
Abstract
The Italian National Reference Center for Echinococcosis (CeNRE, Sassari, Italy) set up a diagnostic protocol of “one-step-PCR” useful for the detection of E. granulosus sensu stricto (E.g.s.s.) and the identification of its genotype (G1–G3). The purpose of this work was to perform the validation of the “PCR E.g.s.s.” method. The procedures were performed employing the criteria of the World Organization for Animal Health as well as of the Italian Accreditation Body (ACCREDIA) based on the Regulation UNI CEI EN ISO/IEC 17025. Positive DNA samples belonging to E. granulosus, E. ortleppi, E. multilocularis, E. canadensis species were used for the experiments. Analytical specificity evidenced primer pairs Cal (Calreticulin l gene of 1001 bp) with an specificity higher respect to Ef1 (Elongation-Factor 1 Alpha gene of 706 bp) and NAD (Dehydrogenase-subunit 1 gene of 219 bp). The analytical sensitivity presented the capability to detect a very low amount of parasite DNA corresponding to a concentration of 12.5 pg/µl; accuracy and precision related to the operator performance, along with repeatability and reproducibility, evidenced high concordance among results and demonstrated an excellent κ values of Cohen. According to the good performance related to the evaluated parameters, the method “PCR E.g.s.s.” was suitable for the validation procedure, and consequently, to be undergone to the accreditation process. In conclusion, the results demonstrated an elevated robustness and reliable features of the “PCR E.g.s.s.” able to perform a rapid diagnosis of E. granulosus in only “one step”, hence, it is likely to avoid the sequencing step.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cystic Echinococcosis (CE) is a neglected zoonotic disease caused by the larval stage of the tapeworm Echinococcus granulosus sensu lato (s.l.) [1, 2]. CE is considered a significant global public health problem with a worldwide distribution and an economic issue [3,4,5]. The World Health Organization [6] has estimated the CE as one of the 17 Neglected Tropical Diseases with global distribution and it is listed among the most severe parasitic diseases in humans, ranking second in the list of food-borne parasites [7, 8]. CE was evaluated at the first Joint Expert Committee on Zoonoses, hence, since 1950 this parasitosis has been recognised as a worldwide public health problem [8].
Echinococcus granulosus s.l. displays a significant variability in terms of genetic diversity, infectivity to humans, pathogenicity, antigenicity, host range, morphology and developing rate [1, 9,10,11]. Molecular characterization studies have already identified several species and genotypes belonging to E. granulosus s.l [12,13,14,15,16,17,18]. Further studies based on mitochondrial genomes and nuclear marker genes discriminated E. granulosus complex into five different species. According to taxonomy, the groups can be classified in different species, as described. E. granulosus sensu stricto (s.s.) (genotypes G1–G3), E. equinus (G4), E. ortleppi (G5), E. canadensis (G6–G8 and G10) and E. felidis [2, 14, 16]. These species are geographically and epidemiologically separated. According to the geographical distribution, E. granulosus s.s. G1–G3, representing the principal causative agent of CE in humans, has a high prevalence in all the Mediterranean basin [1, 4].
The parasite life cycle typically includes intermediate hosts, usually ungulates, and definitive hosts, domestic and wild canids, harboring the adult worm. Intermediate hosts are infected by ingesting eggs released in the feces by definitive hosts. The hydatid cyst, the metacestode larval stage consisting of a fluid-filled cyst, could develop in different organs, mainly liver and/or lungs, and may remain asymptomatic for years [19, 20]. Occasionally humans [3, 21,22,23,24] and rarely other species, as carnivores [25], could act as aberrant hosts, though representing an epidemiological dead end.
Currently diagnosis of CE in animal intermediate hosts is mainly performed at abbatoirs, following the veterinary inspection that permits the detection of hydatid cysts. On the other hand, diagnosis on human beings is mainly performed by means of imaging techniques. These exams permit to establish the localization and the classification of the cyst. According to the guidelines of WHO-IWGE, based on stage-specific cystic, it is possible to distinguish active (CE1, CE2), transitional (CE3a and CE3b) and inactive (CE4 and CE5) cysts [26]. In contrast, differential diagnosis involves infectious lesions and tumors [27].
Imaging techniques are supported by serological analysis, based on the detection of IgG antibodies anti-E. granulosus. Enzyme-linked immunosorbent assays (ELISA) and indirect hemagglutination assay (IHA) are the main immunological tests for CE patients [28]. Diagnosis and follow-up of CE is till now a matter of concern due to several difficulties related with the available additional techniques used to support the findings of imaging techniques [28]. Nevertheless, several disadvantages have been detected, including low specificity and sensitivity by using both ELISA and IHA [29,30,31,32,33,34]. Moreover, these techniques might give rise to cross-reactivity and, consequently, to false positive reaction for sera from patients infected with other pathogens different from Echinococcus [20, 35, 36]. Instead, Immunoblotting technique has higher specificity and sensitivity than ELISA and IHA. For this reason the former is usually used as a confirmatory test [28]. On the other hand, cross-reactivity and false positive reaction has also been detected by analysing sera from animals affected by other cestodes than Echinococcus by means of ELISA and IHA [37, 38]. False negative and a number of different factors, as additional difficulties due to other parasite infection common in sheep, could also be presented [20, 35, 36].
