
Abstract
To identity immunodiagnostic antigen genes, a Plasmodium falciparum (Dd2 clone) expression library was screened using human immune sera. The ring-infected erythrocyte surface antigen (RESA) was isolated: this antigen of the resistant clone presents repeat tandem sequences like the 3D7 clone, albeit in different numbers. RESA has been studied as a marker of genetic diversity, with different sizes being observed in different isolates and clones of Plasmodium falciparum. The native protein was localised in cultures by western-blot and immuno-transmission electron microscopy. The antigenicity of RESA was evaluated by ELISA, using the carboxy-terminal repeat region as antigen. The assay’s sensitivity and specificity were 78.2 and 94% respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Falciparum malaria is one of the most deadly and far-reaching human health problems throughout the tropical world. Antimalarial drugs are now considered to be a powerful treatment. Nevertheless, there are Plasmodium falciparum (P. falciparum) parasites that are resistant to drugs that are currently being used in different parts of the world. Although the protective immune response against intraerythrocytic stages of the falciparum malaria parasite is still not fully understood, immune antibodies have been shown to be associated with reduced parasite prevalence [1].
The malaria parasite, P. falciparum, spends part of its life cycle inside the enucleated erythrocytes of its human host, which are cells that lack their own machinery for protein synthesis and trafficking. As a consequence, an unusual and highly specialised secretory system is needed to target parasite proteins at their correct destinations. Indeed the parasite achieves the targeting of proteins, directing them not only to compartments within its own confines but also to the parasitophorous vacuole (PV) in which it resides and onwards to the PV membrane, the erythrocyte cytoplasm, and the host cell membrane [2, 3].
The antigenic variation is a survival mechanism developed by the malaria parasite P. falciparum in order to allow for the establishment of a chronic infection [4]. After invading human erythrocytes, the malaria parasite, P. falciparum, exports proteins beyond the confines of its own plasma membrane to modify the properties of the host red cell membrane. These modifications are critical to the pathogenesis of malaria. Analysis of the P. falciparum genome sequence has identified a large number of molecules with putative atypical signal sequences. The signals remain poorly characterised, yet a number of molecules with these motifs are localised in the host erythrocyte [5]. Coppel et al. [6] described a cloned polypeptide of P. falciparum representing part of an antigen found at the surface of erythrocytes infected with ring-stage parasites. The ring-infected erythrocyte surface antigen (RESA) contains two separate blocks of tandem repeat sequences that encode antigenic determinants recognised by antibodies in the sera of individuals exposed to malaria [7].
The development of vaccines is currently receiving a major amount of attention in malaria research. As it is not possible for malaria vaccines to be based on the use of killed or attenuated organisms, the vaccines that are being developed are subunit vaccines in which the immunogens consist of defined parasite antigens or antigenic fragments [8]. Fragments of RESA have been used to protect Aotus monkeys against overwhelming infection with P. falciparum. This protein has been identified as potential vaccine component [9]. Several trials of blood (asexual)-stage vaccines are under development, but only one (MSP/RESA, also known as Combination B) has been tested in randomised controlled trials. The MSP/RESA vaccine shows promise as a way of reducing the severity of malaria episodes [10]. The Pexel motif is conserved in Plasmodium spp. Although many exported proteins are prime candidates for vaccine development or therapeutic intervention, their usefulness is limited by antigenic variation and strain-specific differences [11].
Ring-infected erythrocyte surface antigen was detected by immunoelectron microscopy in the erythrocyte membrane infected with ring-stage parasites but not in association with immature parasites within the erythrocyte [12]. RESA is found in all field isolates of P. falciparum, suggesting that it facilitates survival of the parasite in vivo [13]. This protein is produced in the final stages of schizont development and is stored in apical organelles, known as dense granules, within the individual merozoites [14]. Following rupture and reinvasion of a new red blood cell by a merozoite, RESA is secreted into the newly formed PV and then transported, by an unknown mechanism, to the red cell membrane skeleton [15]. The interaction of RESA with erythrocyte spectrin has been shown to stabilise the erythrocyte membrane [16, 17] and may be involved in repair of the host membrane following invasion.
In this report, we describe the cloning of P. falciparum RESA, isolated from cDNA expression constructed using erythrocytic stages of Dd2 resistant clone (chloroquine, mefloquine and pyrimethamine), and confirm its property as an immunogenic molecule for diagnosis of malaria. Genetic diversity was studied, and different degrees of diversity of RESA gene sequences were found among P. falciparum patient’s isolates and different strains. RESA was localised at erythrocytes stages of P. falciparum, Dd2 clone, by immunoelectron microscopy (IEM) and scanning electron microscopy (SEM).
Materials and methods
Parasites
Plasmodium falciparum was obtained from in vitro culture of human blood cells. The following were used: clone 3D7 from West Africa, derived from the clone NF54 [18] and sensitive to most antimalarial drugs; and clone Dd2 from South-east Asia, derived from the Indochina III/CDC clone and resistant to chloroquine, mefloquine and pyrimethamine [19].
