
Abstract
The aim of the study was to produce a single-domain antibody (nanobody) specific for endothelin receptor type B (EDNRB) which has high expression in melanoma. Cultured human melanoma cells were used as antigens to immunize alpacas. After antibody generation was verified in alpaca serum, total RNA was extracted from alpaca lymphocytes and the target VHH fragment was amplified by two-step PCR, cloned in the pCANTAB5E phagemid vector, and used to transform Escherichia coli TG1 cells to obtain a phage-display nanobody library, which was enriched by panning. The results indicated successful construction of a phage-display anti-human melanoma A375 nanobodies library with a size of 1.2 × 108/ml and insertion rate of 80%. After screening, eight positive clones of anti-EDNRB nanobodies were used to infect E. coli HB2151 for production of soluble nanobodies, which were identified by ELISA. Finally, we obtained a high-affinity anti-EDNRB nanobody, which consisted of 119 amino acids (molecular weight: 12.97 kDa) with 22 amino acids in CDR3 and had good affinity in vitro. The results suggest that the nanobody may be potentially used for the treatment of human melanoma.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genetically engineered antibodies represent a new generation of biological drugs that have been increasingly used in clinics. Since the discovery of heavy-chain antibodies in camelids [1] their clinical application has been extensively investigated. The variable domain of a heavy-chain (VHH) antibody has a simple structure with only approximately 400 bp gene fragments. It can be easily obtained in vitro in both prokaryotic and eukaryotic expression systems, which is the major technical advantage of nanobodies over conventional genetically engineered antibodies [2, 3]. A single antigen-specific camelid VHH fragment obtained by genetic engineering has the relative molecular mass of 15 kDa (only 1/10 of that of regular antibodies) and size in the nanometer range; also known as nanobody, it is currently the smallest antibody fragment retaining full functional activity [4]. Therefore, nanobodies have been extensively studied as novel molecular tools for molecular imaging of tumors [5] and immunotherapeutic purposes. Currently, therapeutically relevant nanobodies have been suggested for use in oncology, including in vivo imaging [6], and therapy, and some of them are in advanced stages of clinical investigation [7].
Malignant melanoma is one of the fastest-growing tumors among all cancers; it has a high metastasis rate and accounts for a considerable proportion of deaths due to skin malignancies. Endothelin (EDN), a major paracrine factor in melanocyte biology, plays a role in proliferation and differentiation of melanocytes in response to ultraviolet radiation and is involved in the pathogenesis of melanoma [8]. EDN and its receptor (EDNRB) on the melanocyte surface form the EDN signaling system [9] implicated in survival, proliferation, differentiation, and migration of melanocytes [10]. EDNRB overexpression was detected in most melanoma cell lines and in melanoma metastasis sites, where it was higher than in primary tumors [11]. Furthermore, the upregulation of EDNRB was shown to be associated with melanoma growth and intratumoral angiogenesis [12] and with increased tumor metastasis and invasiveness [13]. EDNRB is also involved in dedifferentiation of mature melanocytes, causing their malignant transformation. It has been suggested that EDNRB can play a role in melanoma spread by influencing the interaction between tumor and normal cells; thus, it is considered to be a potential driver of melanoma development and an important marker of tumor invasiveness [14]. Therefore, it is important to develop methods to detect and block EDNRB expression in melanoma, which should improve cancer diagnosis and therapeutic outcomes.
In this study, we generated anti-EDNRB nanobodies by genetic engineering and performed preliminary analysis of the binding capacity to EDNRB in vitro.
Materials and methods
Human melanoma cell culture and synthesis of antigen polypeptides
Human melanoma cell line A375 (ZQ0042; Shanghai Zhongqiaoxinzhou Biotech) was cultured in DMEM (ZQ-101; Shanghai Zhongqiaoxinzhou Biotech).
After the third generation, cells were collected and sub-packed, and frozen in liquid nitrogen routinely. The cells were recovered at 37 ℃ before being used to immunize animals [15].