In addition, molecular diagnosis of DNA-based analyses is necessary, not only to confirm the diagnosis, but also to have a complete diagnostic picture of CE patients [28]. PCR is the foremost molecular analysis presenting a high specificity and sensitivity that could determine the actual infection status of a subject by detection of DNA from a pathogen agent.
The aim of our study is to validate a new diagnostic protocol of “one-step-PCR” useful for the identification of the agent, the detection of the genus and the specie of E. granulosus, also for the identification of its genotype (G1–G3). Our protocol was performed according to the Italian Accreditation Body (ACCREDIA) procedure, based on the Regulation UNI CEI EN ISO/IEC 17025 [39] and OIE (World Organisation for Animal Health) guidelines [40]. The OIE Terrestrial Manual supplies recommendations on the choice of test methods, provides detailed guidance about tests selection, optimisation, standardisation, and validation. In addition, it involves development and optimisation of several tests that aim to detect antibody, antigen or nucleic acid, statistical approaches to test validation, measurement uncertainty, selection and use of reference samples and panels, validation of diagnostic assays and comparability trials after modification in a test method already validated. Among the different criteria for assay development and validation suggested by OIE, we tested the Method performance by evaluating specifically analytical sensitivity (ASe), analytical specificity (ASp), moreover, operator performance such as repeatability and reproducibility.
Materials and methods
Samples
An operator external to the network prepared the blind sets of samples used in the study for the method and operator performances determination (Table 1). Positive DNA samples were employed for the PCR validation. They comprised, as different Echinococcus species (n = 3), including E. ortleppi, E. multilocularis and E. canadensis, kindly provided by the Italian Istituto Superiore di Sanità (ISS) ready to use, as E. granulosus s.s. (n = 3) extracted during diagnostic routine procedures in our laboratories. DNA negative control samples were represented by DNA (n = 1) extracted during diagnostic routine procedures from an Echinococcus healthy human lymph node and sterile MilliQ water (n = 1).
DNA from E. granulosus s.s. positive samples, extracted during diagnostic routine procedures in our laboratories, were firstly extracted, amplified and sequenced to determine the species and the genotype before to be undergone to the method performance evaluation.
DNA extraction from parasite tissue
DNA positive samples were obtained after hydatid fluid and germinal layer isolation from cystic lesions caused by E. granulosus found in ovine and bovine species, in liver and lung (Table 1). Samples were collected during slaughtering procedures in an abattoir. Germinal layer was placed in a Petri dish together with hydatid fluid and gently scraped to remove protoscoleces. Parasite material was washed twice in PBS (10 min at 1000 g), the surnatant was discarded and the pellet was aliquoted (25 mg) and stocked at − 80 °C pending DNA extraction. DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions.
DNA amplification
DNA was amplified by PCR using specific primers [12] slightly modified [41] (F 5′-TTTTTTGGCCATCCTGAGGTTTAT-3′ e R 5′-TAACGACATAACATAATGAAAATG-3′) that amplified the gene sequence of the enzyme Cytochrome Oxidase subunit I (COX1). An amount of 1 µl from a solution of 25 pmol/µl 1.6 µM (1 pmol/µl final concentration) of the COX1 primers was used for the amplification of DNA from E. granulosus s.s., together with 1 µl of DNA (3 ng/µl final concentration) plus 4 µl milliQ water RNAse-free, 12.5 µl (1X final concentration) of 2x QuantiTect Probe PCR Master Mix (QIAGEN), finally 5.5 µl of milliQ water RNAse-free were added. The amplification cycle was performed with the following protocol, 1 cycle of 15 min at 95 °C, next 40 cycles of 1 min at 94 °C, 30 s at 58 °C, 1 min at 72 °C and 1 cycles of 5 min at 72 °C, finally, the reaction was stopped at 15 °C. The amplicons were stored at 4 °C until they were electrophoresed.
DNA sequencing
The sequence of the diagnostic product of the COX1 primers, resulted of about 446 bp in size [12, 41], was soon determined by Sanger method, after the purification of positive amplicons by means of QIAquick PCR Purification Kit (QIAGEN), in a 20 µl reaction volume containing: 4 µl of BigDye Terminator, 2 µl of BigDye Terminator 5X sequencing buffer, 2 µl of H2O, 1 µl of each primer (Forward or Reverse), finally 10 µl of DNA. The amplification cycling profile was performed by 25 cycles, each consisting of 10 s at 96 °C, 5 s at 57 °C, and 2 min at 60 °C, finally at 4 °C until the samples were purified by chromatographic resin (Sephadex G-50 DNA grade F). Sequencing step was performed by an electrophoretic run in automatic capillary sequencer 3500 (Genetic Analyzer, Applied Biosystems).
Method performance
All samples were analysed blindly by a total of 5 operators belonging to different laboratories and equally involved in validation of the “PCR E.g.s.s.”.