Human sera
Positive sera
A total of 110 sera were collected from individuals infected with IFA-confirmed malaria.
Negative sera
To set suitable cut-off values for the assays, we used: 34 sera from healthy Spanish individuals who had had no contact with endemic areas; and 30 sera from individuals who had other serologically confirmed parasitic diseases, such as leishmaniasis (n = 10), Chagas disease (n = 10) and toxoplasmosis (n = 10).
Equal volumes of sera from individuals infected with malaria, diagnosed by microscopy and IFA, were collected from different geographic areas. These were then pooled and used for screening the cDNA library prepared from P. falciparum haematic stages. All sera were supplied by the Parasitology Department of the National Microbiology Centre (Carlos III Institute of Health).
Cultures
Plasmodium falciparum Dd2 was maintained in culture, in human erythrocytes incubated at 37°C in RPMI 1640 medium with human serum and gas mixture (3% carbon dioxide + 1% oxygen + 96% nitrogen). Fresh human erythrocytes were added at 3–4 day intervals. The parasites continued to reproduce in their normal asexual cycle approximately every 48 h.
Isolates of patients
To study the genetic diversity of P. falciparum, we used RESA isolated from malaria patients in different geographic areas. Sample collection was undertaken in 2006. Diagnosis of P. falciparum was confirmed by semi-nested, multiplex polymerase chain reaction (PCR) [20]. All samples were supplied by the Parasitology Department of the National Microbiology Centre (Carlos III Institute of Health).
Enrichment of parasite-infected red blood cells by the magnetic method
One millilitre of the 10% suspension of erythrocytes was applied to an LD column assembled in a magnetic unit (Miltenyi Biotec), and washed with 20 ml phosphate-buffered saline pH: 7.3 (PBS) to remove non-infected erythrocytes and white blood cells (WBCs). After the effluent from the column became almost colourless, the magnet was removed, the cells retained in the column were eluted with 1 ml PBS, and the parasite-infected red blood cells (PRBC)-enriched fraction was thus obtained. Percentages of PRBCs to total red blood cells were ascertained, and the WBC/PRBC ratio was determined after Giemsa staining of the smear [21].
Extraction of genomic DNA
Plasmodium falciparum genomic DNA (gDNA) was extracted from blood-stage parasites, Dd2 and 3D7 strains cultured in vitro, and isolates of malaria patients. The parasites were resuspended with 400 μl of extraction buffer (100 mM Tris–HCl, pH 7.4, 100 mM NaCl, 10 mM EDTA, pH 8.0). Then 1% SDS and K-Proteinase were added and incubated for 2 h at 55°C. After incubation, RNAse (100 μg/ml) was added and left to react for 1 h at 55°C. 0.1 volume of potassium acetate was added and incubated in ice for 55 min. The DNA was extracted and precipitated with phenol/chloroform and ethanol respectively. The pellet was resuspended in Milli-Q sterile water (Millipore), the DNA concentration was determined by a spectrophotometer, and the samples were stored at −20°C.
cDNA expression library
Total RNA was prepared from P. falciparum erythrocyte stages (Dd2 clone), as previously described [22]. mRNA was extracted as described by Avid and Leder [23]. A sample containing 2.2 μg mRNA was converted into double-stranded cDNA with the ZAP Express cDNA synthesis kit (Stratagene, CA, USA) according to the manufacturer’s instructions, and then ligated to the ZAP expression vector using the λ-ZAP Express library kit (Stratagene). The original library was amplified so as to contain 4.2 × 108 pfu/ml and 31% of non-recombinant phage.
Isolation and sequence analysis of P. falciparum RESA cDNA
The cDNA library was screened using equal volumes of sera from individuals infected with malaria, diagnosed by microscopy and IFA, collected from different geographic areas. The screening yielded several promising signals, 14 of which were purified by repeated cycles of immunoscreening. Recombinant pBK-CMV plasmids were excised from positive phages. The size of the cDNA inserts were determined by PCR with T3 and T7 pBK-CMV primers and EcoRI/XhoI double digestions. The nucleotide sequences were determined by the dideoxy method [24], using the Big-Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer, Langen, Germany) and an Applied Biosystems 377 DNA sequencer (Perkin Elmer). An insert of 1,336 pb, which showed a high degree of similarity to RESA, was selected. DNA sequence and predicted amino acid sequence comparisons were performed with the EMBL and SWISS-PROT databanks, using software packages from Genetic Computer Group [25]. The primers were synthesized by Roche Molecular Biochemicals, Barcelona, Spain. The PCR experiments were performed on a GeneAmp PCR System 2400 (Perkin Elmer, NJ, USA).
