
Abstract
Rapeseed (Brassica napus L.) is an important oil crop in the world. In order to fulfill the requirement of mechanized harvesting and raised production, it is highly desirable to gain a better understanding of the regulatory networks controlling the main agronomic traits of this crop. In this study, we obtained a natural mutant of rapeseed with more main stems named as dt (duo tou, meaning more main stems in Chinese). The dt mutant exhibits abnormal differentiation of stems, increased leaves, and decreased plant height. Phenotype and tissue section analysis showed that abnormal development of the shoot apical meristem (SAM) led to the dt phenotype. Genes that participated in SAM activity maintenance, cytokinin biosynthesis, and signal transduction displayed greatly variation at transcriptional level, which was associated with the high level of cytokinin in the dt mutant. These results provide desirable material for improving the breeding and production of Brassica napus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rapeseed (Brassica napus L.) is an important oil crop in the world, which is a major resource of edible oil and industrial raw material. To improve the production of rapeseed, great efforts have been made recently to elucidate the molecular mechanisms underlying its important agronomic traits, such as plant height, branch number, and silique number which contribute to rapeseed yields (Chen et al. 2007; Tan et al. 2015; Lu et al. 2017).
Stems and branches are the key yield-related traits, which are differentiated from shoot meristem. The ability of the plant continuously generates new tissues depends on the undifferentiated stem cells activity (Aichinger et al. 2012; Sablowski 2007). After early embryo formation, the shoot begins to form a group of stem cells with a constantly divided capacity. With the onset of seed germination, contributions from stem cells gradually differentiate to form the shoot and root apical meristem (SAM, RAM, respectively). The SAM generates aerial organs throughout the lifespan of higher plants, such as stems, leaves, and flowers (Aichinger et al. 2012; Barton 2010; Stahl and Simon 2005). The meristem must maintain a balance between stem cell niches updating and peripheral organ initiation in order to fulfill this function. In the process of differentiation, the morphological structure, physiological status, and endogenous hormonal content of the SAM undergo substantial change (Chickarmane et al. 2012). These changes represent the periodical characteristics of SAM in different development stages, which were selected as important indicators in this process.
The SAM generates branches and leaves during the vegetative developmental stage. During the reproductive developmental stage, flowers are the main products, which are generated from the floral meristem (Ha et al. 2010). Regardless of the developmental stage, it is important to keep the homeostasis between the stem cell activity maintenance and organ differentiation; this employs a complicated signal network (Ha et al. 2010). SAM stem cell activity is regulated by a feedback loop consisted of WUSCHEL (WUS), CLAVAT3 (CLV3), and related genes (Schoof et al. 2000). In the Arabidopsis SAM, WUS is a key regulator of shoot stem cell maintenance. Mutation of WUS results in the disruption of stem cell formation and meristem development termination (Ikeda et al. 2009; Mayer et al. 1998). Ectopic expression of WUS is also known to be sufficient to induce organ primordium formation and stem cell activity in root tissues (Gallois et al. 2004). CLV3 belongs to the CLV3/ESR protein family and demonstrates an expression domain coincident with SAM stem cell distribution and is the only known molecular marker of these cells (Fletcher et al. 1999). In SAM, the expression of WUS is sufficient to activate the transcription of CLV3 (Leibfried et al. 2005), meanwhile, the activated CLV3 could conversely suppress the expression of WUS through two parallel receptor systems which consist of CLAVATA1 (CLV1) and CLAVATA2 (CLV2) (Brand et al. 2000; Clark et al. 1997; Jeong et al. 1999; Ogawa et al. 2008). Other genes also make significant contributions to the establishment of the SAM shape. In Arabidopsis, SHOOT MERISTEM LESS (STM) facilitates stem cell division by restraining cell differentiation. In the STM loss-of-function mutant, SAM cannot generate, which results in fused cotyledon and failed to form other parts of the shoot (Long et al. 1996). The co-expression of WUS and STM produced synergistic effects on meristem, and the phenotype of stm mutant could be partly complemented by the overexpression of WUS, but these two genes regulate SAM in two independent manners (Lenhard et al. 2002). The function of WUS is specifying the identity of stem cells in the organization center; however, STM was expressed to suppress the differentiation of cells throughout the meristem dome. Many transcription factors participate in SAM border formation. CUP-SHAPED COTYLEDON (CUC) genes not only play important roles in SAM formation but also participate in the boundary division between the SAM and newborn organs (Burian et al. 2015; Goncalves et al. 2015; Hibara et al. 2006; Vroemen et al. 2003). The CUC genes are supposed to perform redundantly to inhibit boundary cell proliferation, which is a precondition for organ generation from the SAM. The expression of these CUCs is restricted to the boundary regions owing to the presence of STM and auxin (Aida et al. 1999; Aida et al. 2002; Friml et al. 2004; Furutani et al. 2004; Takada and Tasaka 2002).
