
Abstract
Lamellar ichthyosis (LI, MIM# 242300) is a severe autosomal recessive genodermatosis present at birth in the form of collodion membrane covering the neonate. Mutations in the TGM1 gene encoding transglutaminase-1 are a major cause of LI. In this study molecular analysis of two LI Tunisian patients revealed a common nonsense c.788G>A mutation in TGM1 gene. The identification of a cluster of LI pedigrees carrying the c.788G>A mutation in a specific area raises the question of the origin of this mutation from a common ancestor. We carried out a haplotype-based analysis by way of genotyping 4 microsatellite markers and 8 SNPs flanking and within the TGM1 gene spanning a region of 6 Mb. Haplotype reconstruction from genotypes of all members of the affected pedigrees indicated that all carriers for the mutation c.788G>A harbored the same haplotype, indicating common ancestor. The finding of a founder effect in a rare disease is essential for the genetic diagnosis and the genetic counselling of affected LI pedigrees in Tunisia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autosomal recessive congenital ichthyosis (ARCI) is a rare, clinically and genetically heterogeneous group of cornification diseases. It defines three clinical subtypes which include the spectrum of lamellar ichthyosis (LI; OMIM 242300), congenital ichthyosiform erythroderma (CIE; OMIM 242100) and recently harlequin ichthyosis (HI; OMIM 242500) [1]. The ARCI is characterized by epidermal scaling, often collodion membrane at birth, ectropion, eclabium, alopecia, palmar-plantar hyperkeratosis, hypohidrosis and/or variable erythema [2].
Over the last decade at least six different genes have been implicated in the etiology of ARCI, all of which encode proteins essential for the formation of the skin barrier: TGM1 [3, 4], ABCA12 [5, 6], two lipoxygenase genes (ALOXE3 and ALOX12B) [7, 8], ichthyin (NIPAL4) [9] and CYP4F2 homolog (FLJ39501) [10].
Lamellar ichthyosis (LI) constitutes a rare disease but its prevalence may increase in population which has a high consanguinity owing to fonder effect [11–13]. The most common cause of LI/CIE is inactivating mutations in the TGM1 gene, which encodes transglutaminase-1, an enzyme that crosslinks proteins in the cornified cell envelope [4, 14].
The TGM1 gene has 14,095 bp, including 2,454 bp of coding sequence, and spans 15 exons (GenBankNM_000359.2). It encodes for the transglutaminase-1 (TGase-1) enzyme, which has 817 amino acid residues and a molecular weight of ~89 kDa (GenBank NM_000359.2) [15, 16]. It is a catalytic membrane-bound enzyme involved in the formation of the epidermal cornified cell envelope, which acts as a mechanical barrier to protect against water loss and infectious agents [14]. Up to now, more than 130 TGM1 different pathogenic mutations have been reported in the literature associated with LI/CIE phenotypic spectrum in patients from diverse ethnic backgrounds [1]. TGM1 mutations include missense, nonsense, and splice site mutations. The majority are single-base changes; rarely, insertions or deletions are found.
This study presents detailed molecular analysis of lamellar ichthyosis in two Tunisian patients belonging to two consanguineous families. They present a common nonsense mutation c.788G>A in TGM1 gene. Since it is the only mutation described to be responsible for the LI in Tunisia, we confirmed a founder effect using polymorphic microsatellite markers.
Patients and methods
Two patients belonging to two Tunisian families were born to healthy consanguineous parents. Genealogical investigation was performed and excluded any close relation between both families. Five additional healthy siblings and the parents were also recruited. Informed consent was obtained from patients and control individuals in accordance with the ethics committee of Hedi Chaker Hospital (Sfax, Tunisia). The diagnosis of LI was made on the basis of clinical manifestation; a detailed medical and dermatologic history was obtained from the affected persons. They were examined more than one time over the period of 10 years so that potential changes in phenotype could be evaluated. Blood samples were collected from family members and healthy individuals. Genomic DNA was extracted from the whole blood following a standard phenol–chloroform method [17].
