flavivruses and subsequently supports the growth of WNV. Blocking of apoptosis by a PI3K-dependant mechanism has similarly been observed following viral entry of DENV-2 and JEV, but pharmacological blocking of PI3K had no effect on their viral replication [137].
It is evident that the mTOR signalling pathway is important in Flavivirus pathogenicity, with roles in controlling apoptosis, induction of autophagy and viral replication, but it is unknown why some Flaviviruses upregulate the pathway whereas others, namely ZIKV, downregulate the pathway.
A key factor which separates ZIKV from other related Flaviviruses is its tissue and cell tropism. ZIKV preferentially infects neural progenitor cells of the embryonic brain, resulting in their apoptotic cell death, with the ability to also infect mature neurons to a lesser extent [73, 138, 139]. Conversely, related Flaviviruses such as WNV and JEV, which are responsible in causing encephalitis post-natally, primarily target mature neurons in infected adults [140,141,142]. Interestingly, a study by Brault et al. [143] investigated the cell tropism for ZIKV, WNV and DENV-4 (Dengue Virus Serotype 4) in mouse developing neocortex and found that this tissue supported viral replication of ZIKV and WNV but not of DENV-4. It was further shown that only infection with ZIKV was able to halt cell cycle progression of neural stem cells, whereas WNV infection did not. This would indicate that the ability of ZIKV in causing microcephaly is largely due to its infection of NPCs and subsequent impairing of cell cycle progression, followed by the induction of apoptosis of these cells [138], hence reducing the pool of cortical neural progenitors and consequently reducing the size of the foetal cortex and brain.
Infection of various cells of the brain has been supported in many studies. Van den Pol et al. [113] showed that, following peripheral inoculation of developing new-born normal mice, neural glial cells were substantially infected as an initial target. This was particularly seen in cells with a morphology and GFAP (glial fibrillary acidic protein) antigen expression aligning with that of astrocytes. Infected isolated astrocytes were found throughout the brain, suggesting a widespread occurrence of infected astrocytes and hence many penetrations of ZIKV into the developing brain. This could indicate that not only do astrocytes show the first signs of infection, but they may also be used as a mechanism of further amplifying and distributing infectious viral particles to neighbouring neurons and glia. Interestingly, CMV has also been shown to have a tendency to target astrocytes [144].
In addition to targeting of astrocytes, recent work has also explored the targeting of human neural progenitor cells (hNPCs), the cells which differentiate to produce the neurons and glia of the nervous system during embryonic development, and why these cells appear to be exceptionally vulnerable to infection with the virus.
It has been widely shown that ZIKV is lethal to human neural stem/progenitor cells [57, 73, 129, 145]. These cells express the transcription factor Sox2, a critical factor for initiating neural induction and maintaining the properties displayed by neural progenitor stem cells throughout their differentiation [146]. One study suggested that ZIKV may cause microcephaly through the depletion of the founding population of NPCs expressing Sox2, which, as previously discussed, are highly susceptible to ZIKV infection [147].
Tang et al. [73] showed that human NPCs are highly susceptible to ZIKV infection in vitro, with the infection reaching 65–90% of the cells within 3 days of inoculation. Other cells such as human embryonic stem cells and human induced pluripotent stem cells were able to be infected, but the infection did not spread further than the cells at the colony edge. This establishes that hNPCs, a population of cells that contribute to the developing embryonic brain, act as a direct target for ZIKV. The study concluded that infection of hNPCs with ZIKV leads to attenuated population growth. This was said to be due to caspase 3-mediated apoptosis and dysregulation of the cell cycle, induced by viral infection. Hanners et al. [139] also suggested an apoptotic mode of action as, during their study, they noted the presence of pyknotic nuclei in infected hNPCs, typically coupled with ZIKV antigen and activated caspase 3. In addition, this study found that ZIKV persistently replicates in hNPCs, with one cell line continuing to produce virus particle for up to 28 days post infection. This persistent virus production is supported in other studies; for example, Mlakar et al. [109] observed ZIKV particles present in the brain tissue of foetuses 19 weeks after presumed maternal infection.
In vitro, infection of NPCs with ZIKV leads to impaired proliferation of NPCs and apoptotic cell death [147]. In mouse models, it has been shown that the virus targets NPCs, altering cell division and inducing cell death, and reducing the pool of cortical neural progenitors [74, 148]. This leads to brain developmental defects, including microcephaly, in infected mice.
Further research indicating NPCs as viral targets was carried out during the investigation of neurodevelopmental RNA-binding protein Musashi-1 (MSI1). MSI1 was shown to promote UTR-driven translation of the ZIKV genome in vitro, showing that the protein interacts with the genome of the virus in order to aid or enable its replication and therefore it promotes the viral life cycle [149]. Unsurprisingly, MSI1 was found to be highly expressed in neural progenitors found in the embryonic brain of human foetuses, while being absent in mature neurons. Furthermore, this protein has been shown to be mutated in individuals suffering from recessive primary microcephaly. This suggests that MSI1 is a key protein in both the infection period of ZIKV, as well as in the development of foetal microcephaly.
Zika-encoded NS2A has been found to disrupt mammalian cortical neurogenesis and decrease cell proliferation by degrading the adherens junction proteins that form the adherens junction complex [150]. Also, the capsid protein has been shown to interact with mouse-double-minute-2 (MDM2) homolog, a protein involved in the regulation of p53. This lead to the activation of the p53-mediated apoptosis pathway and hence the death of infected neural cells [151]. Interestingly, the same study found that synthetic mimics of the ZIKV capsid protein were able to induce cell death both in vitro and in vivo. This provides an important insight into the role of the virally encoded proteins in infection and causation of the microcephalic phenotype.
It has been shown that ZIKV persistently infects various trigeminal and dorsal root sensory neurons. Conversely, autonomic sympathetic and parasympathetic neurons were shown to be non-permissive to infection [152]. This is supported by the finding that cells of the peripheral nervous system are much less susceptible to infection than cells of the central nervous system (CNS), with all major CNS cell types being susceptible to infection, particularly oligodendrocytes [153].
This evidence amounts to a consensus that the main target of the Zika virus once it has reached the foetus in utero is neural progenitor/stem cells. The fact that these cells have high expression levels of the proposed entry receptor, AXL, may be critical to the tissue specificity of the viral target. The degeneration of this target cell population via the various proposed mechanisms may explain the fact that the patients with microcephaly have brains three or more standard deviations below the normal size. It is already known that the size of the cerebral cortex is determined by the rate at which neurons and glial cells are produced [154]. It would therefore be plausible to assume that microcephaly in these infected foetuses is caused by cell death and dysregulation in neural progenitor and glial cells. However, questions still remain concerning why not all foetuses develop microcephaly, the exact mechanism behind the susceptibility of neural progenitor cells, the exact receptors involved in viral entry and the role of the immune response.