Genetic diversity in RESA
The study of genetic diversity was based on isolates of malaria patients from different geographic areas and 3D7 and Dd2 clones. DNA (gDNA) was extracted from blood-stage parasites as previously described. PCR amplification was performed for RESA. The primers used, forward primer 5′-ATATAGAAGAAAATGTGC-3′ and reverse primer 5′-ATTCATCATATTCTTCGTT-3′, were conserved in all isolates.
Preparation of rabbit RESA-specific antiserum
Two 3-month-old female New Zealand white rabbits were immunised by subcutaneous injection of 200 μg of the recombinant purified protein (RESACt) in Freund’s complete adjuvant (Difco®). Before the first immunisation, 10 ml were obtained from each rabbit as negative control. Animals were boosted twice with 200 μg of RESACt purified in Freund’s incomplete adjuvant (Difco®), 6 weeks post-intramuscular injection. 15 days after the third immunisation, the animals were bled. The serum obtained will be referred to here as anti-RESACt. Serum titre was determined by ELISA.
SDS-PAGE and western blot analysis
One-dimensional sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-10%PAGE) and western blotting of P. falciparum Dd2 clone protein extract (20 μg) were performed. The separated proteins were transferred to nitrocellulose membranes (Schleicher & Schuell, Protran®) with a semi-dry blotting apparatus (Trans-blot® SD Semi-dry, Bio-Rad®) for 30 min at 10 V. Thereafter, the filters were blocked overnight with PBS containing 3% BSA (PBS–BSA) at 4°C. The following day, they were rinsed in PBS containing 0.05% (w/v) Tween® 20, and incubated with anti-RESACt polyclonal rabbit antiserum diluted 1/500 in the blocking solution, for 1 h at room temperature. After washing, goat anti-rabbit IgG conjugated with peroxidase (H and L chains) (Pierce) diluted 1/5,000 was added and incubated for 1 h at room temperature. The nitrocellulose membrane was washed and the reaction developed with DAB substrate diluted in peroxide buffer (Pierce®), as described in the manufacturer’s instructions.
Immuno-transmission electron microscopy
For immunogold detection of RESA plasmodium protein in ultrathin sections, the cells were fixed in 2% paraformaldehyde, 0.2% glutaraldehyde in 0.1 M Sodium Cacodylate buffer, for 60 min at 4°C, rinsed (2 × 30 min) in chilled 0.1 M Sodium Cacodylate buffer supplemented with 4.5% sacarose, and treated for low-temperature embedding in hydrophilic acrylic resin (Lowicryl K4M—Polyscience, Warrington, PA) to preserve the antigen properties. Briefly, the samples were dehydrated, starting with 50% ethylene glycol at 0°C and followed by a graded ethanol (50–90%) series at −35°C, embedded in a 90% ethanol/Lowicryl series graded through to pure Lowicryl resin, and polymerised with a UV light at −35°C and at room temperature. Ultrathin sections, obtained with an ultramicrotome (Ultratome, LKB), were mounted on 400-mesh nickel grids and processed for immunogold-labelling at room temperature, blocked (3×) in 100 μl drops of blocking buffer (TBS 10 mM Tris–HCl, 9 mg/ml NaCl, pH 7.4; 0.2 mg/ml NaN3; 0.1% Tween 20, 20 mg/ml BSA), incubated for 60 min on a 10 μl drop of primary antibodies (anti-RESA rabbit specific antiserum, 1/5,000 in blocking buffer), washed (3×) in 100 μl drops of washing buffer (TBS–NaN3–Tween; 2 mg/ml BSA), incubated for 60 min on 10 μl drops of secondary antibodies (goat anti-rabbit IgG coupled to 10-nm-diameter colloidal gold particles—Biocell Amersham Pharmacia-Biotech—diluted 1/10 in washing buffer), and washed 3× (washing buffer +0.1% Triton X-100) plus 3× in dd water. To contrast the sections, uranyl acetate (1% in dd water) was used. The samples were analysed in TEM mode under a CM-12 TEM/STEM Philips Electron Microscope.
Expression and purification of carboxyl-terminal fragment (Pfal-RESACt) of P. falciparum RESA
The carboxyl-terminal fragment (RESACt 481-951aa) of the P. falciparum RESA cDNA coding sequence was amplified by PCR with the forward primer 5′-GAGAGAGAGAATTCAAGAAAATGTGC-3′ and the reverse primer 5′-GAGAGAGACTCGAGTTATTCATCAT-3′. The primers were synthesized by Roche Molecular Biochemicals, Barcelona, Spain. The PCR experiments were performed on a GeneAmp PCR System 2400 (Perkin Elmer, NJ, USA). The PCR product of RESACt was directly subcloned in the EcoRI and XhoI sites of the expression plasmid vector pGEX-4T-2 (Amersham-Pharmacia-Biotec, Barcelona, Spain), downstream of the glutathione S-transferase (GST) coding sequence. Recombinant GST-fusion protein was expressed in Escherichia coli BL21, after induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma, Madrid, Spain). Bacterial cultures were lysed using B-PER Bacterial Protein Extractor Reagent (Pierce Cheshire, UK), analysed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the expressed proteins were purified by affinity chromatography with glutathione sepharose 4B, as described by the manufacturer (Amersham-Pharmacia-Biotec). The purified protein was checked by SDS-PAGE and Coomassie blue staining, and protein concentration was determined by bicinchoninic acid (BCA) protein assay (Pierce).