Analytic specificity
The Analytic Specificity (ASp) evaluation was performed by two different operators that compared three PCR tests amplifying the set of 1 negative and 4 positive samples belonging to different species (E. ortleppi, E. multilocularis, E. canadensis, and E. granulosus s.s.). Amplifications were performed by using three different primer pairs for the target sequence of the amplicon for E. granulosus s.s. belonging to G1–G3 genotype. The species and genotype were previously determined by the amplification with COX1 primer pairs [41] and sequencing, as described above. The primer pairs compared corresponded to Ef1a (F 5′: TCCTAACATGCCTTGGTAT-3′ R5′: GTTACAGCCTTGATCACG-3′), that amplified the sequence of Elongation-Factor 1 Alpha of 706 bp [42], Cal (F5′: CAATTTACGGTAAAGCAT-3′ R5′: CCTCATCTCCACTCTCT-3′), that amplified the gene Calreticulin l of 1001 bp, [42], and NAD1 (F5′: GGTTTTATCGGTATGTTGGTGTTAGTG-3′-R5′: CATTTCTTGAAG TTAACAGCATCACG-3′), that amplified the amplicon NADH-dehydrogenase-subunit 1 of 219 bp [43].
PCR amplification protocol was carried out according to References method [42] with minor modification. Briefly, under the following conditions: an initial denaturation step at 95 °C for 15 min followed by 35 cycles of 94 °C for 1 min, 56 °C for 30 s and 72 °C for 1 min and a final extension step at 72 °C of 5 min. Further experiments were performed on different temperature by increasing the annealing step from 56 to 58 °C. After that, each assay was performed using an annealing temperature corresponding to 58 °C.
Analytic sensitivity
Analytic sensitivity (ASe) was determined by two operators calculating the limit of detection (LOD). Two-fold serial dilutions of DNA extracted from E. granulosus were prepared starting from a concentration of 10 ng/µl, then each dilution point was amplified by the PCR E.g.s.s.
Reproducibility
The parameter of reproducibility was assessed by five different operators belonging to two different laboratories. The PCR E.g.s.s. method, with the primers pairs Cal was performed on set samples described in Table 1 by using two sets of different instruments: laminar flow hoods, thermal cycles, micropipettes, electrophoresis powers, electrophoresis chambers and transilluminators. Value of concordance by comparing the expected results and those achieved from the operators was measured by κ coefficient.
Operator performance
Repeatability accuracy and precision
The performance of five operators was evaluated following the amplification in triplicate of a blind set of five samples, that were expected to be three positive to E. granulosus and two negative, as described in Table 1. Samples were analysed by the PCR E.g.s.s. by using the primers pairs Cal, according to the protocol described above. Following a training, subsequently a proficiency test, firstly the accuracy, way in which an observed value of a quantity agrees with the true value, than the precision, measure of the extent to which repeated observations conform, finally, the inter-operator Repeatability were assessed. To determine the concordance degree among the inter-assay results and those obtained from each operator we used Cohen k coefficient (κ), calculated according to Byrt table.
Results
Sequencing of the DNAs extracted from cystic lesions belonging to ovine lungs and bovine liver (Table 1), carried out after the amplification, demonstrated its origin from E. granulosus G1.
Method performance
Analytic specificity
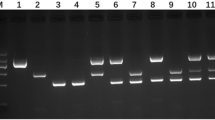
The ASp determined by means of PCRs E.g.s.s., using the three primer pairs Ef1a, Cal and NAD1 by two different operators on the set of 5 blinded samples, resulted higher for the primers Cal (Fig. 1), since they specifically amplified a DNA sequence only for E. granulosus s.s. belonging to G1–G3 genotype. As it is possible to observe in the Fig. 1, the comparison of results did not show a good specificity for the other primers. Species respectively of E. multilocularis and of E. canadensis were amplified in the PCRs E.g.s.s. carried out with Ef1a and NAD1. Further experiments showed that temperature of annealling presented best results for specificity at 58 °C than 56 °C, with a more definite band, and a reduced background.

In the panels A and B are shown respectively the results obtained by each of the two operators concerning the Analytical specificity (ASp) PCR performed on DNA extracted by different species of Echinococcus and amplified by three different primer pairs Egf1a, Nad1 and Cal: the [M] Molecular Marker [DNA Molecular Weight Marker VIII (between 19 and 1114 bp)—Sigma-Aldrich], [lane 1] E. granulosus s.s., [lane 2] E. ortleppi, [lane 3] E. multilocularis, [lane 4] E. canadensis, for Egf1a and Nad1 [lane 5] negative control, [lane 6] positive control, for Cal [lane 5] positive control
Analytic sensitivity
Limit of detection (LOD), determined to evaluate the ASe of the PCR E.g.s.s. by amplifying the two-fold serial dilutions of E. granulosus DNA, corresponded to a concentration of 12.5 pg/µl, since it is still possible to distinguish a signal related to the specific band of 1001 bp (Fig. 2).