Mutation analysis for TGM1 gene
The 15 exons and flanking intron region of the TGM1 gene were tested for mutation in the ARCI patients by sequence analysis. Following DNA extraction, the coding regions and intron–exon boundaries of the TGM1 gene were amplified by polymerase chain reaction (PCR) using generated primers covering the entire coding region (Table 1). PCR was carried out on 50 μl volume samples, in a GeneAmp® PCR system 9700 (Applied Biosystems, Foster City, CA, USA). Each sample contains 100 ng of genomic DNA, 0.4 μM of each primer, 0.2 mM of dNTPs, 1 IU of Amplitaq and 10× PCR Buffer II in a final concentration of 1 × 1.5 mM of MgCl2. The mixture was denatured during 10 min at 95 °C and then followed by 35 cycles: denaturing at 95 °C for 45 s, annealing at 65/60 °C for 45 s and an extension at 72 °C for 45 s; with a final extension at 72 °C for 7 min. Thereafter, the purified amplicons were directly sequenced using a dye terminator cycle sequencing kit V1.1 with an ABI sequence analyser 3100 avant (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s recommendations.
Microsatellites and SNPs screening
For the haplotypic analyses, we screened four microsatellite markers D14S283, D14S275, D14S72 and D14S80. They occur in the 5′ and 3′ flanking region of the TGM1 gene and reported as highly polymorphic STR [12]. Sequences of the primers for genetic markers were obtained from UNISTS (http://www.ncbi.nlm.nih.gov/unists).
Fluorescent dye-labeled microsatellite markers were genotyped for all the family members. We used the True Allele PCR Premix (Applied Biosystems, Foster City, CA) for PCR reactions according to the manufacturer’s instructions. Fluorescently-labelled alleles were analysed on an ABI PRISM 3100-Avant automated Genetic Analyser (Applied Biosystems, Foster City, CA). Genotypes were determined using the GenScan™ software (Applied Biosystems, Foster City, CA). The haplotype co-segregating with the disease was derived from the segregation of markers within the two pedigrees.
Results
Clinical analysis of ARCI patients
We identified two unrelated Tunisian families with two LI patients (MAS and NC). The healthy parents were consanguineous and originated from south Tunisia. Clinical data indicated that the initial presentation was a congenital onset of the disease with a history of collodion presentation and heat intolerance. MAS is a 14-year old son, second child of healthy parents who are first cousins, was born at term. Since his birth he developed a severe lamellar ichthyosis. He had one brother died for unknown reasons. NC is a 21-year old girl was referred to the dermatology department at Hedi Chaker Hospital in Sfax for a severe lamellar ichthyosis since birth, she is the first child of healthy consanguineous parents.
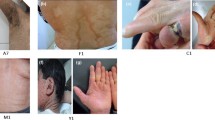
The two patients (MAS and NC) showed a normal intellect. No evidence of congenital hypothyroidism or sensitivity to gluten usually associated to LI Tunisian phenotype was noted. The main clinical features of the two patients are summarized in Table 2 (Table 2; Fig. 1).
One point mutation within TGM1 gene revealed by nucleotide sequencing
Mutation screening of the TGM1 coding region and intron–exon boundaries revealed the c.788G>A nonsense mutation (g.3139 G>A) in the two patients at homozygous state associated with a severe LI phenotype (Fig. 2). It occurs in exon 5 and results in the substitution of tryptophan (TGG) by a stop codon (TGA) at amino acid position 263 (p.W263X). Verification of transmission of this mutation was done for all families members. This variant was absent even in the heterozygous state by direct sequencing in 100 Tunisian control chromosomes.

NC patient’s hands showing palmar keratoderma and brachydactyly

Sequence chromatograms of the TGM1 gene in the region of the c.788G>A mutation, showing a control, carrier and mutant subject. Nucleotide variations are underlined
Founder effect study
According to the prevalence of the c.788G>A mutation among our group of patients and since the studied families sharing the same mutation c.788G>A are all originated from south Tunisia, we test the hypothesis that this mutation arose on a founder ancestor in the distant past. For these reason, we analyzed four polymorphic genetic markers and explored 8 SNPs encompassing approximately 6 Mb in and around the TGM1 gene to construct haplotypes for the mutant and normal TGM1 chromosomes in the families. Patient’s haplotypes showed that the c.788G>A mutation has been transmitted in conserved bloc of 2 Mb. A conserved haplotype encompassing the markers D14S275–D14S283 was found (Fig. 3). This result revealed that the c.788G>A mutation is not the result of separate mutation occurring independently in different individuals, but is the result of a one-time mutation occurring in a common ancestor of all autosomal recessive congenital lamellar ichthyosis families in Tunisia.