Synthesis of peptides derived from the deduced RESA amino acid sequence
Taking into account the partial length of the cloned RESA, the amino acid sequence was deduced and five peptides were designed according to the Jameson–Wolf antigenicity index [26]. The peptides were synthesised by the National Microbiology Centre (Carlos III Institute of Health), and their sequences were as follows: RESA ZR 165EENVEENVEENVEENV180; RESA ZR1 217EENVEENVEENIEENI232; RESA ZR2 221EENVEENIEENIEENA236; RESA ZR3 253EENIEENIEENIEENI268; and RESA ZR4 281EENVEENVEENAEENA296.
ELISA with recombinant protein Pfal-RESACt and peptides RESA ZR, RESA ZR1, RESA ZR2, RESA ZR3 and RESA ZR4
Pfal-RESACt (1 μg/ml, 100 μl/well), GST (as a negative control) and peptides (0.2 μg/ml, 100 μl/well) were diluted in coating buffer (0.1 M carbonate/bicarbonate pH 9.6) and then used to coat 96 well plates (MaxiSorp® 2, flat bottom, Nunc) overnight at 4°C. Unbound antigen was removed by washing the wells with phosphate-buffered saline, pH: 7.3 (PBS) containing 0.05% (w/v) Tween® 20. Free binding sites were blocked with 3% (w/v) bovine serum albumin (BSA) (Sigma) 3% diluted in PBS containing 0.01% (w/v) sodium azide for 1 h at 37°C. After washing, 100 μl of experimental serum samples (diluted 1/100) per well were added and incubated for 1 h at 37°C. Each sample was run in duplicate. After washing, 100 μl per well of goat anti-human IgG conjugated with peroxidase (H and L chains, Pierce®) diluted 1/5,000 were added and incubated for 1 h at 37°C. The plates were washed and 100 μl/well of substrate (ABTS in 10 ml citrate–phosphate–perborate buffer) was added and the plates were incubated for 15 min at room temperature. Absorbencies were read at 405 nm on a microplate reader (Mios, Merk®). A sample was considered positive if the specific optical density (OD) value was greater than the cut-off values. The cut-off value was obtained with the mean value of 34 negative serum samples plus 3 standard deviations (SD) (cut-off values: Pfal-RESACt = 0.123).
Results and discussion
Bioinformatic analysis
Ring-infected erythrocyte surface antigen proteins possess a PEXEL motif, a pentameric motif present in the N-terminal portion. It consists of a positively charged, hydrophilic amino acid in position 1 (Arg or Lys), a hydrophobic amino acid in position 3 (Leu or Ile), and another, less conserved amino acid in position 5 (predominantly Asp, Glu or Gln), with no charged amino acids in positions 2 and 4 [11]. The motif is required for export of soluble proteins in P. falciparum (supporting data in [11]), being needed to transport proteins across the parasitophorous vacuole membrane and into the erythrocyte.
This sequence is functionally conserved in the genus Plasmodium, indicating the presence of a conserved export mechanism across the parasitophorous vacuole membrane in malaria parasites. The PEXEL sequence has made it possible to predict the export into the host erythrocyte of proteins that are likely to be important in terms of erythrocyte remodelling and virulence. The availability of genome sequences from many different species of the genus Plasmodium now provides an opportunity for genus-wide discovery of exported proteins and identification of specific protein domains representing conserved functions in these different organisms [27].
The PHIST protein family, identified by Sargeant et al. [27], is organised into three subfamilies. Of these, PHISTc is entirely shared between the P. falciparum and P. vivax lineages, and, in single-copy form, among the three species of the P. berghei lineage. The PHISTb subfamily appears to contain only two members in P. vivax and P. knowlesi, but has radiated extensively in P. falciparum. The PHISTb subfamily contains a P. falciparum-specific subgroup including RESA, which is distinguished by the additional presence of a DnaJ domain. In eukaryotic cells, proteins carrying a J-domain act as co-chaperones, regulating the activity of 70 kDa heat-shock proteins (HSP70s). Three classes of J-proteins (also called HSP40s) are distinguished by the presence and nature of the three domains originally identified in the bacterial protein DnaJ. Type-I members contain all three domains, namely, the J-domain (including a highly conserved HPD motif essential for interaction between the protein and HSP70), a glycine-rich domain and a carboxy-terminal zinc-finger domain.