Discussion
In this report we have provided an overview of our emerging insight into the link between Zika virus infection of pregnant women and the development of microcephaly in foetuses.
The virus infects the adult human host predominantly through the bite of an infected Aedes mosquito. In most cases, infected adults are asymptomatic, but some infected individuals may present with fever, maculopapular rash, joint pain, headache or conjunctivitis, or, in severe cases, may develop Guillain–Barré syndrome.
The virus must overcome the immune response of the host in order to replicate and sustain infection within host cells, before crossing the placental barrier and moving to the target site of the foetal brain. It appears as though ZIKV has evolved various mechanisms to do this.
Firstly, it has been shown that the virus is able to upregulate and downregulate various genes, mostly those involved in the cell cycle, differentiation (for example astrocyte differentiation), apoptosis and antiviral pathways. Most of the genes affected by the virus are expressed predominantly in neural cell types such as neural progenitor cells and oligodendrocytes. Interestingly, some genes with altered expression as a result of ZIKV infection are also implicated in MCPH. This gives rise to the question as to whether the virus is able to interact with genes that are already known to be associated with the congenital microcephaly phenotype.
Mechanisms of evasion of the host immune response are largely encoded within the ZIKV genome in the form of the non-structural proteins produced following cleavage of the polyprotein. Currently, the NS5 protein has been shown to inhibit type I interferon signalling by degrading the transcription factor STAT2, and NS1 and NS4B have been shown to inhibit RIG-I like receptor signalling, suppressing type I IFN induction. It is likely that other non-structural proteins encoded in the genome also have further roles to play in the immune evasion strategies of the virus, as with related viruses such as Dengue, HCV and WNV, however, these have not yet been firmly established.
In order to reach the cellular targets in the foetal brain (Fig. 6), the virus must survive and replicate within initial cellular targets such as skin cells, and ZIKV has been shown to infect cells of the dermis and epidermis, e.g. skin fibroblasts.

Source figure adapted from [155]
Summary of mechanisms causing impaired brain development and a microcephalic phenotype following infection with Zika virus. ZIKV enters through skin cells, eventually reaching the target population of neural progenitor cells with varying effects.
There have been various receptors proposed to be the entry receptors for ZIKV. The main candidate receptors are AXL and Tyro3. It is thought that the virus attaches to factors (such as polysaccharides) on the cell surface via its envelope (E) protein. This allows the virus particles to be concentrated at the cell surface, thus facilitating their interaction with entry receptors such as AXL. Although the evidence in support of AXL being the major entry receptor is abundant, one study conducted in mice did find that the presence or absence of TAM receptors did not seem to impact on viral infection. However, this study may just indicate that AXL is not the sole receptor responsible for cell entry of the virus, and that other receptors may also carry out this function. At this point, it is still thought that AXL is the main receptor implicated in virus entry, especially due to the high expression levels in cells that are highly targeted by the virus such as neural progenitor cells.
Following initial infection, it is likely that the virus reaches the placenta through viraemia. When at the placental barrier, the virus must cross in order to infect the foetus and be detected in foetal tissue and amniotic fluid, though the exact mechanism has not yet been established. It has, however, been shown that the virus is able to infect Hofbauer cells (placental macrophages) and amniotic epithelial cells. Transmigrating ZIKV-infected monocytes may also act as “Trojan horses” for disseminating the virus and enabling its penetration through endothelial barriers [156].
Once across the placental barrier, the virus then reaches its target population, neural progenitor cells. Here, the virus disrupts these and related cells, such as radial glia. This may be through inducing autophagy and apoptosis, disrupting cellular development or disrupting differentiation and proliferation. Neural progenitor cells have been shown to be highly susceptible to ZIKV infection. These cells are those which differentiate to produce cortical neurons. The fewer neural progenitor cells available, the fewer cortical neurons produced and therefore the smaller the cerebral cortex and the smaller the brain, resulting in microcephaly.
Conclusions
Following the Public Health Emergency of International Concern declaration by WHO regarding ZIKV in February 2016, much progress has been made in terms of understanding the mechanism by which the virus infects human cells and leads to hampered brain development and microcephaly in foetuses. Evidence from varying sources investigating multiple model systems all consistently point to the conclusion that: vertical transmission of ZIKV from pregnant females to their unborn foetuses via the trans-placental route targets neural progenitor cells, and other neuronal cells such as radial glia, in the developing brain, causing death, apoptosis and decreased proliferation of these cells, and resulting in impaired brain development and therefore a microcephalic phenotype.
Unanswered questions
Despite the advancements made already in understanding the impact of ZIKV infection, there are still many questions to be answered before its mechanisms are fully understood, allowing the possibility to create an effective vaccine or treatment for infection.
Firstly, the question remains as to why only a small number of foetuses born of infected mothers display microcephaly. Why aren’t all foetuses affected? Is infection strain-specific or perhaps specific to different human populations? Are there mutations within the ZIKV genome that are required in order to cause microcephaly? Or is this to do with the mother’s own immune system and therefore the ability of the virus to cross the placental barrier? Are there geographical factors, such as previous infection with other viruses such as dengue, that have a part to play in the likelihood of ZIKV infection? How long does the virus persist in the host? If a woman becomes pregnant some years following initial infection, is the foetus still at risk of developing microcephaly? Questions of epidemiology are difficult to answer, and some would require a global retrospective study to provide any solutions. Studies comparing various strains of the virus and their effects on various cells derived from different human populations may aid in answering some of these questions, determining specific molecular and cellular vulnerability.
Secondly, is it possible to pinpoint infection to a specific viral protein or Zika-derived small RNA? How would these individual components elicit specified effects on specific cellular properties? Studies investigating these specific interactions would aid in understanding the pathogenesis of ZIKV infection and in developing therapeutics to target this interaction.
Thirdly, why are neural progenitor cells so susceptible to infection with the virus? What factors and pathways mediate this entry and infection? Are NPCs particularly susceptible due to their direct interaction with the cerebrospinal fluid, or specifically due to their receptor profile and high expression of ZIKV entry receptors? Finding the specific interaction that allows the virus to infect cells, particularly NPCs, may be essential in developing therapeutics for ZIKV infection.
Finally, what role does the host immune system play in the infection with ZIKV? Does the virus manipulate immune cells in order to facilitate its destructive path through the body? What role do glia and microglia play in brain development deficits exhibited during ZIKV infection?
It is clear that there is a relationship between Zika virus infection and microcephaly, but more research needs to be done to establish the facts and therefore provide methods of controlling or limiting disease caused by ZIKV.