PCR performed to measure the LOD for the analytical sensitivity (ASe) assay. [Lane 1] 10 ng/µl, [lane 2] 5 ng/µl, [lane 3] 2.5 ng/µl, [lane 4] 1 ng/µl, [lane 5] 0.5 ng/µl, [lane 6] 0.25 ng/µl, [lane 7] 0.10 ng/µl, [lane 8] 0.075 ng/µl, [lane 9] 0.050 ng/µl, [lane 10] 0.025 ng/µl, [lane 11] 0.0125 ng/µl, [lane 12] 0.0062 ng/µl. The lower concentration of DNA amplified by the PCR E.g.s.s. corresponded to 0.0125 ng/µl [lane 11]
Reproducibility
The assessment of the reproducibility performed by means of the PCR E.g.s.s., by 5 operators belonging to two different laboratories, using different set of instruments, displayed a high agreement value of κ. It was demonstrated by the comparison of the gels obtained by each technician, that didn’t displayed any relevant difference.
Operator performance
Repeatability accuracy and precision
Analysed DNA samples, described in Table 1, as expected, resulted three positive to E. granulosus and two negative. The results from the triplicate analysis performed by the entire assay procedure of the PCR E.g.s.s. presented high concordance. Inter-operator repeatability and Accuracy related to the five operators belonging to different laboratories showed an excellent κ coefficient value (1.00) (Table 2), according to the Byrt evaluation scale, between the results expected and those achieved from each operator. In addition, the precision was confirm.
Discussion
The PCR, named PCR E.g.s.s., set up in our laboratories, is direct to the detection of E. granulosus s.s., through the amplification of a gene named Cal, that amplified the gene Calreticulin l of 1001 bp. This amplicon is distinctive of genus, species and genotype of E. granulosus G1–G3. This diagnostic technique is capable to avoid the sequencing of the DNA sample, consequently it is suitable for a “one step detection” of the pathogenic agent in the routine practice, as a simple and quick method for diagnosis in humans and animals.
Our trials were carried out on E. granulosus s.s. samples from CE affected ovine and bovine. However, PCR E.g.s.s. could be performed on different other species after detection of protoscolices DNA, for the diagnosis of E. granulosus s.s. G1–G3 genotype.
Although the amplification of the nuclear CAL gene consents to identify E. granulosus s.s. G1–G3 genotype, by this, and other genes, it is not possible to differentiate between these two genotypes [44]. At the moment, there is no suitable method for the identification and differentiation among these genotypes G1 and G3, apart from sequencing of almost the entire mitochondrial genome or amplifying same mitochondrial markers genes [44]. Indeed, according to a recent study on the phylogenetic network the Nad5 gene fragment (680 bp) permitted to clear distinguish between the genotypes G1 and G3 instead, other mitochondrial marker genes, such as COX1 (366 bp) and/or NAD1 (471 bp) [12, 13], are able to distinguish between G1 and G3 genotypes when associated to Nad5 [44]. To develop and validate a test is a procedure that lead to improve the performance of the methods and consists on evaluation of the feasibility, development and standardization, determination of the characteristics and a continuous monitoring maintenance and improvement of the assay [40]. According to the OIE guidelines, a lack of standardization of diagnostic methods has been carried a disagreement among the results obtained by different laboratories and a lower sensitivity and specificity [40]. Here we describe the determination of several parameters for the validation of the new assay, the PCR E.g.s.s., for the diagnosis of E. granulosus G1–G3.
ASp evaluation by the comparison of the three primer pairs Ef1a, Cal and NAD1, showed the best results for the primers Cal. On the other hand, the PCRs E.g.s.s. carried out by means of Ef1a and NAD1 did not show a good specificity. As it is demonstrated by the very weak band for E. multilocularis, other than that for E. granulosus s.s., amplified by Ef1a primer pairs, being the lower specificity reported by primer pairs NAD1 that amplified for all the DNA species tested, apart from E. ortleppi. Moreover, Analytic Specificity was lightly increased after utilising a higher temperature of annealing. The approach to detect the ASe by analysing triplicate series of DNA dilutions by PCR E.g.s.s., evidenced the capability of the techniques to amplify and detect a very low amounts of parasite DNA. Our results showed that the lower amount of DNA detected still presenting a signal had a concentration of 12.5 pg/µl (Fig. 2). This amount confirmed the high sensitivity of our method of amplification, even respect to that reported in previous study, in which the Authors by using different primers detect a lower quantity of DNA [45].
According to these results the assay represents an ideal tool for the detection of low levels of parasite DNA. Based on the high sensitivity of the PCR E.g.s.s., performed on the DNA extracted by protoscolices, we believe that it is possible to detect a positive signal not only in simple biological matrices containing abundant parasitic material such as, worms, germinal layer, but also complex matrices [46, 47]. Heterogeneous samples, for instance, soil, vegetables, host intestinal mucosa, faeces and eggs due to the presence of inhibitors and scarce parasitic material shows serious difficulties to detect DNA material [28, 48,49,50,51]. High ASp and ASe demonstrated that PCR E.g.s.s. is a suitable analysis developed to detect E. granulosus s.s., and to perform a differential diagnosis of E. granulosus sensu lato [s.l.] and E. multilocularis. Moreover, this technique is very useful for the identification of species and genotypes, specifically to discriminate G1–G3, endemic in the Mediterranean basin [52], with no necessity for DNA sequence analysis.