Total RNA was extracted from A375 cells by the TRIzol method; cDNA was synthesized with the PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara Bio, USA) according to the manufacturer’s instructions and used as a template to amplify the EDNRB coding region by PCR with primers EDNRB-F and EDNRB-B (Table 1). The region showing strong immunogenic properties was analyzed by sequencing. (China Peptides Co., Ltd. Shanghai, China). The antigenic polypeptide PTedn with the sequence CLLHPVQKTAFMQFYK-NH2 was synthesized by China Peptides Co., Ltd. (Shanghai, China). EDNRB is a sevenfold transmembrane protein, and PTedn is located in the extracellular domain, between the 255th and 270th amino acids in EDNRB (Fig. 1).

The analysis result for extracellular region of EDNRB
Immunization of alpacas
Housing and care of alpacas and the study protocol were approved by the Animal Experimentation Ethics Committee of Shanxi Agricultural University (2017). The number of the alpaca is A006.
For the initial and subsequent immunizations, A375 cells (2.8 × 106) were mixed with complete or incomplete Freund’s adjuvants (Sigma-Aldrich), respectively, and injected in the neck and back skin of alpacas at several sites to ensure antigen entrance into the reflux pathway of different lymph nodes. A total of four immunizations were performed at 2-week intervals [16]. Before the first and fourth immunizations, 10 ml blood was drawn from each animal to obtain serum, which was checked for immune response to the antigen by ELISA plate coated with 3 μg/ml PTedn.
Construction of the anti-human melanoma nanobodies library
On the 10th day after the fourth immunization, 50 ml of blood was collected from the jugular vein of alpacas, treated with anticoagulant, and lymphocytes were isolated using Lymphocyte Separation Medium (Solarbio Science & Technology Co. Ltd., Beijing, China). Total lymphocyte RNA was extracted by TRIzol and cDNA was obtained using the Takara PrimeScript™ II 1st Strand cDNA Synthesis Kit. Variable domains of conventional antibodies and the heavy-chain antibody located between the leader zone and CH2 region (1000 bp and 600 bp, respectively) were amplified by PCR using primers CALL001 and CALL002 (Table 1) (Els Pardon 2014) and the products were separated by electrophoresis in 1% agarose gels. The 600-bp fragment was purified using the Gel Extraction Kit (CWbio, Beijing, China) according to the manufacturer’s instructions, inserted into the pMD19-T Simple vector (Takara Bio), and used to transform Escherichia coli DH5α. Thirty randomly selected clones were sequenced and primers VHH-F and VHH-R (Table 1) designed based on the sequence information were used to re-amplify the nanobody-encoding gene with all the extracted 600-bp fragments as a template to obtain the VHH fragments of 400 bp. The 400-bp fragments were purified by electrophoresis in 1% agarose gels using the Gel Extraction Kit (CWbio).
At least 10 µg VHH was digested with FastDigest SfiI (Thermo Fisher Scientific, Shanghai, China) and purified with the QIAquick PCR purification kit (QIAGEN, Germany); the phagemid vector pCANTAB5E was also cut with FastDigest SfiI and purified with the Gel Extraction Kit. To ensure complete digestion, the purified VHH fragments and the vector were cut again with FastDigest SfiI, purified, and 1 µg of the vector and 3 µg of the fragment were ligated using T4 DNA Ligase (Thermo Fisher Scientific) overnight at 4 ℃. The reaction products were purified and used to transform 1.2 ml of freshly prepared electrocompetent E. coli TG1 cells (Bio-View Shine Biotechnology, Beijing, China) at 1.8 kV, 25 μf, 200 ω, for 5 ms. The transformed bacteria were incubated in 20 ml SOC medium for 1 h at 37 ℃ with shaking at 200 rpm and 100 μl of the cultured cells were serially diluted (10−1–10−5 cells/ml) in 2× YT medium. Then, 100 μl of every dilution was spread on a plate containing 2 × YTAG medium (2× YT supplemented with 100 μg/ml ampicillin and 2% glucose) to calculate the effective library size, and 50 isolated bacterial colonies were randomly selected to perform colony PCR and sequencing to determine the insertion rate. The remaining cultures were centrifuged at 5000×g for 5 min, re-suspended in 8 ml of SOC medium, plated onto 39 2× YTAG plates (150-mm), and incubated overnight at 30 ℃. The bacteria were scraped from the surface, mixed with 20% glycerol in culture medium, and stored at − 80 ℃ as a primary anti-human melanoma nanobodies library.