References
WHO (2016) Screening, assessment and management of neonates and infants with complications associated with Zika virus exposure in utero rapid advice guideline. World Health Organization, Geneva
Woods CG, Bond J, Enard W (2005) Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet 76(5):717–728. https://doi.org/10.1086/429930
Devakumar D, Bamford A, Ferreira MU, Broad J, Rosch RE, Groce N, Breuer J, Cardoso MA, Copp AJ, Alexandre P, Rodrigues LC, Abubakar I (2018) Infectious causes of microcephaly: epidemiology, pathogenesis, diagnosis, and management. Lancet Infect Dis 18(1):e1–e13. https://doi.org/10.1016/S1473-3099(17)30398-5
Cox J, Jackson AP, Bond J, Woods CG (2006) What primary microcephaly can tell us about brain growth. Trends Mol Med 12(8):358–366. https://doi.org/10.1016/j.molmed.2006.06.006
Nicholas AK, Khurshid M, Desir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A, Passemard S, Misson JP, Lindsay S, Gergely F, Dobyns WB, Roberts E, Abramowicz M, Woods CG (2010) WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet 42(11):1010–1014. https://doi.org/10.1038/ng.682
Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, Karbani G, Jafri H, Rashid Y, Mueller RF, Markham AF, Woods CG (2002) Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet 71(1):136–142. https://doi.org/10.1086/341283
Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, Mannan J, Karbani G, Corry P, Levene MI, Mueller RF, Markham AF, Lench NJ, Woods CG (1998) Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet 63(2):541–546
Kousar R, Hassan MJ, Khan B, Basit S, Mahmood S, Mir A, Ahmad W, Ansar M (2011) Mutations in WDR62 gene in Pakistani families with autosomal recessive primary microcephaly. BMC Neurol 11:119. https://doi.org/10.1186/1471-2377-11-119
Moynihan L, Jackson AP, Roberts E, Karbani G, Lewis I, Corry P, Turner G, Mueller RF, Lench NJ, Woods CG (2000) A third novel locus for primary autosomal recessive microcephaly maps to chromosome 9q34. Am J Hum Genet 66(2):724–727. https://doi.org/10.1086/302777
Jamieson CR, Govaerts C, Abramowicz MJ (1999) Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15. Am J Hum Genet 65(5):1465–1469. https://doi.org/10.1086/302640
Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG (2002) ASPM is a major determinant of cerebral cortical size. Nat Genet 32(2):316–320. https://doi.org/10.1038/ng995
Jamieson CR, Fryns JP, Jacobs J, Matthijs G, Abramowicz MJ (2000) Primary autosomal recessive microcephaly: MCPH5 maps to 1q25-q32. Am J Hum Genet 67(6):1575–1577. https://doi.org/10.1086/316909
Leal GF, Roberts E, Silva EO, Costa SM, Hampshire DJ, Woods CG (2003) A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2. J Med Genet 40(7):540–542
Kumar A, Girimaji SC, Duvvari MR, Blanton SH (2009) Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet 84(2):286–290. https://doi.org/10.1016/j.ajhg.2009.01.017
Hussain MS, Baig SM, Neumann S, Nurnberg G, Farooq M, Ahmad I, Alef T, Hennies HC, Technau M, Altmuller J, Frommolt P, Thiele H, Noegel AA, Nurnberg P (2012) A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am J Hum Genet 90(5):871–878. https://doi.org/10.1016/j.ajhg.2012.03.016
Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau-Sturk T, Beis J, Dumas N, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Patry L, Rideout AL, Thomas A, Orr A, Hoffmann I, Michaud JL, Awadalla P, Meek DC, Ludman M, Samuels ME (2010) Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet 87(1):40–51. https://doi.org/10.1016/j.ajhg.2010.06.003
Brown NJ, Marjanovic M, Luders J, Stracker TH, Costanzo V (2013) Cep63 and cep152 cooperate to ensure centriole duplication. PLoS ONE 8(7):e69986. https://doi.org/10.1371/journal.pone.0069986
Awad S, Al-Dosari MS, Al-Yacoub N, Colak D, Salih MA, Alkuraya FS, Poizat C (2013) Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum Mol Genet 22(11):2200–2213. https://doi.org/10.1093/hmg/ddt072
Hussain MS, Baig SM, Neumann S, Peche VS, Szczepanski S, Nurnberg G, Tariq M, Jameel M, Khan TN, Fatima A, Malik NA, Ahmad I, Altmuller J, Frommolt P, Thiele H, Hohne W, Yigit G, Wollnik B, Neubauer BA, Nurnberg P, Noegel AA (2013) CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum Mol Genet 22(25):5199–5214. https://doi.org/10.1093/hmg/ddt374
Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, Tatarakis A, Mahajan MA, Samuels HH, Shi Y, Golden JA, Mahajnah M, Shenhav R, Walsh CA (2012) Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151(5):1097–1112. https://doi.org/10.1016/j.cell.2012.10.043
Basit S, Al-Harbi KM, Alhijji SA, Albalawi AM, Alharby E, Eldardear A, Samman MI (2016) CIT, a gene involved in neurogenic cytokinesis, is mutated in human primary microcephaly. Hum Genet 135(10):1199–1207. https://doi.org/10.1007/s00439-016-1724-0
Alakbarzade V, Hameed A, Quek DQ, Chioza BA, Baple EL, Cazenave-Gassiot A, Nguyen LN, Wenk MR, Ahmad AQ, Sreekantan-Nair A, Weedon MN, Rich P, Patton MA, Warner TT, Silver DL, Crosby AH (2015) A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat Genet 47(7):814–817. https://doi.org/10.1038/ng.3313
Khan MA, Rupp VM, Orpinell M, Hussain MS, Altmuller J, Steinmetz MO, Enzinger C, Thiele H, Hohne W, Nurnberg G, Baig SM, Ansar M, Nurnberg P, Vincent JB, Speicher MR, Gonczy P, Windpassinger C (2014) A missense mutation in the PISA domain of HsSAS-6 causes autosomal recessive primary microcephaly in a large consanguineous Pakistani family. Hum Mol Genet 23(22):5940–5949. https://doi.org/10.1093/hmg/ddu318
Morris-Rosendahl DJ, Kaindl AM (2015) What next-generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol Cell Probes 29(5):271–281. https://doi.org/10.1016/j.mcp.2015.05.015