The method demonstrated a high capacity to remain unaffected by small, but deliberate, variations in method parameters as verified by Reproducibility evaluation. The high agreement value and consistency of results using different series of instrument by several operators from different laboratories has been assessed by the Excellent value of the coefficient κ. The study displayed that the analytical procedure showed a high robustness, as measured during the development of the assay method.
According to the Regulation UNI CEI EN ISO/IEC 17043 [53], our results regarding the performance of operator allows the habilitation of the five operators, as demonstrated by the Excellent value of the coefficient κ that has been displayed by the inter-operator repeatability (Table 2). Moreover, the evaluation measure of the intra-assay precision for the five operators did not show any relevant variation on the execution of the assay in triplicate, as well as the conformity of the precision measure.
The use of reliable measurement instruments and validated is of critical implication for the analysis of results and the clinical practice. Hence, it is crucial to estimate and compare the repeatability and reproducibility of such assay, mostly earlier than they become broadly applied in clinical diagnosis as screening tool.
Biomolecular tests could be used to confirm the diagnosis, in addition, they can be very useful tools for the identification of Echinococcus spp. and genotypes [28].
The global annual costs related to CE, according with WHO, are estimated to be US $ 3 billion [6, 7]. In addition, the average incidence rates in Italy is correlated to a cost of more than Euro 53 million, according to the Hospital Discharge Records (HDRs) in 2001–2014, and more than 3000 disability-adjusted life years (DAYLs). Nevertheless, the highest annual average of costs and DAYLs were observed in Italian Islands (Sardinia and Sicily) [54]. Development and validation of a suitable and quick molecular diagnostic tools is significant not only for routine diagnostic testing of E. granulosus, but also for large scale detection on a wide range of samples and for epidemiological surveillance, particularly useful in endemic areas.
Moreover, DNA analysis is a suitable mass-screening, it can be performed to detect infections at intra vitam and post mortem, are safe for laboratory personnel and are cost-effective. In addition, PCR has increasingly been recognized as a complementary diagnostic tool for echinococcosis since several years [55, 56]. In conclusion, consistent with the conformity of the assessed parameters such as analytical and diagnostic performance, conformity of precision and the Excellent κ coefficient values obtained for operator performance, repeatability and reproducibility, the technique PCR E.g.s.s. demonstrated an elevated robustness and reliable features. As additional advantage, the PCR E.g.s.s. allows to perform a quicker diagnosis of E. granulosus G1–G3 in only “one step”, hence, it is likely to avoid the sequencing step.
References
Romig T, Ebi D, Wassermann M (2015) Taxonomy and molecular epidemiology of Echinococcus granulosus sensu lato. Vet Parasitol 213:76–84. https://doi.org/10.1016/j.vetpar.2015.07.035
Nakao M, Yanagida T, Konyaev S, Lavikainen A, Odnokurtsev VA, Zaikov VA, Ito A (2013) Mitochondrial phylogeny of the genus Echinococcus (Cestoda: Taeniidae) with emphasis on relationships among Echinococcus canadensis genotypes. Parasitology 140:1625–1636 https://doi.org/10.1017/S0031182013000565
Eckert J, Gemmell MA, Meslin F-X, Pawłowski ZS (2001) WHO/OIE manual on echinococcosis in humans and animals: a public health problem of global concern. World Organization for Animal Health, Paris, pp. 265
Alvarez Rojas CA, Romig T, Lightowlers MW (2014) Echinococcus granulosus sensu lato genotypes infecting humans-review of current knowledge. Int J Parasitol 44(1):9–18. https://doi.org/10.1016/j.ijpara.2013.08.008
Marcinkute A, Šarkunas M, Moks E, Saarma U, Jokelainen P, Bagrade G, Laivacuma S, Strupas K, Sokolovas V, Deplazes P (2015) Echinococcus infections in the Baltic region. Vet Parasitol 213:121–131. https://doi.org/10.1016/j.vetpar.2015.07.032
World Health Organization (2015) Investing to overcome the global impact of neglected tropical diseases: third WHO report on neglected tropical diseases. WHO, Geneva, pp. 95–99