Unlike PHISTb and PHISTc, PHISTa is entirely P. falciparum-specific. It is difficult to assign possible functions to PHIST proteins because none of the members of this family has previously been described (apart from the unusual RESA), and the helical structure of the prototypical PHIST domain appears dissimilar to any known structure. It is clear, however, that whereas PHISTc performs a function which is common to the genus Plasmodium but is expanded in the P. falciparum and P. vivax lineages, the function of PHISTa is specific to P. falciparum. Proteomic data show a consistent presence of PHIST proteins in parasite-infected erythrocytes [27].
Isolation and analysis of P. falciparum RESA cDNA
To identify immunodiagnostic antigen genes, a screening of 5 × 104 clones from P. falciparum cDNA library (Dd2 clone) with a mix of malaria patient sera yielded 35 promising signals. Of these, 14 were purified, excised into pBK-CMV phagemid and sequenced. All corresponded to the same molecule but with different sizes. The largest insert consisted of 1,336 bp, with a putative open reading frame of 936 bp which coded for the putative protein with 317 amino acid residues. The molecule displayed a possible start codon, stop codon and UTR 3′. The primary structure amino acid sequence showed a central domain which contains 34 imperfect repeats with a consensus sequence EENV/I/A (positions 165–300). This domain comprises four repeats of the tetramer sequence that is repeated seven times in block.
The majority of malaria antigens contain short sequence repeats which encode antigenic epitopes that are naturally immunogenic. Synthetic peptides have been used to confirm the natural immunogenicity of epitopes encoded within two blocks of related repeats in the antigen RESA. A 16-amino acid peptide, comprising four repeats of the tetramer sequence EENV, induced antibodies that were reactive with the native molecule. Detailed analyses of these anti-peptide antisera indicate that short sequence repeats express more than one epitope, some of which may cross-react with other repeat structures [28].
Alignments of the P. falciparum DNA sequence, and the amino acid sequence predicted with the Genbank/EMBL and SWISSPROT databanks show similarity to the protein RESA (3D7 clone) in 14 promising signals. The other DNA sequences display similarity to CSP (circumsporozoite protein), MESA (mature parasite-infected erythrocyte surface antigen), HSP (heat shock protein) 70 and RHOPTRY (rhoptry protein). Comparison between the incomplete protein and the different proteins published in databanks indicated a 44–98% match (Fig. 1). The remarkable level of antigenic diversity among different strains of P. falciparum [29, 30] reflects, at least in part, the repeat structure of these with an incomplete sequence of the exon 2 from the Dd2 RESA. The RESA gene shows two exons separated by one intron [31]. The exon 2 of both clones presents repeat tandem sequences but in different numbers. P. falciparum contains polypeptides with sequence repeats, the extent of which is sometimes extraordinary. These repeats are immunodominant in the sense that much of the human antibody response is directed against them. Many non-immune individuals have been observed to have very high levels of antibodies to many repetitive antigens [32].

Alignment of different antigens RESA in different isolates of P. falciparum and P. yoelii. Comparison of RESA sequences: RESA (Dd2), incomplete sequence of resistant clone to chloroquine, mefloquine and pyrimethamine, coming from Southeast of Asia; RESA (3D7), sensible clone coming from African West; 7 RESA-like genes, isolated 3D7 (RESA hypothetical protein, RESA precursor, RESA-like protein, RESA-like protein putative, RESA-like protein truncated); RESA (NF7), coming from Ghana; RESA (FC27), coming from Papua Nueva Guinea and Plasmodium yoelii RESA. In boxes are marked identical amino acids
The genes encoding the three PHIST subfamilies are located in subtelomeric regions of all chromosomes other than chromosome 3 [27].
Genetic diversity in RESA
Genetic diversity was studied using PCR, with primers selected from highly conserved regions that flanked the tandem repeat regions (variable region) of the highly polymorphic cell surface antigen, RESA. We used 28 positive P. falciparum-infected samples of malaria patients from different geographical areas (in Africa—mainly Equatorial Guinea—Asia and America). Of these, 21, which came from Equatorial Guinea, Nigeria, Mali, Gabon and Ivory Coast, were shown to be positive by PCR. Variation in the size of the amplified products was observed in different isolates of P. falciparum and in the two clones used (3D7 and Dd2; Fig. 2). Several studies use the same methodology with different polymorphic cell surface antigens (MSP-1, MSP-2, CSP and RESA) from positive P. falciparum-infected field samples. Nevertheless, variation in the size of the amplified products was observed using MSP-1 and MSP-2 specific primers in different field isolates of P. falciparum, but CSP and RESA displayed no variation in size of the amplified products [33]. Some differences were, however, observed in the size of amplified fragments. Other authors, using MSP-1 and MSP-2, have reported that amplified products varied in size and that there was specific pattern for each isolate [34].