The pedigrees of the two Tunisian families showing the inheritance of the c.788G>A mutation. The same founder haplotype containing mutation is framed for the two patients. (del is a deletion of GGGGAGTCCAGGGCTCCCGGAG of rs41294726)
Discussion
In the present study, we report two Tunisian LI patients associated with nonsense c.788G>A mutation in the TGM1 gene predicting an amino acid substitution of tryptophan for a stop codon (p.W263X). It introduces a premature stop codon after nearly one-third of the TGase-1 polypeptide and leads to a truncated enzyme lacking the majority of the active site. To the best of our knowledge, this homozygous mutation is not prevalent worldwide, being found in only two Tunisian LI patients [14, 18, 19] and suggested to be a recurrent mutation in Tunisian patients affected by severe LI.
The clinical phenotypes observed in our patients correspond to those that have been described as lamellar ichthyosis in the literature [20]. It is noteworthy that the previous Tunisian patients described by Esposito et al. and Hennies et al. [18, 19], additionally suffered from congenital hypothyroidism or from severe gluten sensitive enteropathy respectively. Moreover, Oji et al. [21] report a relatively mildly affected girl from France with compound heterozygous genotype c.788G>A/c.919C>G and abnormal in vivo TGase-1 activity in affected skin areas.
Several reports suggest a link between thyroid diseases and ichthyosis. Firstly, in patients affected by both autoimmune thyroid disease and acquired ichthyosis, thyroid hormone replacement resulted in improvement of ichthyosis [20, 22]. Moreover, TGase-1 deficient LI was also found to be associated to congenital hypothyroidism in a child of consanguineous parents [23]. In such case, ichthyosis and hypothyroidism could be co-inherited, since the TGM1 and TSHR genes maps to the same chromosome localization (14q). This inter-familial variability, in clinical phenotype, encountered in LI patients sharing the same mutation could be due to differences in environmental factors or to the genetic background (influence of modifier gene) or both [24].
With regard to the nonsense nature of the c.788G>A mutation, two hypotheses may be evoked: either the translation of a truncated transglutaminase I protein lacking catalytic domain (core domain), or possibly the degradation of the nonsense mRNA that manifests as a complete absence of transglutaminase I protein. The most probable hypothesis is that the resulting nonsense mRNA is probably destroyed by the quality control surveillance mechanism NMD (nonsense-mediated mRNA decay) according to the PTC position rule (>50–55 nt upstream of the last exon–exon junction), which is detected in our patient at position c.788 [25].
Previously, using 32P labelled TGM1 probes, mRNA was shown to be absent from cultured keratinocytes taken from a patient homozygous for the nonsense mutation R127X [26]. Northern blotting of tissue taken from a skin biopsy and cultured keratinocytes from a patient homozygous for the truncating mutation c.1297delT showed absent TGase-1 mRNA in vitro [27]. NMD, which decreases the amount of cellular mRNA with premature termination codons (PTCs), could be the mechanism that explains these observations [25].
High carrier rates are usually attributed to a founder effect in endogamic and consanguineous populations [28]. They are usually evidenced by conservation of haplotypes with directly associated markers. Since the c.788G>A mutation has not been described elsewhere, it seems to be specific to the Tunisian population. According to such finding and in agreement with the prevalence of this mutation reported in our study and the previous ones [18, 19], we demonstrated that a clustering of g.3139 G>A mutation in two Tunisian families is due to a common founder ancestor. In fact, the analysis of 4 microsatellite markers and 8 SNPs flanking or within the TGM1 gene revealed a common haplotype carried by the patients sharing the same g.3139 G>A mutation. Hence, we suggest that this founder haplotype which ranges over 2 Mb could be the same for the two patients already reported and sharing the same mutation. In addition, the two families carrying the g.3139 G>A mutation are originated from southern Tunisia. Such rural communities are characterized by a high consanguinity and endogamy rates which supports our findings. Evidence for a founder effect of TGM1 gene has been also reported in American and Spanish populations [2, 29]. Given that it was the only founder mutation described so far in Tunisia, the c.788G>A mutation could be the only variation responsible for LI in Tunisian population.