Panning for anti-EDNRB positive recombinant phage-displayed nanobodies
The binding-elution-amplification method was used to perform the panning. Three rounds of panning for recombinant anti-EDNRB phage-displayed nanobodies were performed according to a published protocol [4] with some modifications. 100 μl of the primary library cells were serially diluted (10−1–10−8) in 2× YT medium. Then, 100 μl of every dilution was spread on a plate containing 2× YTAG medium to calculate the amount of primary library.
Rescue of recombinant phages from the primary anti-human melanoma nanobodies library
Primary library cells (600 μl, about 1.1 × 1012) were incubated in 2× YTAG medium at 37 ℃ with shaking until OD600 reached 0.4 and infected with 1.5 × 1012 plaque-forming units (PFU) of M13KO7 helper phage (Bio-View Shine Biotek) at 37 ℃ without shaking for 30 min and then with shaking for 30 min. After centrifugation at 3600×g for 10 min, the pellet was re-suspended in 100 ml 2× YT medium supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin, incubated at 30 ℃ with shaking overnight, centrifuged at 11,000×g for 10 min at 4 ℃ and the supernatant (about 100 ml) containing recombinant phages was transferred to a new tube.
Re-infection of E. coli TG1 with the recombinant phage
The supernatant was mixed with 20 ml PEG/NaCl solution (200 g PEG600 and 146.1 g NaCl in 1 L sterile water), incubated on ice for 70 min, and centrifuged at 11,000×g for 30 min at 4 °C. The precipitate containing recombinant phages was resuspended in 2 ml PBS, serially diluted (10−7–10−13) in 2× YT medium, and 10 μl of each dilution was incubated with 190 μl log phase E. coli TG1 for 30 min at 37 ℃; after that, 100 μl of infected cultures was plated on 2× YTAG solid medium to calculate the number of the input recombinant phages. Then, 1.5 ml of the recombinant phage suspension was mixed with 1 ml blocking buffer (3% Difco Skim Milk in PBS [MPBS]), incubated at room temperature for 1 h, and added to MaxiSorp immune test tubes (Nunc, The Kingdom of Denmark), which were pre-coated with 2 ml of PTedn peptide (20 μg/ml in 0.05 M Na2CO3/Na2HCO3, pH 9.6) overnight at 4 °C, washed with PBS, and blocked with 4 ml of MPBS for 2 h to eliminate non-specific binding sites. After incubation at room temperature for 2 h, the supernatant was removed and tubes were washed 10 times with PBST and 10 times with PBS, and the recombinant phage was eluted by shaking at 300 rpm with 2 ml TEA solution for 15 min; then, 2 ml of 1 M Tris–HCl (pH 7.4) was added for neutralization. The eluted phage particles were incubated with 20 ml log-phase E. coli TG1 at 37 ℃ for 30 min and the infected bacteria were serially diluted (10–1 to 10–5) in 2× YT medium; then, 100 μl of every dilution was spread on 2 × YTAG plates to calculate the number of the output recombinant phages. The remaining cultures were plated on five 2× YTAG plates (150-mm) and incubated at 30 ℃ overnight; bacterial cells were scraped, mixed with 20% glycerol in culture medium, and stored at − 80 ℃ as the first anti-EDNRB nanobodies library.