Cytokinin (CK) positively regulates cell division and control the size of the SAM (Shani et al. 2006). The spatial regulation of CK biosynthesis and cellular responses to CK in the central domain are important for SAM formation (Chickarmane et al. 2012; Kieber and Schaller 2014). A study in Arabidopsis indicated that mutations in ARABIDOPSIS HISTIDINE KINASE2 (AHK2), AHK3, and AHK4, which encode CK receptor histidine kinase, resulted in a smaller SAM size, a decreased rate of leaf primordium germination, and the suppression of new leaf generation (Nishimura et al. 2004). Ectopic expression of cytokinin oxidase (CKX) in Arabidopsis and tobacco, which encode the CK-degrading enzyme, could also result in a smaller meristem and decreased leaf initiation (Werner et al. 2003; Werner et al. 2001). In serious cases, the meristem of transgenic plants leads to disruption eventually, which confirms the necessity of plenty CK to the development of SAM.
In the past decades, considerable progress had been achieved on the plant SAM research (Barton 2010; Wang et al. 2018). However, huge space still exists in the regulation network. In this study, we present a B. napus mutant of NY12 (Ningyou 12) background, which exhibits an abnormal development of SAM. Basing on the more main stems phenotype, the mutant was named as dt (dou tou), which means “more main stems” in Chinese. RNA sequencing (RNA-seq) was adopted to uncover the mechanism of these changes at the transcriptional level. Genes participate in SAM regulation and basic processes of metabolism displayed significant variation, which provides insights into the development of the B. napus SAM and the molecular bases for the formation of plant architecture.
Materials and methods
Plant materials and growth conditions
B. napus plants in this study (dt and NY12) were grown in the experimental field of Jiangsu University. All collected samples were frozen in liquid nitrogen immediately after picked from the plant and stored at − 80 °C before use. Seedlings were grown in 7 cm × 7 cm × 7.5 cm pots containing nutrient soil in an incubator under the controlled condition: the light was provided for 16 h per day, the temperature is 25 °C during the light and 20 °C in the night, the relative air humidity is kept on 55%. Shoot apexes were collected at 2 cm from the tip of the stem when the first flower opened. For RNA-seq, shoot apexes were collected from three individuals as biological replicates.
Tissue section
For paraffin sections, fresh samples were soaked in formaldehyde-acetic acid-alcohol (FAA) stationary liquid overnight and dehydrated in a gradient ethanol solution. Tissue sections (7 μm) were performed using a microtome (Leica RM 2155, Hesse-Darmstadt, Germany), stained with 1% of safranine overnight and 0.5% of fast green for 30 s. The sections were photographed using an OLYMPUS microscope (BX53, Tokyo, Japan).
RNA extraction, sequencing, and data analysis
The total RNA of the collected samples of dt mutant and NY12 were extracted using TRIzol Reagent (Life technologies, New York, USA) according to the manufacturer’s instruction. One hundred milligrams of the shoot apex was used for RNA extraction. The purified RNA was detected and qualified using OneDrop OD-1000+ spectrophotometer (RockGene, Shanghai, China). Library construction was performed according to the standard protocol. The sequencing of the library was performed using the BGISeq-500 platform. The experiment was repeated three times with independent samples. The raw data were filtered to trim adaptor sequences and to remove low-quality sequences (Q < 20) with > 10% uncertain (N) bases. The NCBI database was used to annotate gene function. Differentially expressed genes (DEGs) were screened according to the NOIseq method (Tarazona et al. 2011). Transcripts reach the criterion of log2 fold change ≥ 1 (or ≤ − 1) and probability ≥ 0.8 were selected as DEGs. Gene ontology (GO) classification of DEGs was performed using the gene ontology database. The GO enrichment analysis was performed by GO::TermFinder software. The pathway enrichment analysis of the DEGs was searched for the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The clean data was deposited in NCBI SRA section with BioProject No. PRJNA449176. Significantly differently expressed genes and all differentially expressed genes are listed in Supplemental Table 1 and Supplemental Table 2, respectively.
Cytokinin content detection
Shoot apexes were collected at the same time when the RNA-seq samples were collected. More than three shoot apexes were mixed together and ground to powder in liquid nitrogen. About 500 μg of ground samples was used for CK detection. Solid Phase Extraction-Liquid Chromatogram-Tandem Mass Spectrometry (SPE-LC-MS/MS) was used to detect the content of CKs in dt and NY12. This step was performed on the plant hormone platform of the Institute of Genetics and Developmental Biology, Chinese Academy of Science (http://www.genetics.ac.cn/).
Quantitative real-time PCR analysis
Quantitative real-time PCR (qPCR) was performed to detect the expression pattern of selected genes in dt and NY12 in order to validate the result of RNA-seq. The RNA from 100 mg frozen shoot apex samples were isolated using TRIzol reagent (Life technologies, New York, USA) following the protocol of the supplier. Five hundred nanogram of total RNA was used to synthesize the first strand cDNA using HiScript Q RT SuperMix for qPCR kit (Vazyme, Nanjing, China). ABI 7300 Real-Time PCR Detection System (Applied Biosystems, Carlsbad, USA) was used to perform the qPCR reaction with SYBR Green Master Mix (Vazyme, Nanjing, China). Gene-specific primers were designed using Primer premier 5.0 based on the corresponding sequence of selected genes. The sequence of primers is listed in Supplemental Table 3. Standard curve and melting curve methods were used to check the efficiency and specificities of primers after all qPCR reactions. All qPCRs were performed in triplicate in a final volume of 20 μL. The ACTIN gene was used as an internal reference gene. 2−ΔΔCt method was adopted to calculate the relative expression level of genes.