Kadir R, Harel T, Markus B, Perez Y, Bakhrat A, Cohen I, Volodarsky M, Feintsein-Linial M, Chervinski E, Zlotogora J, Sivan S, Birnbaum RY, Abdu U, Shalev S, Birk OS (2016) ALFY-controlled DVL3 autophagy regulates Wnt signaling determining human brain size. PLoS Genet 12(3):e1005919. https://doi.org/10.1371/journal.pgen.1005919
Roberts E, Hampshire DJ, Pattison L, Springell K, Jafri H, Corry P, Mannon J, Rashid Y, Crow Y, Bond J, Woods CG (2002) Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J Med Genet 39(10):718–721
Coyaud E, Ranadheera C, Cheng D, Goncalves J, Dyakov BJA, Laurent EMN, St-Germain J, Pelletier L, Gingras AC, Brumell JH, Kim PK, Safronetz D, Raught B (2018) Global interactomics uncovers extensive organellar targeting by Zika virus. Mol Cell Proteom 17(11):2242–2255. https://doi.org/10.1074/mcp.TIR118.000800
Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, Satkamp L, Newton B, Huttenhain R, Petit MJ, Baum T, Everitt A, Laufman O, Tassetto M, Shales M, Stevenson E, Iglesias GN, Shokat L, Tripathi S, Balasubramaniam V, Webb LG, Aguirre S, Willsey AJ, Garcia-Sastre A, Pollard KS, Cherry S, Gamarnik AV, Marazzi I, Taunton J, Fernandez-Sesma A, Bellen HJ, Andino R, Krogan NJ (2018) Comparative Flavivirus-Host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell 175(7):1931–1945. https://doi.org/10.1016/j.cell.2018.11.028
Scaturro P, Stukalov A, Haas DA, Cortese M, Draganova K, Plaszczyca A, Bartenschlager R, Gotz M, Pichlmair A (2018) An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 561(7722):253–257. https://doi.org/10.1038/s41586-018-0484-5
Golubeva VA, Nepomuceno TC, Gregoriis G, Mesquita RD, Li X, Dash S, Garcez PP, Suarez-Kurtz G, Izumi V, Koomen J, Carvalho MA, Monteiro ANA (2020) Network of interactions between ZIKA virus non-structural proteins and human host proteins. Cells. https://doi.org/10.3390/cells9010153
Ahmad I, Baig SM, Abdulkareem AR, Hussain MS, Sur I, Toliat MR, Nurnberg G, Dalibor N, Moawia A, Waseem SS, Asif M, Nagra H, Sher M, Khan MMA, Hassan I, Rehman SU, Thiele H, Altmuller J, Noegel AA, Nurnberg P (2017) Genetic heterogeneity in Pakistani microcephaly families revisited. Clin Genet 92(1):62–68. https://doi.org/10.1111/cge.12955
Higgins J, Midgley C, Bergh AM, Bell SM, Askham JM, Roberts E, Binns RK, Sharif SM, Bennett C, Glover DM, Woods CG, Morrison EE, Bond J (2010) Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol 11:85. https://doi.org/10.1186/1471-2121-11-85
Chen JF, Zhang Y, Wilde J, Hansen KC, Lai F, Niswander L (2014) Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat Commun 5:3885. https://doi.org/10.1038/ncomms4885
Thornton GK, Woods CG (2009) Primary microcephaly: do all roads lead to Rome? Trends Genet 25(11):501–510. https://doi.org/10.1016/j.tig.2009.09.011
Szabo N, Pap C, Kobor J, Svekus A, Turi S, Sztriha L (2010) Primary microcephaly in Hungary: epidemiology and clinical features. Acta Paediatr 99(5):690–693. https://doi.org/10.1111/j.1651-2227.2009.01666.x
Gonzalez C, Saunders RD, Casal J, Molina I, Carmena M, Ripoll P, Glover DM (1990) Mutations at the asp locus of Drosophila lead to multiple free centrosomes in syncytial embryos, but restrict centrosome duplication in larval neuroblasts. J Cell Sci 96(Pt 4):605–616
Zhong X, Liu L, Zhao A, Pfeifer GP, Xu X (2005) The abnormal spindle-like, microcephaly-associated (ASPM) gene encodes a centrosomal protein. Cell Cycle 4(9):1227–1229. https://doi.org/10.4161/cc.4.9.2029
Evans PD, Anderson JR, Vallender EJ, Gilbert SL, Malcom CM, Dorus S, Lahn BT (2004) Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet 13(5):489–494. https://doi.org/10.1093/hmg/ddh055
Kouprina N, Pavlicek A, Mochida GH, Solomon G, Gersch W, Yoon YH, Collura R, Ruvolo M, Barrett JC, Woods CG, Walsh CA, Jurka J, Larionov V (2004) Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biol 2(5):E126. https://doi.org/10.1371/journal.pbio.0020126
Montgomery SH, Mundy NI (2012) Evolution of ASPM is associated with both increases and decreases in brain size in primates. Evolution 66(3):927–932. https://doi.org/10.1111/j.1558-5646.2011.01487.x
Paramasivam M, Chang YJ, LoTurco JJ (2007) ASPM and citron kinase co-localize to the midbody ring during cytokinesis. Cell Cycle 6(13):1605–1612. https://doi.org/10.4161/cc.6.13.4356
Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB (2006) Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA 103(27):10438–10443. https://doi.org/10.1073/pnas.0604066103
Dick GW, Kitchen SF, Haddow AJ (1952) Zika virus. I. isolations and serological specificity. Trans R Soc Trop Med Hyg 46(5):509–520
Fontes CA, Dos Santos AA, Marchiori E (2016) Magnetic resonance imaging findings in Guillain-Barre syndrome caused by Zika virus infection. Neuroradiology 58(8):837–838. https://doi.org/10.1007/s00234-016-1687-9
Leis AA, Stokic DS (2016) Zika virus and Guillain-Barre syndrome: is there sufficient evidence for causality? Front Neurol 7:170. https://doi.org/10.3389/fneur.2016.00170
Oehler E, Watrin L, Larre P, Leparc-Goffart I, Lastere S, Valour F, Baudouin L, Mallet H, Musso D, Ghawche F (2014) Zika virus infection complicated by Guillain–Barre syndrome—case report, French Polynesia, December 2013. EuroSurveillance 19(9):20720
Adibi JJ, Marques ET Jr, Cartus A, Beigi RH (2016) Teratogenic effects of the Zika virus and the role of the placenta. Lancet 387(10027):1587–1590. https://doi.org/10.1016/S0140-6736(16)00650-4
Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, Guillaumot L, Griggs A, Bel M, Lambert AJ, Laven J, Kosoy O, Panella A, Biggerstaff BJ, Fischer M, Hayes EB (2009) Zika virus outbreak on Yap Island, federated stsates of Micronesia. N Engl J Med 360(24):2536–2543. https://doi.org/10.1056/NEJMoa0805715
Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, Sall AA (2013) Musso D (2014) Zika virus, French polynesia, South pacific. Emerg Infect Dis 20(6):1085–1086. https://doi.org/10.3201/eid2006.140138