The second panning was performed with recombinant phages from the first library as described above with the following modifications: PTedn peptide concentration was reduced to 14 μg/ml and washing with PBST and PBS was increased to 20 times, respectively. In the third panning performed with the second library, PTedn concentration was 7 μg/ml and the number of washings with PBST and PBS was also 20. The input/output (I/O) ratio, which should have a downward trend, was calculated to evaluate each panning result using the following equations:
I = The number of single colony in the input plate 10−13 ÷ (5 × 10–13) × 103 × 1.5, O = The number of single colony in the output plate 10−1 × 102 × 24.
Screening of antigen-positive recombinant phages in enriched clones
The method of the fourth scouring screen was the same as that of the third. The difference was that the phage-infected bacteria were serially diluted and each gradient was coated with three 150 mm plates. This process (the fourth screen) was carried out twice and 192 randomly selected colonies from the 150 mm plates were inoculated into two 96-tube clusters containing 200 μl 2× YTAG medium per tube and incubated at 30 ℃ for 8 h with shaking at 200 rpm. Then, 20 μl of each cell suspension was transferred to new 96-tube clusters with 180 μl 2× YTAG medium per tube, incubated at 30 ℃ for 1 h at 200 rpm, and infected with M13KO7 helper phage for 30 min. Plates were then incubated at 37 ℃ with shaking at 200 rpm for 40 min, centrifuged at 1800 ×g for 10 min, and pellets were resuspended in 400 μl 2× YT medium containing 100 μg/ml ampicillin and 50 μg/ml kanamycin. Plates were incubated at 30 ℃ with shaking overnight and centrifuged at 2200 ×g for 10 min; 250 μl of phage supernatant from each tube was transferred to two new 96-tube clusters and blocked with 250 μl of PBS with 3% BSA at room temperature for 1 h.
ELISA to detect antigen-positive recombinant phages
Blocked recombinant phage supernatants (100 μl) were added to 96-well clear flat-bottom polystyrene High Bind microplates (Corning, USA) coated with 3 μg/ml Ptedn (to screen nanobody clones) or with 3 μg/ml BSA (negative control) for 1 h at 37 ℃. Microplates were rinsed 10 times with PBS/0.1% Tween 20 (PBST), incubated with 100 μl/well of horseradish peroxidase (HRP)-conjugated anti-M13 antibody (1:5000; GE Healthcare, USA) for 1 h at 37 ℃, rinsed 10 times with PBST, and 100 μl/well of TMB single-component substrate solution (Solarbio Science & Technology) was added in the dark for 10 min at room temperature. After the reaction was terminated with equal volume of 1 M H2SO4, optical density (OD) was measured at 450 nm and the positive clones with the highest OD values were selected for sequencing.
Infection of E. coli HB2151 and production of soluble antibodies
Phage supernatants (2 μl) of each selected clone were incubated with 400 μl of log-phase E. coli HB2151 (Bio-View Shine Biotechnology) for 40 min at 30 ℃, and 30 μl of each mixture was plated on solid SOBAG-N medium (SOB with 100 μg/ml ampicillin, 2% glucose, and 100 μg/ml nalidixic acid) overnight at 30 ℃. A single colony from each plate was transferred to 5 ml of freshly prepared 2× YT medium containing 100 μg/ml ampicillin, 2% glucose, and 100 μg/ml nalidixic acid and incubated overnight at 30 ℃ with shaking at 200 rpm. Then, 5 ml of culture was added to 50 ml fresh 2× YTAG medium, incubated for 1 h at 30 ℃ with shaking, centrifuged at 1500×g for 20 min at room temperature, and cells were resuspended in 50 ml of fresh 2× YT medium containing 100 μg/ml ampicillin and 1 mM IPTG and incubated at 30 ℃ with shaking overnight. Every culture was divided into two tubes, centrifuged at 1500 ×g for 20 min at room temperature, and supernatants containing extracellular soluble antibodies (designated L1) were filtered through 0.45-μm filters and stored at − 80 ℃.