Genetic diversity of RESA gene by PCR from P. falciparum genomic DNA. The position of molecular markers is indicated on the left (marker 100-pb ladder, Biotools®; M). Genomic DNA from Equatorial Guinea isolates (1–6), Nigeria (8–17) Mali (A, E), Costa de Marfil (B, C), Gabón (D). Genomic DNA from resistant clone (Dd2), sensible clone (3D7). Negative controls (−). Variation in the size of the amplified products was observed in different isolates and used clones (3D7 and Dd2)
Localisation of RESA native protein in extract cultures from P. falciparum
The immunogenicity of recombinant RESA was analysed using extracts cultured from P. falciparum Dd2 clone, with sera from rabbits immunised with the recombinant polypeptide. As shown in Fig. 3, sera from polypeptide-immunised rabbits immunoprecipitated a polypeptide of approximately 155 kDa, the apparent molecular mass previously found for RESA by SDS-PAGE.

Western blot of protein extracts of Dd2 clone. Incubation with 1:500 dilution of antiserum from rabbits before immunization (P), sera from rabbits immunized with the recombinant polypeptide at 1:500 dilution (I). Marker prestained SDS-PAGE Standards, broad range. Biorad® (M)
Ultrastructural localisation of the RESA native protein by immuno-transmission electron microscopy
Immuno-transmission electron microscopy (immuno-TEM) was used to analyse the ultrastructural localisation of RESA (Dd2 clone). Whereas all the rabbit antisera prepared against recombinant purified protein (RESACt) reacted with ultra-thin sectional material (the PRBC-enriched fraction) containing asexual stages of the Dd2 clone, the pre-immune serum, used as control, yielded a negative result. The gold particles were distributed over the infected erythrocyte’s surface (Fig. 4), confirming the presence of the RESA protein of P. falciparum (Dd2 clone). This also suggests that these antibodies have the potential to recognise the RESA protein of a resistant clone in its natural form and that they could be used in capture assays for diagnosis of malaria.

Ultrastructural localization of RESA protein in different stages of P. falciparum. Transmission electron microscopy: immunogold-labelling (10 nm gold particles, arrows) in ultra-thin sections. a General overview of a parasitized erythrocyte. b The RESA protein is localized in the infected erythrocyte’s surface
Transmission electron microscopy in ultrathin sections provides a higher resolution than does scanning electron microscopy (SEM) and is very useful for studying the inner subcellular ultrastructure or localisation of molecules. Yet the amount of specific epitopes really exposed to reagents is generally not very high [35].
Expression of GST fusion Pfal-RESACt
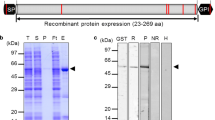
The open reading frame of Pfal-RESACt was subcloned into the pGEX-4T-2 expression vector to produce GST fusion proteins. Escherichia coli cells (BL21) transformed with pGEX-4T-2–Pfal-RESACt yielded a single fusion protein with a molecular mass of about 45 kDa, 19 kDa corresponding to the carboxyl-terminal fragment of RESA and 26 kDa to GST. Bacterial cultures transformed with pGEX-4T-2–Pfal-RESACt were lysed, and fusion proteins were purified by affinity chromatography with glutathione sepharose 4B (Fig. 5). The size obtained for Pfal-RESA (Dd2 clone) was in agreement with the size of RESA described for other clones [12].

Expression and purification of carboxyl-terminal fragment (Pfal-RESACt) of P. falciparum RESA Bacterial cultures were lysed, analysed by SDS-PAGE and the expressed protein was purified by affinity chromatography. The expression of Pfal-RESACt is on the left, 2nd and 3rd lane: positive control of induction, non-recombinant vector; 4th and 5th lane: recombinant vector. The purification of Pfal-RESACt is on the right. M: marker prestained SDS-PAGE Standards, broad range. Biorad®; NI no induced sample, I induced sample, L cells lysate; 1 and 2 elution of fusion protein
Antigenicities of P. falciparum
The antigenicity of the RESA fragment was evaluated by ELISA. When sera from humans clinically immune to malaria were examined by ELISA for antibodies to recombinant P. falciparum RESA using Pfal-RESACt as antigen, the assay’s sensitivity was 78.2%. When the recombinant protein was tested with sera from patients with other parasitic diseases, such as leishmaniasis, Chagas disease and toxoplasmosis, specificities of 94% were obtained. In contrast to results with recombinant P. falciparum RESA, 0% sensitivities were obtained when the synthetic peptides (RESA ZR, RESA ZR1, RESA ZR2, RESA ZR3 and RESA ZR4) were tested with serum samples from humans clinically immune to malaria. Serum samples from Spanish donors never exposed to malaria were used as controls, and none of the human sera recognised GST. It may be significant that the highest sensitivities were detected with recombinant protein, which contains the carboxy-terminal repeat region of the molecule. This is in line with what other authors previously described, i.e., in order to identify the main antigenic determinants of a recombinant P. falciparum glutamate-rich protein (GLURP), the non-repetitive amino-terminal region, the central repeat region and the carboxy-terminal repeat region were produced as separate products and the antibody response was almost exclusively directed against the carboxy-terminal repeat region [36]. This immunodominance may be related to the highly repetitive nature of the carboxy-terminal region [37]. It is very important to underline the fact that the immunogenic capacity of a peptide depends, not only on intrinsic chemical properties of the peptide, but also on many complex interactions with various elements of the host immune system, such as the host immunoglobulin repertoire, self-tolerance, and various cellular and regulatory mechanisms definable only in the context of the host [38]. This would explain the failure to recognise the peptides.