FAO/WHO. Multicriteria-Based Ranking for Risk Management of Food-Borne Parasites (2014) Microbiological Risk Assessment Series (MRA) no 23. WHO Press Report of a Joint FAO/WHO Expert Meeting
Mableson HE, Okello A, Picozzi K, Welburn SC (2014) Neglected zoonotic diseases-the long and winding road to advocacy. PLoS Negl Trop Dis 8:e2800. https://doi.org/10.1371/journal.pntd.0002800
Eckert J, Thompson RC, Bucklar H, Bilger B, Deplazes P (2001) Efficacy evaluation of epsiprantel (Cestex) against Echinococcus mutilocularis in dogs and cats. Berl Munch Tierarztl Wochenschr 114(3–4):121–126
Thompson RC (2008) The taxonomy, phylogeny and transmission of Echinococcus. Exp Parasitol 119(4):439–446. https://doi.org/10.1016/j.exppara.2008.04.016
Gholami S, Irshadullah M, Mobedi I (2011) Rostellar hook morphology of larval Echinococcus granulosus isolates from the Indian buffalo and Iranian sheep, cattle and camel. J Helminthol 85(3):239–245. https://doi.org/10.1017/S0022149X10000520
Bowles J, Blair D, McManus DP (1992) Genetic variants within the genus Echinococcus identified by mitochondrial DNA sequencing. Mol Biochem Parasitol 54(2):165–173. https://doi.org/10.1016/0166-6851(92)90109-W
Bowles J, Blair D, McManus DP (1994) Molecular genetic characterization of the cervid strain (‘northern form’) of Echinococcus granulosus. Parasitology 109(Pt 2):215–221. https://doi.org/10.1017/S0031182000076332
Lavikainen A, Lehtinen MJ, Meri T, Hirvelä-Koski V, Meri S (2003) Molecular genetic characterization of the Fennoscandian cervid strain, a new genotypic group (G10) of Echinococcus granulosus. Parasitology 127(Pt 3):207–215
Nakao M, McManus DP, Schantz PM, Craig PS, Ito A (2007) A molecular phylogeny of the genus Echinococcus inferred from complete mitochondrial genomes. Parasitology 134(Pt 5):713–722. https://doi.org/10.1017/S0031182006001934 (Epub 2006 Dec 11)
Hüttner M, Nakao M, Wassermann T, Siefert L, Boomker JD, Dinkel A, Sako Y, Mackenstedt U, Romig T, Ito A (2008) Genetic characterization and phylogenetic position of Echinococcus felidis (Cestoda: Taeniidae) from the African lion. Int J Parasitol 38(7):861–868. https://doi.org/10.1016/j.ijpara.2007.10.013 (Epub 2007 Nov 4)
Saarma U, Jõgisalu I, Moks E, Varcasia A, Lavikainen A, Oksanen A, Simsek S, Andresiuk V, Denegri G, González LM, Ferrer E, Gárate T, Rinaldi L, Maravilla P (2009) A novel phylogeny for the genus Echinococcus, based on nuclear data, challenges relationships based on mitochondrial evidence. Parasitology 136(3):317–328. https://doi.org/10.1017/S0031182008005453
Knapp J, Nakao M, Yanagida T, Okamoto M, Saarma U, Lavikainen A, Ito A (2011) Phylogenetic relationships within Echinococcus and Taenia tapeworms (Cestoda: Taeniidae): an inference from nuclear protein-coding genes. Mol Phylogenet Evol 61(3):628–638. https://doi.org/10.1016/j.ympev.2011.07.022
Chiboub H, Boutayeb F, Wahbi S, El Yacoubi M, Ouazzani N, Hermas M (2001) Echinococcosis of the pelvic bone: four cases. Rev Chir Orthop Reparatrice Appar Mot 87(4):397–401
Zhang W, Wen H, Li J, Lin R, McManus DP (2012) Immunology and immunodiagnosis of cystic echinococcosis: an update. Clin Dev Immunol 2012:101895. https://doi.org/10.1155/2012/101895 (Epub 2011 Dec 25)
Moks E, Jõgisalu I, Saarma U, Talvik H, Järvis T, Valdmann H (2006) Helminthologic survey of the wolf (Canis lupus) in Estonia, with an emphasis on Echinococcus granulosus. J Wildl Dis 42(2):359–365. https://doi.org/10.7589/0090-3558-42.2.359
Moks E, Jõgisalu I, Valdmann H, Saarma U (2008) First report of Echinococcus granulosus G8 in Eurasia and a reappraisal of the phylogenetic relationships of ‘genotypes’G5-G10. Parasitology 135(5):647–654. https://doi.org/10.1017/S0031182008004198
Deplazes P, van Knapen F, Schweiger A, Overgaauw PA (2011) Role of pet dogs and cats in the transmission of helminthic zoonoses in Europe, with a focus on echinococcosis and toxocarosis. Vet Parasitol 182(1):41–53. https://doi.org/10.1016/j.vetpar.2011.07.014
Laurimaa L, Davison J, Süld K, Plumer L, Oja R, Moks E, Keis M, Hindrikson M, Kinkar L, Laurimäe T, Abner J, Remm J, Anijalg P, Saarma U (2015) First report of highly pathogenic Echinococcus granulosus genotype G1 in dogs in a European urban environment. Parasit Vectors 26(8):182. https://doi.org/10.1186/s13071-015-0796-3
Bonelli P, Masu G, Dei Giudici S, Pintus D, Peruzzu A, Piseddu T, Santucciu C, Cossu A, Demurtas N, Masala G (2018) Cystic echinococcosis in a domestic cat (Felis catus) in Italy. Parasite 25:25. https://doi.org/10.1051/parasite/2018027 (Epub 2018 May 4)