The remaining pellets were washed twice with PBS and one of the two was resuspended first in 0.5 ml of ice-cold 1 × TES and then in 0.75 ml of ice-cold 0.2 × TES, vortexed, incubated on ice for 30 min, and centrifuged at 12,000×g for 15 min at 4 ℃. The supernatant containing soluble antibodies from the periplasm (L2) was transferred to a clean tube and stored at − 80 ℃.
The second pellet was resuspended in 0.5 ml PBS, boiled for 5 min, incubated on ice for 30 min, centrifuged at 12,000×g for 15 min at 4 ℃, and supernatant containing intracellular soluble nanobodies (L3) was transferred to a clean tube and stored at − 80 ℃.
Testing of soluble antibodies
Immunoreactivity of supernatants L1, L2, and L3 was tested by ELISA. ELISA was performed as described in chapter 2.4.2 (Re-infection of E.coli TG1 with the recombinant phages) except that the number of washes time was six. L1, L2, and L3 were used as primary antibodies and HRP-E-tag antibody (1:5000; Abcam, UK) as a secondary antibody because the VHH gene was followed with the E-tag peptide-encoding sequence containing the amber termination codon in the expression vector.
Production and purifying of nanobody
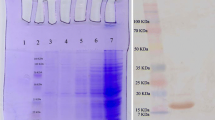
The VHH fragments with the highest OD450 was amplified and inserted into the pColdI vector [17], and used to transform Escherichia coli BL21. The 5 ml of Escherichia coli BL21 were incubated at 37 ℃ with shaking overnight and added in 500 ml of fresh LB medium containing 100 μg/ml ampicillin and incubated at 37 ℃ with shaking until the OD600 reached 0.4. Then added IPTG (0.2 mM), and incubated at 16 ℃ with shaking overnight and centrifuged at 5000× g for 10 min at 4 °C. The precipitates were washed twice with PBS and resuspended with 50 ml ddH2O. After ultrasonication (100 W, 30 min) and centrifugation (12,000 ×g for 15 min at 4 °C), the purified nanobody was obtained by HisTrap™ HP (GE, US) and verified by SDS-PAGE.
Testing of nanobody
Immunoreactivity of the purified nanobody was tested by ELISA and western blotting. ELISA was performed as described in chapter 2.4.4 (ELISA to detect antigen-positive recombinant phages). The purified nanobody and the serial dilution were used as primary antibodies and HRP-His-tag antibody (1:10,000; CWbio, China) as a secondary antibody because the VHH gene was followed with the His-tag peptide-encoding sequence in the expression vector.
The testing of KD value [18]: the PTedn peptides were serially diluted (1.5−1–1.5−18) with the initial concentration: 40 nM. 50 μl of each dilution and 50 μl of Nb4 (0.01 mg/ml) were mixed and incubated at 37 ℃ for 30 min. ELISA was performed as described in chapter 2.4.4 (ELISA to detect antigen-positive reconbinant phages). 90 μl of the mixture were used as primary antibodies and incubated at room temperature for 10 min. PBS was used as a negative control for the primary antibody and a blank control coated with PBS. The obtained OD450 value is imported into analysis software prism 6 for analysis and calculation of KD value.
Western blotting: PTedns were separated by SDS-PAGE on a 12% polyacrylamide gel, transferred to nitrocellulose membrane (BOSTER, USA), and the strip was cut and incubated with blocking buffer (5% Difco Skim Milk in Tris-buffered saline/0.1%Tween-20 [TBST]) at room temperature for 2 h and incubated with the threefold dilution of the purified nanobody and then with HRP-His-tag antibody (1:10,000; CWbio, China). After washing six times with TBST, signals were developed using an enhanced chemiluminescence kit and their intensity was analyzed. (Positive control: the primary antibody: rabbit anti-EDNRB antibody [1:1000 Bioss, China]; secondary antibody: HRP-Goat anti-rabbit IgG antibody [1:30,000; CWbio, China]).