In this study, we found that the human immune response against RESA is primarily directed against the carboxy-terminal portion containing the repeat region.
With regard to the effect on parasites, less is known about antibodies to the non-repeat regions of antigens, such as Pf 155/RESA. In one study, rabbits were immunised with synthetic peptides corresponding to three different non-repeated sequences of the antigen and, though all antisera reacted strongly with the corresponding synthetic peptides, they reacted only weakly with full-length peptides [39]. These results indicate that, unlike the findings reported by our study, non-repeat sequences of Pf 155/RESA are immunogenic.
References
Wajanarogana S, Prasomrothanakul T, Udomsangpeth R, Tungpradabkul S (2006) Construction of a human functional single-chain variable fragment (scFv) antibody recognizing the malaria parasite Plasmodium falciparum. Biotechnol Appl Biochem 44:55–61
Taraschi TF, Trelka D, Martinez S, Schneider T, O’Donell ME (2001) Vesicle-mediated trafficking of parasite proteins to the host cell cytosol and erythrocyte surface membrane in Plasmodium falciparum infected erythrocytes. Int J Parasitol 31:1381–1391
Van Doren GG, Waller RF, Joiner KA, Roos DS, McFAden GI (2000) Traffic jams: protein transport in Plasmodium falciparum. Parasitol Today 16:421–427
Mok BW, Ribacke U, Winter G, Yip BH, Tan CS, Fernández V, Chen Q, Nilsson P, Wahigrn M (2007) Comparative transcriptomal analysis of isogenic Plasmodium falciparum clones of distinct antigenic and adhesive phenotypes. Mol Biochem Parasitol 151:184–192
Rug M, Wickham ME, Foley M, Cowman AF, Tilley L (2004) Correct promoter control is needed for trafficking of the ring-infected erythrocyte surface antigen to the host cytosol in transfected malaria parasites. Infect Immun 72:6095–6105
Coppel RL, Cowman AF, Anders RF, Bianco AE, Saint RB, Lingelbach KR, Kemp DJ, Brown GV (1984) Immune sera recognize on erythrocytes a Plasmodium falciparum antigen composed of repeated amino acid sequences. Nature 310:789
Cowman AF, Coppel RL, Saint RB, Favaloro J, Crewther PE, Stahl HD, Bianco AE, Brown RF, Anders RF, Kemp DJ (1984) The RESA polypeptide of Plasmodium falciparum contains two separate blocs of tandem repeats encoding antigenic epitopes that are naturally immunogenic in man. Mol Biol Med 2:207–221
Perlmann H, Perlman P, Berzins K, Wahlin B, Troye-Blomberg M, Hagstedt M, Andesson I, Hogh B, Petersen E, Bjorkman A (1989) Dissection of the human antibody response to the malaria antigen Pf155/RESA into epitope specific components. Immunol Rev 112:115–132
Anders RF, Murray LJ, Thomas LM, Davern KM, Brown GV, Kemp DJ (1987) Structure and function of candidate vaccine antigens in Plasmodium falciparum. Biochem Soc Symp 53:103–114
Graves P, Gelband H (2006) Vaccines for preventing malaria (blood-stage). Cochrane Database Syst Rev 18:CD006199
Marti M, Good RT, Rug M, Knuepfer E, Cowman AF (2004) Targeting malaria virulence and remodelling proteins to the host erythrocyte. Science 306:1930–1933
Brown GV, Culvenor FG, Crewther PE, Bianco AE, Coppel RL, Saint RB, Stahl HD, Kemp DJ, Anders RF (1985) Localization of the ring-infected erythrocyte surface antigen (RESA) of Plasmodium falciparum in merozoites and ring-infected erythrocytes. J Exp Med 162:774–779
Perlmann HK, Berzins K, Wahlin B, Udomsangpetch R, Ruanfjirachuporn W, Wahlgren M, Perlmann PH (1987) Absence of antigenic diversity in Pf 155, a major parasite antigen in membranes of erythrocytes infected with Plasmodium falciparum. J Clin Microbiol 25:2347–2354
Aikawa M, Torii M, Sjolander A, Berzins K, Perlmann P, Miller LH (1990) Pf155/RESA antigen is localized in dense granules of Plasmodium falciparum merozoites. Exp Parasitol 71:326–329
Culvenor JG, Day KP, Anders RF (1991) Plasmodium falciparum ring-infected erythrocyte surface antigen is released from merozoite dense granules after erythrocyte invasion. Infect Immun 59:1183–1187
Da Silva E, Foley M, Dkyzewsju AR, Murray LJ, Anders RF, Tilley L (1994) The Plasmodium falciparum protein RESA interacts with the erythrocyte cytoskeleton and modifies erythrocyte thermal stability. Mol Biochem Parasitol 66:59–69