ECDC (2015) Rapid risk assessment: Zika virus epidemic in the Americas: potential association with microcephaly and Guillain-Barré syndrome. European Centre for Disease Prevention and Control, Stockholm
Zanluca C, Melo VC, Mosimann AL, Santos GI, Santos CN, Luz K (2015) First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz 110(4):569–572. https://doi.org/10.1590/0074-02760150192
Kuno G, Chang GJ (2007) Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 152(4):687–696. https://doi.org/10.1007/s00705-006-0903-z
Goertz GP, Abbo SR, Fros JJ, Pijlman GP (2018) Functional RNA during Zika virus infection. Virus Res 254:41–53. https://doi.org/10.1016/j.virusres.2017.08.015
Wang A, Thurmond S, Islas L, Hui K, Hai R (2017) Zika virus genome biology and molecular pathogenesis. Emerg Microbes Infect 6(3):e13. https://doi.org/10.1038/emi.2016.141
Ye J, Zhu B, Fu ZF, Chen H, Cao S (2013) Immune evasion strategies of flaviviruses. Vaccine 31(3):461–471. https://doi.org/10.1016/j.vaccine.2012.11.015
Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao-Lormeau VM, Coulpier M, Misse D, Jouvenet N, Tabibiazar R, Gressens P, Schwartz O, Amara A (2017) Axl mediates ZIKA Virus entry in human glial cells and modulates innate immune responses. Cell Rep 18(2):324–333. https://doi.org/10.1016/j.celrep.2016.12.045
Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein AR (2016) Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 18(5):591–596. https://doi.org/10.1016/j.stem.2016.03.012
Perera-Lecoin M, Meertens L, Carnec X, Amara A (2013) Flavivirus entry receptors: an update. Virusess 6(1):69–88. https://doi.org/10.3390/v6010069
Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, Choe H (2017) AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci USA 114(8):2024–2029. https://doi.org/10.1073/pnas.1620558114
Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ (2016) The 3.8 a resolution cryo-EM structure of Zika virus. Science 352(6284):467–470. https://doi.org/10.1126/science.aaf5316
Heinz FX, Auer G, Stiasny K, Holzmann H, Mandl C, Guirakhoo F, Kunz C (1994) The interactions of the flavivirus envelope proteins: implications for virus entry and release. Arch Virol Suppl 9:339–348
Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB (2012) A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135(Pt 5):1348–1369. https://doi.org/10.1093/brain/aws019
Ahlfors K, Ivarsson SA, Bjerre I (1986) Microcephaly and congenital cytomegalovirus infection: a combined prospective and retrospective study of a Swedish infant population. Pediatrics 78(6):1058–1063
Gerardin P, Samperiz S, Ramful D, Boumahni B, Bintner M, Alessandri JL, Carbonnier M, Tiran-Rajaoefera I, Beullier G, Boya I, Noormahomed T, Okoi J, Rollot O, Cotte L, Jaffar-Bandjee MC, Michault A, Favier F, Kaminski M, Fourmaintraux A, Fritel X (2014) Neurocognitive outcome of children exposed to perinatal mother-to-child Chikungunya virus infection: the CHIMERE cohort study on Reunion Island. PLoS Negl Trop Dis 8(7):e2996. https://doi.org/10.1371/journal.pntd.0002996
O'Leary DR, Kuhn S, Kniss KL, Hinckley AF, Rasmussen SA, Pape WJ, Kightlinger LK, Beecham BD, Miller TK, Neitzel DF, Michaels SR, Campbell GL, Lanciotti RS, Hayes EB (2006) Birth outcomes following West Nile Virus infection of pregnant women in the United States: 2003–2004. Pediatrics 117(3):e537–545. https://doi.org/10.1542/peds.2005-2024
Sinha SK, Kaveggia E, Gordon MC (1972) The incidence of cytomegalovirus among mentally retarded and microcephalic children in a state institution. J Ment Defic Res 16(2):90–96
Teissier N, Fallet-Bianco C, Delezoide AL, Laquerriere A, Marcorelles P, Khung-Savatovsky S, Nardelli J, Cipriani S, Csaba Z, Picone O, Golden JA, Van Den Abbeele T, Gressens P, Adle-Biassette H (2014) Cytomegalovirus-induced brain malformations in fetuses. J Neuropathol Exp Neurol 73(2):143–158. https://doi.org/10.1097/NEN.0000000000000038
von der Hagen M, Pivarcsi M, Liebe J, von Bernuth H, Didonato N, Hennermann JB, Buhrer C, Wieczorek D, Kaindl AM (2014) Diagnostic approach to microcephaly in childhood: a two-center study and review of the literature. Dev Med Child Neurol 56(8):732–741. https://doi.org/10.1111/dmcn.12425
Lazear HM, Diamond MS (2016) Zika virus: new clinical syndromes and Its emergence in the western hemisphere. J Virol 90(10):4864–4875. https://doi.org/10.1128/JVI.00252-16
da Costa CF, da Silva AV, do Nascimento VA, de Souza VC, Monteiro D, Terrazas WCM, Dos Passos RA, Nascimento S, Lima JBP, Naveca FG (2018) Evidence of vertical transmission of Zika virus in field-collected eggs of Aedes aegypti in the Brazilian Amazon. PLoS Negl Trop Dis 12(7):e0006594. https://doi.org/10.1371/journal.pntd.0006594
Avsic Zupanc T, Petrovec M (2016) Zika: an old virus with a new face. Zdr Varst 55(4):228–230. https://doi.org/10.1515/sjph-2016-0031
Smith DW, Mackenzie J (2016) Zika virus and Guillain-Barre syndrome: another viral cause to add to the list. Lancet 387(10027):1486–1488. https://doi.org/10.1016/S0140-6736(16)00564-X
Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, Christian KM, Didier RA, Jin P, Song H, Ming GL (2016) Zika virus infects human cortical neural progenitors and attenuates Their growth. Cell Stem Cell 18(5):587–590. https://doi.org/10.1016/j.stem.2016.02.016
Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin CF, Xu Z (2016) Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19(1):120–126. https://doi.org/10.1016/j.stem.2016.04.017
Lossia OV, Conway MJ, Tree MO, Williams RJ, Goldthorpe SC, Srinageshwar B, Dunbar GL, Rossignol J (2018) Zika virus induces astrocyte differentiation in neural stem cells. J Neurovirol 24(1):52–61. https://doi.org/10.1007/s13365-017-0589-x
Der SD, Zhou A, Williams BR, Silverman RH (1998) Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA 95(26):15623–15628