The nanobody was further examined for binding to melanoma cells by immunohistochemistry. Paraffinized melanoma sections (obtained from Doctor Hao Huang) were subjected to dewaxing, antigen repair, depigmentation in 15% hydrogen peroxide (three times for 10 min), and then incubated in 3% hydrogen peroxide at room temperature for 30 min. After washing with PBS, the sections were blocked with 5% BSA at 37 °C for 30 min and incubated with the purified nanobody or the non-induced supernatant of ultrasonic wave disruption (negative control) or PBS (blank control) and then with HRP-His-tag antibody (1:100; CWbio, China). DAB was used as an HRP chromogenic substrate.
Results
Validation of immunoreactivity in immune serum of alpacas
To produce single-domain antibodies (nanobodies) specific for EDNRB, alpacas were immunized with cultured human melanoma cells used as antigens. Analysis by ELISA revealed strong reactivity of immune sera as evidenced by OD450 values at 1:1000, 1:5000, 1:10,000 and 1:50,000 dilutions, which were significantly higher than those of pre-immune sera (Fig. 2), indicating successful immunization.

The ELISA result of serum
Construction of the anti-human melanoma nanobodies library
The VHH fragment was generated by two-step PCR (Fig. 3). Using the serial dilution method, we estimated the size of the anti-human melanoma nanobodies library as 1.2 × 108/ml. PCR analysis of 50 isolated bacterial colonies indicated that the insertion rate of the nanobody library was 80% (Fig. 4). The final volume of the primary library was 40 ml. There were 736 colonies on the plate with dilution of 10−8 to calculate the amount of primary library as follows: 736 ÷ (40 × 10−8) × 103/ml = 1.84 × 1012/ml.

PCR Amplification of VHH

Colony PCR of anti-Melanoma nanobodies immunolibrary
Screening of antigen-binding recombinant phages from enriched clones
The result of four screening were listed in Table 2. After the fourth screening, the I/O ratio was at the same exponential level (Table 2). The eight positive recombinant phage clones, with an OD450 higher than 0.5 (Fig. 5), were selected for BLAST sequencing, which revealed that eight clones had the VHH sequence. Sequence analysis showed that three clones had high sequence similarity and one clone was shorter than others. In comparison with the VHH sequence published by David R Maass et al. [19], five different sequences had correct CDR sequences (Fig. 6). The short sequence that only had part of framework 3 and CDR3 was abandoned. The remaining five different antibody-positive recombinant phage clones were used to infect E. coli HB2151 for antibody expression.

ELISA result of the fourth screening

The sequence analysis of five correct nanobodies
Affinity evaluation of recombinant antibodies
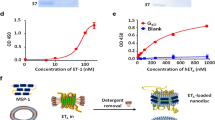
Soluble recombinant antibodies from the extracellular (L1), periplasmic (L2), and intracellular fractions (L3) were analyzed for immune reactivity by ELISA. The results revealed significantly higher OD450 values for the recombinant antibody Nb4 compared to the other nanobodies (Fig. 7). Therefore, Nb4 was selected for further production and purification (Fig. 8). 1.1 mg Nb4 were obtained from the 500 ml after purification. The purified nanobodies (0.1 mg/ml) were verified by ELISA, western blotting, and immunohistochemistry. The ELISA results (Fig. 9) showed significantly higher OD450 values for the purified nanobody and for the tenfold dilution compared to the positive control, negative control, and blank groups. The 100-fold dilution (1 μg/ml) had a significantly high binding activity. In the testing of KD value, it was found that the curve was generated, each point was basically located on both sides of the curve, R2 was 0.9972, indicated that the curve fitting was correct. The Kd value was 0.9723. (Fig. 10). In western blotting, the nanobody demonstrated the same signal as the positive group (Fig. 11). The immunohistotochemistry showed positive reaction (brown staining) with the surface of human melanoma cells, whereas no staining was observed for negative and blank controls (Fig. 12).