Brunetti E, Kern P, Vuitton DA, Writing Panel for the WHO-IWGE (2010) Expert consensus for the diagnosis and treatment of cystic and alveolar echinococcosis in humans. Acta Trop 114(1):1–16. https://doi.org/10.1016/j.actatropica.2009.11.001
Pawlowski Z, Eckert J, Vuitton DA, Ammann RW, Kern P, Craig P, Dar KF, De Rosa F, Filice C, Gottstein B, Grimm F, Macpherson N, Sato N, Todorov T, Uchino J, von Sinner W, Wen H (2001) Echinococcosis in humans: clinical aspects, diagnosis and treatment. In: Eckert J, Gemmel MA, Meslin F-X, Pawlowski ZS (eds) WHO/OIE manual on echinococcosis in humans and animals: a public health problem of global concern. World Organisation for Animal Health and World Health Organization, Paris, pp 20–71
Siles-Lucas M, Casulli A, Conraths FJ, Müller N (2017) Laboratory diagnosis of Echinococcus spp. in human patients and infected animals. Adv Parasitol Cap 3(96):159–257. https://doi.org/10.1016/bs.apar.2016.09.003
Zhang W, McManus DP (2006) Recent advances in the immunology and diagnosis of echinococcosis. FEMS Immunol Med Microbiol 47(1):24–41. https://doi.org/10.1111/j.1574-695X.2006.00060.x
Carmena D, Benito A, Eraso E (2006) Antigens for the immunodiagnosis of Echinococcus granulosus infection: an update. Acta Trop 98(1):74–86. https://doi.org/10.1016/j.actatropica.2006.02.002
Barnes TS, Deplazes P, Gottstein B, Jenkins DJ, Mathis A, Siles-Lucas M, Torgerson PR, Ziadinov I, Heath DD (2012) Challenges for diagnosis and control of cystic hydatid disease. Acta Trop 123(1):1–7. https://doi.org/10.1016/j.actatropica.2012.02.066
Sarkari B, Rezaei Z (2015) Immunodiagnosis of human hydatid disease: where do we stand? World J Methodol 5(4):185–195
Manzano-Román R, Sánchez-Ovejero C, Hernández-González A, Casulli A, Siles-Lucas M (2015) Serological diagnosis and follow-up of human cystic Echinococcosis: a new hope for the future? Biomed Res Int 2015:428205
Lissandrin R, Tamarozzi F, Piccoli L, Tinelli C, De Silvestri A, Mariconti M, Meroni V, Genco F, Brunetti E (2016) Factors influencing the serological response in hepatic Echinococcus granulosus infection. Am J Trop Med Hyg 94(1):166–171
Mohammadzadeh T, Sako Y, Sadjjadi SM, Sarkari B, Ito A (2012) Comparison of the usefulness of hydatid cyst fluid, native antigen B and recombinant antigenB8/1 for serological diagnosis of cystic echinococcosis. Trans R Soc Trop Med Hyg 106(6):371–375
Tamarozzi F, Sako Y, Ito A, Piccoli L, Grisolìa A, Itoh S, Gatti S, Meroni V, Genco F, Brunetti E (2013) Recombinant AgB8/1 ELISA test vs. commercially available IgG ELISA test in the diagnosis of cystic echinococcosis. Parasite Immunol 35(12):433–440
McManus DP (2015) Immunodiagnosis of sheep infections with Echinococcus granulosus: in 35 years where have we come? Parasite Immunol 36(3):125–130
Craig P, Mastin A, van Kesteren F, Boufana B (2015) Echinococcus granulosus: epidemiology and state-of-the-art of diagnostics in animals. Vet Parasitol 213(3–4):132–148
UNI CEI EN ISO/IEC 17025:2018 (2018) General requirements for the competence of testing and calibration laboratories. ISO, Geneva
OIE - Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (2018) Volumes 1 and 2 ISBN 978-92-9044-878-5. http://www.oie.int/standard-setting/terrestrial-manual/access-online/
Bart JM, Morariu S, Knapp J, Ilie MS, Pitulescu M, Anghel A, Cosoroaba I, Piarroux R (2006) Genetic typing of Echinococcus granulosus in Romania. Parasitol Res 98(2):130–137
Boubaker G, Macchiaroli N, Prada L, Cucher MA, Rosenzvit MC, Ziadinov I, Deplazes P, Saarma U, Babba H, Gottstein B, Spiliotis M (2017) A Multiplex PCR for the simultaneous detection and genotyping of the Echinococcus granulosus complex. PLoS Negl Trop Dis 7(1):e2017. https://doi.org/10.1371/journal.pntd.0002017 (Epub 2013 Jan 17)
Liu CN, Lou ZZ, Li L, Yan HB, Blair D, Lei MT, Cai JZ, Fan YL, Li JQ, Fu BQ, Yang YR, McManus DP, Jia WZ (2015) Discrimination between E. granulosus sensu stricto, E. multilocularis and E. shiquicus using a multiplex PCR assay. PLoS Negl Trop Dis 9(9):e0004084. https://doi.org/10.1371/journal.pntd.0004084 (eCollection 2015 Sep)
Kinkar L, Laurimäe T, Acosta-Jamett G, Andresiuk V, Balkaya I, Casulli A, Gasser RB, González LM7, Haag KL, Zait H, Irshadullah M, Jabbar A, Jenkins DJ, Manfredi MT, Mirhendi H, Rostami-Nejad MS, Pierangeli NB, Ponce-Gordo F, Rehbein S, Sharbatkhori M, Kia EB, Simsek S, Soriano SV, Sprong H, Šnábel V, Umhang G, Varcasia A, Saarma U, Oudni-M’rad M (2018) Distinguishing Echinococcus granulosus sensu stricto genotypes G1 and G3 with confidence: a practical guide. Infect Genet Evol 64:178–184. https://doi.org/10.1016/j.meegid.2018.06.026