In summary, the conservation of a single haplotype surrounding the c.788G>A mutation suggested that this ancestor haplotype has a single origin. The finding of a founder effect in a unique recurrent mutation in a rare disease is essential for the implementation of protocols for genetic diagnosis, for genetic counselling of affected pedigrees and is fundamental to search for new therapies.
References
Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, Coudiere P, DiGiovanna JJ, Elias P, Fischer J, Fleckman P, Gina M, Harper J, Hashimoto T, Hausser I, Hennies HC, Hohl D, Hovnanian A, Ishida-Yamamoto A, Jacyk WK, Leachman S, Leigh I, Mazereeuw-Hautier J, Milstone L, Morice-Picard F, Paller AS, Richard G, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H (2010) Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 63(4):607–641
Herman ML, Farasat S, Steinbach PJ, Wei MH, Toure O, Fleckman P, Blake P, Bale SJ, Toro JR (2009) Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: summary of mutations (including 23 novel) and modeling of TGase-1. Hum Mutat 30(4):537–547
Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SP, Ponec M, Bon A, Lautenschlager S, Schorderet DF, Hohl D (1995) Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science 267(5197):525–528
Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, Hashem N, Compton JG, Bale SJ (1995) Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet 9(3):279–283
Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, Sawamura D, Shimizu H (2005) Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 115(7):1777–1784
Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, Stephens KG, Sybert VP, Mallory SB, North BV, Witt DR, Sprecher E, Taylor AE, Ilchyshyn A, Kennedy CT, Goodyear H, Moss C, Paige D, Harper JI, Young BD, Leigh IM, Eady RA, O’Toole EA (2005) Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet 76(5):794–803
Jobard F, Lefèvre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Ozgüc M, Lathrop M, Prud’homme JF, Fischer J (2002) Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet 11(1):107–113
Eckl KM, Krieg P, Küster W, Traupe H, André F, Wittstruck N, Fürstenberger G, Hennies HC (2005) Mutation spectrum and functional analysis of epidermis-type lipoxygenases in patients with autosomal recessive congenital ichthyosis. Hum Mutat 26(4):351–361
Lefèvre C, Bouadjar B, Karaduman A, Jobard F, Saker S, Ozguc M, Lathrop M, Prud’homme JF, Fischer J (2004) Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet 13(20):2473–2482
Lefèvre C, Bouadjar B, Ferrand V, Tadini G, Mégarbané A, Lathrop M, Prud’homme JF, Fischer J (2006) Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet 15(5):767–776
Fachal L, Rodríguez-Pazos L, Ginarte M, Toribio J, Salas A, Vega A (2012) Multiple local and recent founder effects of TGM1 in Spanish families. PLoS One 7(4):e33580
Rodríguez-Pazos L, Ginarte M, Fachal L, Toribio J, Carracedo A, Vega A (2011) Analysis of TGM1, ALOX12B, ALOXE3, NIPAL4 and CYP4F22 in autosomal recessive congenital ichthyosis from Galicia (NW Spain): evidence of founder effects. Br J Dermatol 165(4):906–911
Pigg M, Gedde-Dahl T Jr, Cox D, Hausser I, Anton-Lamprecht I, Dahl N (1998) Strong founder effect for a transglutaminase 1 gene mutation in lamellar ichthyosis and congenital ichthyosiform erythroderma from Norway. Eur J Hum Genet 6(6):589–596
Farasat S, Wei MH, Herman M, Liewehr DJ, Steinberg SM, Bale SJ, Fleckman P, Toro JR (2009) Novel transglutaminase-1 mutations and genotype-phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J Med Genet 46(2):103–111