ELISA for E.coli HB2151 products

SDS-PAGE of purification

ELISA result of the purified nanobody

The result of KD value testing

The result of Western bollting

The result of immunohistochemical
Discussion
In this study, we constructed a phage display library of a nanobodies against human melanoma generated in alpaca.
As a result, we obtained a high-affinity nanobody against EDNRB, Nb4. It consisted of 119 amino acids (molecular weight: 12.97 kDa) with 22 amino acids in CDR3 and showed good affinity in vitro.
Melanoma is a highly malignant tumor originating from melanin-producing cells, and its morbidity has been rising over the past decade. Although melanoma diagnosis and treatment have improved in recent years, metastatic melanoma still has poor prognosis [20]. In addition to traditional treatment, including surgery, radiotherapy, and chemotherapy, new methods such as gene therapy and tumor vaccines demonstrate high specificity, effectiveness, and safety, have fewer side effects and are well tolerated by patients. Targeted therapy is considered a new treatment for melanoma. Different monoclonal antibodies and inhibitors of melanin formation have been studied for the treatment of metastatic and unresected melanoma [20, 21]. EDNRB, a surface receptor on melanocytes known to participate in melanin production and transport, demonstrates synergistic/enhancing effects when used with many factors playing a key role in the melanin pathway [22], and it was shown that EDNRB antagonists can slow the growth of melanoma cells and increase their apoptosis [23]. Previous studies indicate that EDNRB antagonists can prevent the progression of EDN-induced melanoma [24], inhibit tumor growth [25], and stabilize metastatic disease [26], indicating that the blockade of EDNRB signaling is a clinically effective approach to treat melanoma [27].
Therefore, the nanobody anti-EDNRB with affinity activity is a potential new antagonist with utility in melanoma therapy. On the other hand, it can be used for related research as a imaging agent.
References
Hammers-Casterman C, Atarhouch T, Muyldermans S, Robinson G et al (1993) Naturally occurring antibodies devoid of light chains. Nature 363(6428):446–448
Siontorou CG (2013) Nanobodies as novel agents for disease diagnosis and therapy. Int J Nanomed 8:4215–4227
Goethals LR, Bos TJ, Baeyens L et al (2014) Camelid reporter gene imaging: a generic method forin vivocell tracking. EJNMMI Res 4(1):1–8
Pardon E, Laeremans T, Triest S, Rasmussen SGF et al (2014) A general protocol for the generation of nanobodies for structural biology. Nat Protoc 9(3):674–693
Bannas P, Lenz A, Kunick V, Well L, Fumey W, Rissiek B et al (2015) Molecular imaging of tumors with nanobodies and antibodies: timing and dosage are crucial factors for improved in vivo detection. Contrast Media Mol Imaging 10(5):367–378
Xavier C, Blykers A, Vaneycken I, D’Huyvetter M, Heemskerk J, Lahoutte T et al (2016) (18)F-nanobody for PET imaging of HER-2 over expressing tumors. Nucl Med Biol 43:247–252
Kaczmarek V, Holle J, Astudillo R et al (2019) Caplacizumab for relapsing thrombotic thrombocytopenic purpura. Pediatric Nephrol 34:1625–1628
Takeshi S, Masashi Y, Tomoh M (1992) Molecular characterization of endothelin receptors. Tips 13(3):103–108
Kedzierski RM, Yanagisawa M (2001) Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol 41(1):851–876
Garcia RJ, Ittah A, Mirabal S, Figueroa J, Kos L (2008) Endothelin 3 induces skin pigmentation in a keratin-driven inducible mouse model. J Investig Dermatol 128(1):131–142
Mangahas CR, Cruz GVD, Friedman-Jiménez G, Jamal S (2005) Endothelin-1 induces cxcl1 and cxcl8 secretion in human melanoma cells. J Gen Intern Med 20(5):307–311
Grimshaw JM (2007) Endothelins and hypoxia-inducible factor in cancer. Endocr Related Cancer 14(2):233–244