Dinkel A, Njoroge EM, Zimmermann A, Wälz M, Zeyhle E, Elmahdi IE, Mackenstedt U, Romig T (2004) A PCR system for detection of species and genotypes of the Echinococcus granulosus-complex, with reference to the epidemiological situation in eastern Africa. Int J Parasitol 34(5):645–653
Trachsel D, Deplazes P, Mathis A (2007) Identification of taeniid eggs in the faeces from carnivores based on multiplex PCR using targets in mitochondrial DNA. Parasitology 134(Pt 6):911–920 (Epub 2007 Feb 9)
Barazesh A, Sarkari B, Ebrahimi S, Hami M (2018) DNA extraction from hydatid cyst protoscolices: comparison of five different methods. Vet World 11(2):231–234. https://doi.org/10.14202/vetworld.2018.231-234 (Epub 2018 Feb 22)
Ito A (2002) Serologic and molecular diagnosis of zoonotic larval cestode infections. Parasitol Int 51(3):221–235
Abbasi I, Branzburg A, Campos-Ponce M, Abdel Hafez SK, Raoul F, Craig PS, Hamburger J (2003) Copro-diagnosis of Echinococcus granulosus infection in dogs by amplification of a newly identified repeat DNA sequence. Am J Trop Med Hyg 69:324–330
Stefanić S, Shaikenov BS, Deplazes P, Dinkel A, Torgerson PR, Mathis A (2004) Polymerase chain reaction for detection of patent infections of Echinococcus granulosus (“sheep strain”) in naturally infected dogs. Parasitol Res 92(4):347–351 (Epub 2004 Jan 16)
Boufana BS, Campos Ponce M, Naidich A, Buishi I, Lahmar S, Zeyle E, Jenkins DJ, Combes B, Wen H, Xiao M, Nakao M, Ito A, Qiu J, Craig PS (2008) Evaluation of three PCR Assay for the identification of the sheep strain (Genotype 1) of the Echinococcus granulosus in canid feces and parasite tissue. Am J Trop Med Hyg 78(5):777–783
Deplazes P, Rinaldi L, Alvarez Rojas CA, Torgerson PR, Harandi MF, Romig T, Antolova D, Schurer JM, Lahmar S, Cringoli G, Magambo J, Thompson RC, Jenkins EJ (2017) Global distribution of alveolar and cystic echinococcosis. Adv Parasitol 95:315–493 (Epub Jan 20. Review)
UNI CEI EN ISO/IEC 17043:2010 (2010) Conformity assessment—general requirements for proficiency testing. ISO/IEC, Switzerland
Piseddu T, Brundu D, Stegel G, Loi F, Rolesu S, Masu G, Ledda S, Masala G (2017) The disease burden of human cystic echinococcosis based on HDRs from 2001 to 2014 in Italy. PLoS Negl Trop Dis 11(7):e0005771. https://doi.org/10.1371/journal.pntd.0005771 (eCollection 2017 Jul)
Ito A, Okamoto M, Ishiguro T, Ma L, Suzuki H, Yasui A, Shigeta H, Matsuura T, Hosokawa T, Chai JJ (1998) Short report: an imported case of cystic echinococcosis in Japan diagnosed by imaging and serology with confirmation of Echinococcus granulosus-specific DNA sequences. Am J Trop Med Hyg 58(6):790–792
Myjak P, Nahorski W, Pietkiewicz H, von Nickisch-Rosenegk M, Stolarczyk J, Kacprzak E, Felczak-Korzybska I, Szostakowska B, Lucius R (2003) Molecular confirmation of human alveolar echinococcosis in Poland. Clin Infect Dis 37(8):e121–e125 (Epub 2003 Sep 25. Office International des Epizooties 2003)
Acknowledgements
The Authors acknowledge Dr Edoardo Pozio [European Union Reference Laboratory for Parasites, Department of Infectious Diseases, Istituto Superiore di Sanità (ISS), Rome].
Funding
This work was partially funded by Fondazione di Sardegna (https://www.fondazionedisardegna.it/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors has a financial or other relationship with other people or organizations that may inappropriately influence this work.
Ethical approval
All procedures performed in the study involving human biological material were in accordance with the ethical standards of the institutional and/or national ethic committee. A written informed consent was obtained from patients at the time of hospitalization. The Istituto Zooprofilattico Sperimentale of Sardinia has been authorized to analysed human samples by the ethics committee of the Local Health Authority of Sassari (Comitato di Bioetica, ASL N. 1, Sassari - Prot n° 1136), following the request of the National Health Service doctors, since the 26th of March 2013.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Santucciu, C., Masu, G., Mura, A. et al. Validation of a one-step PCR assay for the molecular identification of Echinococcus granulosus sensu stricto G1–G3 genotype. Mol Biol Rep 46, 1747–1755 (2019). https://doi.org/10.1007/s11033-019-04625-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-04625-w