Kim HC, Idler WW, Kim IG, Han JH, Chung SI, Steinert PM (1991) The complete amino acid sequence of the human transglutaminase K enzyme deduced from the nucleic acid sequences of cDNA clones. J Biol Chem 266(1):536–539
Kim IG, McBride OW, Wang M, Kim SY, Idler WW, Steinert PM (1992) Structure and organization of the human transglutaminase 1 gene. J Biol Chem 267(11):7710–7717
Kawasaki E, Erlich H (1990) Polymerase chain reaction and analysis of cancer cell markers. J Natl Cancer Inst 82(10):806–807
Hennies HC, Raghunath M, Wiebe V, Vogel M, Velten F, Traupe H, Reis A (1998) Genetic and immunohistochemical detection of mutations inactivating the keratinocyte transglutaminase in patients with lamellar ichthyosis. Hum Genet 102(3):314–318
Esposito G, De Falco F, Brazzelli V, Montanari L, Larizza D, Salvatore F (2009) Autosomal recessive congenital ichthyosis and congenital hypothyroidism in a Tunisian patient with a nonsense mutation in TGM1. J Dermatol Sci 55(2):128–130
Kurtoğlu S, Caksen H, Erdoğan R, Kisaarslan AF (1998) Collodion baby concomitant with congenital hypothyroidism: a patient report and review of the literature. J Pediatr Endocrinol Metab 11(4):569–573
Oji V, Hautier JM, Ahvazi B, Hausser I, Aufenvenne K, Walker T, Seller N, Steijlen PM, Küster W, Hovnanian A, Hennies HC, Traupe H (2006) Bathing suit ichthyosis is caused by transglutaminase-1 deficiency: evidence for a temperature-sensitive phenotype. Hum Mol Genet 15(21):3083–3097
Brazzelli V, Prestinari F, Barbagallo T, Bellani E, Calcaterra V, Larizza D, Lauriola MM, Borroni G (2005) Acquired ichthyosis and hypertrichosis due to autoimmune thyroiditis: therapeutic response to thyroxine replacement. Pediatr Dermatol 22(5):447–449
Small K, Ginsburg H, Greco MA, Sarita-Reyes C, Kupchik G, Blei F (2008) More than skin deep: a case of congenital lamellar ichthyosis, lymphatic malformation, and other abnormalities. Lymphat Res Biol 6(1):39–44
Génin E, Feingold J, Clerget-Darpoux F (2008) Identifying modifier genes of monogenic disease: strategies and difficulties. Hum Genet 124(4):357–368
Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5(2):89–99
Huber M, Yee VC, Burri N, Vikerfors E, Lavrijsen AP, Paller AS, Hohl D (1997) Consequences of seven novel mutations on the expression and structure of keratinocyte transglutaminase. J Biol Chem 272(34):21018–21026
Huber M, Rettler I, Bernasconi K, Wyss M, Hohl D (1995) Lamellar ichthyosis is genetically heterogeneous–cases with normal keratinocyte transglutaminase. J Invest Dermatol 105(5):653–654
Louhichi N, Medhaffar M, Hadjsalem I, Mkaouar-Rebai E, Fendri-Kriaa N, Kanoun H, Yaïch F, Souissi T, Elloumi M, Fakhfakh F (2010) Congenital factor XIII deficiency caused by two mutations in eight Tunisian families: molecular confirmation of a founder effect. Ann Hematol 89(5):499–504
Shevchenko YO, Compton JG, Toro JR, DiGiovanna JJ, Bale SJ (2000) Splice-site mutation in TGM1 in congenital recessive ichthyosis in American families: molecular, genetic, genealogic, and clinical studies. Hum Genet 106(5):492–499
Acknowledgments
The authors are indebted to the patients and their families who participated in this study for their constant support and motivation. This work was supported by the Tunisian Ministry of Higher Education and Scientific Research.
Conflict of interests
None of the authors has any conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Louhichi, N., Hadjsalem, I., Marrakchi, S. et al. Congenital lamellar ichthyosis in Tunisia is caused by a founder nonsense mutation in the TGM1 gene. Mol Biol Rep 40, 2527–2532 (2013). https://doi.org/10.1007/s11033-012-2333-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-012-2333-1