Bellahcène A, Castronovo V, Ogbureke KUE, Fisher LW, Fedarko NS (2008) Small integrin-binding ligand n-linked glycoproteins (siblings): multifunctional proteins in cancer. Nat Rev Cancer 8(3):212–226
Demunter A, Wolf-Peeters CD, Degreef H, Stas M, Oord JJVD (2001) Expression of the endothelin-b receptor in pigment cell lesions of the skin. Virchows Archiv Int J Pathol 438(5):485–491
Sondak VK, Sosman JA (2003) Results of clinical trials with an allogenic melanoma tumor cell lysate vaccine: melacine. Semin Cancer Biol 13(6):409–415
Nemunaitis J, Sterman D, Jablons D, Fox B, Maples P, Hamilton S et al (2004) Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. J Natl Cancer Inst 96(7):558
Navid HL, Latifi-Navid S, Zahri S (2015) A new generation of cold-shock vector derived from pcold i (pcold i-lz) for expression of recombinant peptides and proteins. Govaresh 20(3):168–177
Yu X, Pegram CN, Bigner DD et al (2017) Development and validation of a cell-based fluorescent method for measuring antibody affinity. J Immunol Methods 442:49–53
Maass David R, Jorge S, Anton P, Shoemaker Charles B (2007) Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHH). J Immunol Methods 324:13–25
Franklin C, Livingstone E, Roesch A, Schilling B, Schadendorf D (2016) Immunotherapy in melanoma: recent advances and future directions. Eur J Surg Oncol 43(3):604–611
Asundi J, Lacap JA, Clark S, Nannini M, Roth L, Polakis P (2014) Mapk pathway inhibition enhances the efficacy of an anti-endothelin b receptor drug conjugate by inducing target expression in melanoma. Mol Cancer Ther 13(6):1599–1610
Fang D, Leishear K, Nguyen TK, Finko R, Cai K, Fukunaga M et al (2006) Defining the conditions for the generation of melanocytes from human embryonic stem cells. Stem Cells 24(7):1668–1677
Berger Y, Bernasconi CC, Juillerat-Jeanneret L (2006) Targeting the endothelin axis in human melanoma: combination of endothelin receptor antagonism and alkylating agents. Exp Biol Med 231(6):1111–1119
Spinella F, Rosanò L, Di CV, Decandia S, Nicotra MR, Natali PG et al (2007) Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cells. Cancer Res 67(4):1725–1734
Bagnato A (2004) Endothelin b receptor blockade inhibits dynamics of cell interactions and communications in melanoma cell progression. Cancer Res 64(4):1436–1443
Kefford R, Beith MN, Hazel GAV, Millward M, Trotter JM, Wyld DK et al (2007) A phase ii study of bosentan, a dual endothelin receptor antagonist, as monotherapy in patients with stage iv metastatic melanoma. Invest New Drugs 25(3):247–252
Smith MP, Rowling EJ, Miskolczi Z, Ferguson J, Spoerri L, Haass NK et al (2017) Targeting endothelin receptor signalling overcomes heterogeneity driven therapy failure. EMBO Mol Med 9(8):1011–1029
Funding
The study was supported by a Special Fund for the Aid Program for Innovation Team in Shanxi Agricultural University (Grant No. CXTD201201).
Author information
Authors and Affiliations
Contributions
Each author participated sufficiently in the study: LJ is responsible for implementation of this research and drafting of the manuscript. Professor CD and Professor RF are responsible for conception and design. SQ is responsible for data analysis. Each author has approved the final version of the manuscript. Doctor HH (Taigu Chengshi Hospital) participate in the collection of experimental materials. Doctor Ying Niu (Taigu Chengshi Hospital) participate in the data analysis.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, L., Dong, C., Fan, R. et al. A high affinity nanobody against endothelin receptor type B: a new approach to the treatment of melanoma. Mol Biol Rep 47, 2137–2147 (2020). https://doi.org/10.1007/s11033-020-05313-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05313-w