Pagni S, Fernandez-Sesma A (2012) Evasion of the human innate immune system by dengue virus. Immunol Res 54(1–3):152–159. https://doi.org/10.1007/s12026-012-8334-2
Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL (2016) The IFITMs inhibit Zika virus replication. Cell Rep 15(11):2323–2330. https://doi.org/10.1016/j.celrep.2016.05.074
Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC (2016) Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17(12):1766–1775. https://doi.org/10.15252/embr.201642627
Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, Garcia-Sastre A (2016) Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19(6):882–890. https://doi.org/10.1016/j.chom.2016.05.009
Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, Yang H, Cui J (2017) Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov 3:17006. https://doi.org/10.1038/celldisc.2017.6
Beatty PR, Puerta-Guardo H, Killingbeck SS, Glasner DR, Hopkins K, Harris E (2015) Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci Transl Med 7(304):304ra141. https://doi.org/10.1126/scitranslmed.aaa3787
Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, Hume DA, Stacey KJ, Young PR (2015) Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med 7(304):304ra142. https://doi.org/10.1126/scitranslmed.aaa3863
Wilson JR, de Sessions PF, Leon MA, Scholle F (2008) West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J Virol 82(17):8262–8271. https://doi.org/10.1128/JVI.00226-08
Erdtmann L, Franck N, Lerat H, Le Seyec J, Gilot D, Cannie I, Gripon P, Hibner U, Guguen-Guillouzo C (2003) The hepatitis C virus NS2 protein is an inhibitor of CIDE-B-induced apoptosis. J Biol Chem 278(20):18256–18264. https://doi.org/10.1074/jbc.M209732200
Yang XJ, Liu J, Ye L, Liao QJ, Wu JG, Gao JR, She YL, Wu ZH, Ye LB (2006) HCV NS2 protein inhibits cell proliferation and induces cell cycle arrest in the S-phase in mammalian cells through down-regulation of cyclin a expression. Virus Res 121(2):134–143. https://doi.org/10.1016/j.virusres.2006.02.004
Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A (2012) DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8(10):e1002934. https://doi.org/10.1371/journal.ppat.1002934
Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL (2012) Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8(6):e1002780. https://doi.org/10.1371/journal.ppat.1002780
Dalrymple NA, Cimica V, Mackow ER (2015) Dengue virus NS proteins inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation: dengue virus serotype 1 NS4A is a unique interferon-regulating virulence determinant. MBio 6(3):e00553. https://doi.org/10.1128/mBio.00553-15
Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, Shu HB, Zhong J (2013) Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol 59(1):52–58. https://doi.org/10.1016/j.jhep.2013.03.019
Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437(7062):1167–1172. https://doi.org/10.1038/nature04193
Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A (2005) Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol 79(13):8004–8013. https://doi.org/10.1128/JVI.79.13.8004-8013.2005
Nitta S, Sakamoto N, Nakagawa M, Kakinuma S, Mishima K, Kusano-Kitazume A, Kiyohashi K, Murakawa M, Nishimura-Sakurai Y, Azuma S, Tasaka-Fujita M, Asahina Y, Yoneyama M, Fujita T, Watanabe M (2013) Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 57(1):46–58. https://doi.org/10.1002/hep.26017
Liu WJ, Chen HB, Wang XJ, Huang H, Khromykh AA (2004) Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J Virol 78(22):12225–12235. https://doi.org/10.1128/JVI.78.22.12225-12235.2004
Liu WJ, Wang XJ, Mokhonov VV, Shi PY, Randall R, Khromykh AA (2005) Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J Virol 79(3):1934–1942. https://doi.org/10.1128/JVI.79.3.1934-1942.2005
Banatvala JE, Brown DW (2004) Rubella. Lancet 363(9415):1127–1137. https://doi.org/10.1016/S0140-6736(04)15897-2
Chambers TJ, Hahn CS, Galler R, Rice CM (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688. https://doi.org/10.1146/annurev.mi.44.100190.003245
Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Despres P, Amara A, Yssel H, Misse D (2015) Biology of Zika virus infection in human skin cells. J Virol 89(17):8880–8896. https://doi.org/10.1128/JVI.00354-15
Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM (1997) Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med 3(8):866–871
Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW, Zarski JP, Drouet E (2002) Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology 292(1):162–168. https://doi.org/10.1006/viro.2001.1232
Hilgard P, Stockert R (2000) Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology 32(5):1069–1077. https://doi.org/10.1053/jhep.2000.18713
Kroschewski H, Allison SL, Heinz FX, Mandl CW (2003) Role of heparan sulfate for attachment and entry of tick-borne encephalitis virus. Virology 308(1):92–100
Lee E, Pavy M, Young N, Freeman C, Lobigs M (2006) Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antiviral Res 69(1):31–38. https://doi.org/10.1016/j.antiviral.2005.08.006
Okamoto K, Kinoshita H, Parquet Mdel C, Raekiansyah M, Kimura D, Yui K, Islam MA, Hasebe F, Morita K (2012) Dengue virus strain DEN2 16681 utilizes a specific glycochain of syndecan-2 proteoglycan as a receptor. J Gen Virol 93(Pt 4):761–770. https://doi.org/10.1099/vir.0.037853-0
Zhang L (2010) Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Prog Mol Biol Transl Sci 93:1–17. https://doi.org/10.1016/S1877-1173(10)93001-9
Lemke G, Rothlin CV (2008) Immunobiology of the TAM receptors. Nat Rev Immunol 8(5):327–336. https://doi.org/10.1038/nri2303
Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G (2007) TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131(6):1124–1136. https://doi.org/10.1016/j.cell.2007.10.034
Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, Lemke G, Schwartz O, Amara A (2012) The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12(4):544–557. https://doi.org/10.1016/j.chom.2012.08.009
Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, Vizjak A, Pizem J, Petrovec M, Avsic Zupanc T (2016) Zika virus associated with microcephaly. N Engl J Med 374(10):951–958. https://doi.org/10.1056/NEJMoa1600651
Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, Gaitsch HF, Parnell LA, Cao B, Mysorekar IU, Rothlin CV, Fikrig E, Diamond MS, Iwasaki A (2017) TAM receptors are not required for Zika virus infection in mice. Cell Rep 19(3):558–568. https://doi.org/10.1016/j.celrep.2017.03.058
Briant L, Despres P, Choumet V, Misse D (2014) Role of skin immune cells on the host susceptibility to mosquito-borne viruses. Virology 464–465:26–32. https://doi.org/10.1016/j.virol.2014.06.023
Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, Schinazi RF, Chakraborty R, Suthar MS (2016) Zika virus infects human placental macrophages. Cell Host Microbe 20(1):83–90. https://doi.org/10.1016/j.chom.2016.05.015
van den Pol AN, Mao G, Yang Y, Ornaghi S, Davis JN (2017) Zika virus targeting in the developing brain. J Neurosci 37(8):2161–2175. https://doi.org/10.1523/JNEUROSCI.3124-16.2017
Blackburn D, Sargsyan S, Monk PN, Shaw PJ (2009) Astrocyte function and role in motor neuron disease: a future therapeutic target? Glia 57(12):1251–1264. https://doi.org/10.1002/glia.20848
Kozak RA, Majer A, Biondi MJ, Medina SJ, Goneau LW, Sajesh BV, Slota JA, Zubach V, Severini A, Safronetz D, Hiebert SL, Beniac DR, Booth TF, Booth SA, Kobinger GP (2017) MicroRNA and mRNA dysregulation in astrocytes infected with Zika virus. Viruses. https://doi.org/10.3390/v9100297
Bell TM, Field EJ, Narang HK (1971) Zika virus infection of the central nervous system of mice. Arch Gesamte Virusforsch 35(2):183–193
Bhatnagar J, Rabeneck DB, Martines RB, Reagan-Steiner S, Ermias Y, Estetter LB, Suzuki T, Ritter J, Keating MK, Hale G, Gary J, Muehlenbachs A, Lambert A, Lanciotti R, Oduyebo T, Meaney-Delman D, Bolanos F, Saad EA, Shieh WJ, Zaki SR (2017) Zika virus RNA replication and persistence in brain and placental tissue. Emerg Infect Dis 23(3):405–414. https://doi.org/10.3201/eid2303.161499
Calvet G, Aguiar RS, Melo ASO, Sampaio SA, de Filippis I, Fabri A, Araujo ESM, de Sequeira PC, de Mendonca MCL, de Oliveira L, Tschoeke DA, Schrago CG, Thompson FL, Brasil P, Dos Santos FB, Nogueira RMR, Tanuri A, de Filippis AMB (2016) Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 16(6):653–660. https://doi.org/10.1016/S1473-3099(16)00095-5
Martines RB, Bhatnagar J, de Oliveira Ramos AM, Davi HP, Iglezias SD, Kanamura CT, Keating MK, Hale G, Silva-Flannery L, Muehlenbachs A, Ritter J, Gary J, Rollin D, Goldsmith CS, Reagan-Steiner S, Ermias Y, Suzuki T, Luz KG, de Oliveira WK, Lanciotti R, Lambert A, Shieh WJ, Zaki SR (2016) Pathology of congenital Zika syndrome in Brazil: a case series. Lancet 388(10047):898–904. https://doi.org/10.1016/S0140-6736(16)30883-2
Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA (1992) The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med 326(10):663–667. https://doi.org/10.1056/NEJM199203053261003
Kawasaki H, Kosugi I, Sakao-Suzuki M, Meguro S, Arai Y, Tsutsui Y, Iwashita T (2015) Cytomegalovirus initiates infection selectively from high-level beta1 integrin-expressing cells in the brain. Am J Pathol 185(5):1304–1323. https://doi.org/10.1016/j.ajpath.2015.01.032
Paisley JE, Hinckley AF, O'Leary DR, Kramer WC, Lanciotti RS, Campbell GL, Hayes EB (2006) West Nile virus infection among pregnant women in a northern Colorado community, 2003 to 2004. Pediatrics 117(3):814–820. https://doi.org/10.1542/peds.2005-1187
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI (2013) The global distribution and burden of dengue. Nature 496(7446):504–507. https://doi.org/10.1038/nature12060
Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Harris E, Pereira L (2018) Zika virus replicates in proliferating cells in explants from first-trimester human placentas, potential sites for dissemination of infection. J Infect Dis 217(8):1202–1213. https://doi.org/10.1093/infdis/jix552
Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB (2016) Type III interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 19(5):705–712. https://doi.org/10.1016/j.chom.2016.03.008
Reyes L, Wolfe B, Golos T (2017) Hofbauer Cells: placental macrophages of fetal origin. Results Probl Cell Differ 62:45–60. https://doi.org/10.1007/978-3-319-54090-0_3
Halstead SB (1989) Antibody, macrophages, dengue virus infection, shock, and hemorrhage: a pathogenetic cascade. Rev Infect Dis 11(Suppl 4):S830–839
Rakic P (2003) Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex 13(6):541–549
Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK (2016) Zika virus impairs growth in human neurospheres and brain organoids. Science 352(6287):816–818. https://doi.org/10.1126/science.aaf6116
Foster KG, Fingar DC (2010) Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem 285(19):14071–14077. https://doi.org/10.1074/jbc.R109.094003
Franke TF (2008) PI3K/Akt: getting it right matters. Oncogene 27(50):6473–6488. https://doi.org/10.1038/onc.2008.313
Yu JS, Cui W (2016) Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143(17):3050–3060. https://doi.org/10.1242/dev.137075
Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, Ge J, Wang S, Goldman SA, Zlokovic BV, Zhao Z, Jung JU (2016) Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 19(5):663–671. https://doi.org/10.1016/j.stem.2016.07.019
Asif A, Manzoor S, Tuz-Zahra F, Saalim M, Ashraf M, Ishtiyaq J, Khalid M (2017) Zika virus: immune evasion mechanisms, currently available therapeutic regimens, and vaccines. Viral Immunol 30(10):682–690. https://doi.org/10.1089/vim.2017.0046