Foley M, Tilley L, Sawyer WH, Anders RF (1991) The ring-infected erythrocyte surface antigen of Plasmodium falciparum associates with spectrin in the erythrocyte membrane. Mol Biochem Parasitol 46:137–147
Rosario V (1981) Cloning of naturally occurring mixed infections of malaria parasites. Science 212(4498):1037–1038
Guinet F, Dvorak JA, Fujioka H, Keister DB, Muratova O, Kaslow DC, Aikawa M, Vaidya AB, Wellems (1996) TEA developmental defect in Plasmodium falciparum male gametogenesis. J Cell Biol 135(1):269–278
Rubio JM, Benito A, Roche J, Berzosa PJ, Garcia ML, Mico M, Edu M, Alvar J (1999) Semi-nested, multiplex polymerase chain reaction for detection of human malaria parasites and evidence of Plasmodium vivax infection in Equatorial Guinea. Am J Trop Med Hyg 60:183–187
Trang DT, Huy NT, Kariu T, Tajima K, Kamei K (2004) One-step concentration of malarial parasite-infected red blood cells and removal of contaminating white blood cells. Malar J 17:3–7
Rangel-Aldao R, Comach G, Mendoza A (1987) In vitro translation of T. cruzi antigens recognised by human chagasic sera. J Parasitol 73:855–857
Aviv H, Leder P (1972) Purification of biologically active globin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc Natl Acad Sci USA 69:1408–1412
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Devereux JR, Haeberli PL, Smithies O (1984) A comprehensive set of sequence analysis programmes for the VAX. Nucleic Acids Res 12:387–395
Jameson BA, Wolf H (1988) The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci 4(1):181–186
Sargeant TJ, Marti M, Caler E, Carlton JM, Simpson K, Speed TP, Cowman AF (2006) Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol 7(2):R12
Anders RF, Shi PT, Scanlon DB, Leach SJ, Coppel RL, Brown GV, Stahl HD, Demp DJ (1986) Antigenic repeat structures in proteins of Plasmodium falciparum. Ciba Found Symp 111:164–183
Fenton B, Walker A, Walliker D (1985) Protein variation in clones of Plasmodium falciparum detected by two dimensional electrophoresis. Mol Biochem Parasitol 16:173–183
Tait A (1981) Analysis of protein variation in Plasmodium falciparum by two-dimensional gel electrophoresis. Mol Biochem Parasitol 2:205–218
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Half D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419(6906):498–511
Anders RF (1986) Multiple cross-reactivities amongst antigens of Plasmodium falciparum impair the development of protective immunity against malaria. Parasite Immunol 8:529–539
Sidhu AB, Madhubala R (2000) Plasmodium falciparum: detection and strain identification of Indian isolates by polymerase chain reaction. Southeast Asian J Trop Med Public Health 31:213–218
Heidari A, Keshavarz H, Brokni M, Jelinek T (2007) Genetic diversity in merozoite surface protein (MSP)-1 and MSP-2 genes of Plasmodium falciparum in a major endemic region of Iran. Korean J Parasitol 45:59–63
Herrera MI, Santa Maria I, de Andres R, Najera R (1988) Localization of human immunodeficiency virus antigens in infected cells by scanning/transmission-immunogold techniques. Ultrastruct Pathol 12(4):439–447
Theisen M, Vuust J, Gottschau A, Jepsen S, How B (1995) Antigenicity and immunogenicity of recombinant glutamate-rich protein of Plasmodium falciparum expressed in Escherichia coli. Clin Diagn Lab Immunol 2:30–34
Anders RF, Coppel RL, Brown GV, Kemp DJ (1988) Antigens with repeated amino acid sequences from asexual blood stages of Plasmodium falciparum. Prog Allergy 41:148–172
Van Regenmortel MHV (2001) Antigenicity and immunogenicity of synthetic peptides. Biologicals 29:209–213
Siddique AB, Iqbal J, Ahlborg N, Wahlin FB, Perlmann P, Berzins K (1998) Antibodies to nonrepeat sequences of antigen Pf 155/RESA of Plasmodium falciparum inhibit parasite growth in vitro. Parasitol Res 84:485–491
Acknowledgments
The study was funded by the Instituto de Salud Carlos III, Spanish Research Network of Tropical Diseases (RICET; RD06/0021/0000), Ministry of Science and Innovation of Spain.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moyano, E.M., González, L.M., Cuevas, L. et al. Molecular cloning and characterisation of the RESA gene, a marker of genetic diversity of Plasmodium falciparum . Mol Biol Rep 37, 2893–2902 (2010). https://doi.org/10.1007/s11033-009-9849-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-009-9849-z