Jordan TX, Randall G (2017) Dengue virus activates the AMP kinase-mTOR axis to stimulate a proviral lipophagy. J Virol. https://doi.org/10.1128/JVI.02020-16
Shives KD, Beatman EL, Chamanian M, O'Brien C, Hobson-Peters J, Beckham JD (2014) West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J Virol 88(16):9458–9471. https://doi.org/10.1128/JVI.01323-14
Lee CJ, Liao CL, Lin YL (2005) Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J Virol 79(13):8388–8399. https://doi.org/10.1128/JVI.79.13.8388-8399.2005
Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, Rana TM (2016) Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19(2):258–265. https://doi.org/10.1016/j.stem.2016.04.014
Hanners NW, Eitson JL, Usui N, Richardson RB, Wexler EM, Konopka G, Schoggins JW (2016) Western Zika virus in human fetal neural progenitors persists long term with partial cytopathic and limited immunogenic effects. Cell Rep 15(11):2315–2322. https://doi.org/10.1016/j.celrep.2016.05.075
Brown AN, Kent KA, Bennett CJ, Bernard KA (2007) Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology 368(2):422–430. https://doi.org/10.1016/j.virol.2007.06.033
Samuel MA, Diamond MS (2005) Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol 79(21):13350–13361. https://doi.org/10.1128/JVI.79.21.13350-13361.2005
Sharma M, Bhattacharyya S, Sharma KB, Chauhan S, Asthana S, Abdin MZ, Vrati S, Kalia M (2017) Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J Gen Virol 98(5):1027–1039. https://doi.org/10.1099/jgv.0.000792
Brault JB, Khou C, Basset J, Coquand L, Fraisier V, Frenkiel MP, Goud B, Manuguerra JC, Pardigon N, Baffet AD (2016) Comparative analysis between flaviviruses reveals specific neural stem cell tropism for Zika virus in the mouse developing neocortex. EBioMedicine 10:71–76. https://doi.org/10.1016/j.ebiom.2016.07.018
van den Pol AN, Robek MD, Ghosh PK, Ozduman K, Bandi P, Whim MD, Wollmann G (2007) Cytomegalovirus induces interferon-stimulated gene expression and is attenuated by interferon in the developing brain. J Virol 81(1):332–348. https://doi.org/10.1128/JVI.01592-06
Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, Tang W, Terskikh AV, Shresta S, Gleeson JG (2016) Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 19(5):593–598. https://doi.org/10.1016/j.stem.2016.08.005
Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, Knoblich JA, Lachmann R, Paabo S, Huttner WB, Treutlein B (2015) Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 112(51):15672–15677. https://doi.org/10.1073/pnas.1520760112
Souza BS, Sampaio GL, Pereira CS, Campos GS, Sardi SI, Freitas LA, Figueira CP, Paredes BD, Nonaka CK, Azevedo CM, Rocha VP, Bandeira AC, Mendez-Otero R, Dos Santos RR, Soares MB (2016) Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep 6:39775. https://doi.org/10.1038/srep39775
Wu KY, Zuo GL, Li XF, Ye Q, Deng YQ, Huang XY, Cao WC, Qin CF, Luo ZG (2016) Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res 26(6):645–654. https://doi.org/10.1038/cr.2016.58
Chavali PL, Stojic L, Meredith LW, Joseph N, Nahorski MS, Sanford TJ, Sweeney TR, Krishna BA, Hosmillo M, Firth AE, Bayliss R, Marcelis CL, Lindsay S, Goodfellow I, Woods CG, Gergely F (2017) Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 357(6346):83–88. https://doi.org/10.1126/science.aam9243
Yoon KJ, Song G, Qian X, Pan J, Xu D, Rho HS, Kim NS, Habela C, Zheng L, Jacob F, Zhang F, Lee EM, Huang WK, Ringeling FR, Vissers C, Li C, Yuan L, Kang K, Kim S, Yeo J, Cheng Y, Liu S, Wen Z, Qin CF, Wu Q, Christian KM, Tang H, Jin P, Xu Z, Qian J, Zhu H, Song H, Ming GL (2017) Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell 21(3):349–358. https://doi.org/10.1016/j.stem.2017.07.014
Teng Y, Liu S, Guo X, Liu S, Jin Y, He T, Bi D, Zhang P, Lin B, An X, Feng D, Mi Z, Tong Y (2017) An integrative analysis reveals a central role of P53 activation via MDM2 in Zika virus infection induced cell death. Front Cell Infect Microbiol 7:327. https://doi.org/10.3389/fcimb.2017.00327
Swartwout BK, Zlotnick MG, Saver AE, McKenna CM, Bertke AS (2017) Zika virus persistently and productively infects primary adult sss. Pathogens. https://doi.org/10.3390/pathogens6040049
Cumberworth SL, Barrie JA, Cunningham ME, de Figueiredo DPG, Schultz V, Wilder-Smith AJ, Brennan B, Pena LJ, de Oliveira F, Franca R, Linington C, Barnett SC, Willison HJ, Kohl A, Edgar JM (2017) Zika virus tropism and interactions in myelinating neural cell cultures: CNS cells and myelin are preferentially affected. Acta Neuropathol Commun 5(1):50. https://doi.org/10.1186/s40478-017-0450-8
Haydar TF, Kuan CY, Flavell RA, Rakic P (1999) The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex 9(6):621–626
Wen Z, Song H, Ming GL (2017) How does Zika virus cause microcephaly? Genes Dev 31(9):849–861. https://doi.org/10.1101/gad.298216.117
Ayala-Nunez NV, Follain G, Delalande F, Hirschler A, Partiot E, Hale GL, Bollweg BC, Roels J, Chazal M, Bakoa F, Carocci M, Bourdoulous S, Faklaris O, Zaki SR, Eckly A, Uring-Lambert B, Doussau F, Cianferani S, Carapito C, Jacobs FMJ, Jouvenet N, Goetz JG, Gaudin R (2019) Zika virus enhances monocyte adhesion and transmigration favoring viral dissemination to neural cells. Nat Commun 10(1):4430. https://doi.org/10.1038/s41467-019-12408-x
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest relwated to the contents of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

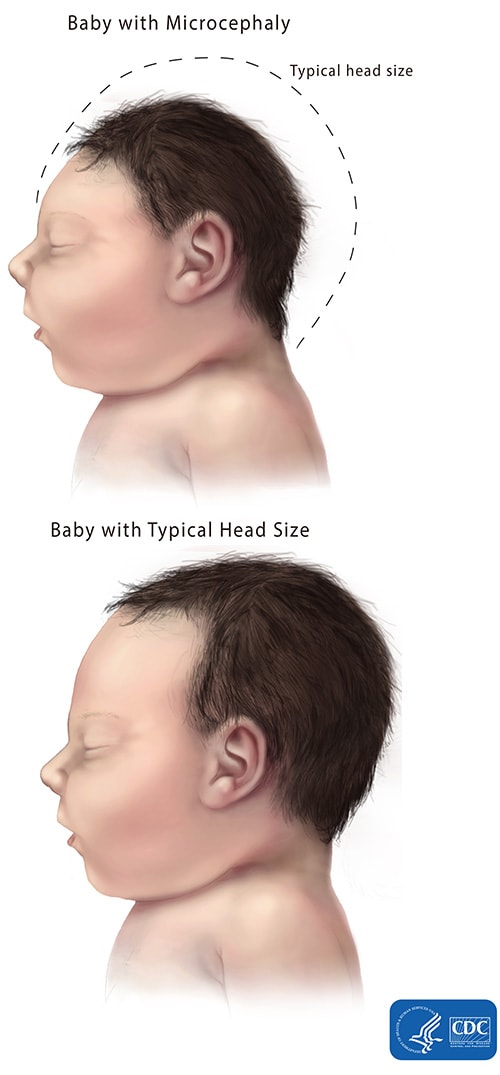{kind=link}
Cite this article
Rombi, F., Bayliss, R., Tuplin, A. et al. The journey of Zika to the developing brain. Mol Biol Rep 47, 3097–3115 (2020). https://doi.org/10.1007/s11033-020-05349-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05349-y