Results
Phenotypic characterization of the dt mutant
The dt mutant was found in the experimental field of NY12 background. Homozygote was obtained through self-crossing, and the individuals from which no segregation of phenotypic traits were reserved. Compared to the wild type, dt displayed many distinct phenotypes. At the vegetative development stage, seedlings of dt generate more leaves than wild type (Fig. 1a, h). At the reproductive stage after bolting, dt generated multiple main stems instead of only one in the wild type (Fig. 1b–g). In addition, part of flowers in dt mutant displayed abnormal differentiation: multiple flowers were generated from a single peduncle in the dt mutant, while only one flower per peduncle in wild type (Fig. 1d, e). At the flowering stage, dt exhibited obviously lower plant height than the wild type (Fig. 1f–i). These results indicated that the aerial part of dt displayed a significant difference from the wild type in organ differentiation and plant architecture.

Comparisons of wild type (left) and dt mutant (right) phenotypes. a Increased leaves in the dt mutant seedlings compared to the wild type, bar = 3 cm. b–c The stem phenotype of wild type (b) and dt (c) in bolting stage, bar = 3 cm. d–e The phenotype of wild type (d) and dt (e) in flower, bar = 1 cm. f–g The phenotype of wild type (f) and dt (g) in the flowering period, bar =10 cm. h–i Quantification of leaf number (h) and plant height (i) among dt mutant and wild type seedlings. Error bars represent ± standard deviation from ten biological replicates. *** indicates significant difference with the wild type at P < 0.001 (t test)
Abnormal development of SAM in dt mutant
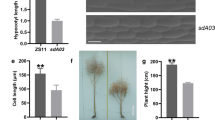
The gradual formation of the plant aerial part mainly depends on the activity of SAM (Lyndon 1999). The changes of leaf number at the seedling stage were tracked to reflect the activity of SAM. The number of leaves in dt mutant significantly exceeds that of the wild type at 15 days after germination (Fig. 2). After further development of the plant, the difference between dt and wild type became quite pronounced (Fig. 2), which suggests a stronger activity of SAM in dt mutant.

Comparison of the leaf dynamic variation between dt mutant and wild type. DAG, day after germination. Error bars represent ± standard deviation from ten biological replicates. *** indicates significant difference with the wild type at P < 0.001 (t test)
To reveal the reason of generating more leaves in dt mutant, tissue sections analysis of 3-week-old shoots from dt and wild type were performed. The development of wild type cotyledons and leaves displayed a symmetric manner (Fig. 3a). However, as compared to wild type, shoots in dt exhibited an irregular shape and more than one SAM in the shoot area (Fig. 3b, d). Further observation showed that parts of the SAM were salient, unlike the regular, arc-shaped structure observed in the wild type (Fig. 3e, f). Therefore, the generation of more leaves and more main stems phenotype are formed by irregular shape and increased number of SAM.

The abnormal development of SAM in dt. a–b Shoot phenotypes of the wild type (a) and dt (b) at 3 weeks, bar = 3 cm. c–d Longitudinal section of wild type (c) and the dt mutant (d) shoots at 3 weeks, bar = 200 μm. e–f Enlargement of the red panels in (c) and (d), respectively. bar = 100 μm
RNA-seq analysis of shoot apex in dt mutant
To investigate the molecular mechanism underlying abnormal development of SAM in dt, transcriptomic analyses of SAM in dt and NY12 were performed using a high-throughput RNA-seq approach. The transcriptome of the dt mutant SAM was compared with that of wild type to identify DEGs. One thousand nineteen significant DEGs between dt and NY12 were identified according to the NOIseq method (Tarazona et al. 2011), in which 426 genes were upregulated and 593 genes were downregulated in dt (Supplemental Fig. 1).
GO enrichment analysis was performed to classify the biological function of DEGs; 696 of the 1019 DEGs were annotated successfully. In order to discover the significantly over-represented functional categories, an enrichment analysis was performed according to the molecular function and biological processes (Supplemental Fig. 2). Five GO terms were enriched in the molecular function category (“nucleic acid binding transcription factor activity,” “hydrolase activity,” “DNA binding,” “structural molecule activity,” and “tetrapyrrole binding”). As for biological processes, 23 subcategories were enriched. “Gene expression,” “response to stress,” and “single-multicellular organism process” were dominant. Go analysis indicated that DEGs are mainly involved in gene expression regulation and stress response, function as structural molecule, and DNA binding. Therefore, the transcription factor prediction of the 74 DEGs in “nucleic acid binding transcription factor activity” category was performed. Twenty-eight of them were identified as eight types of transcription factors, which belong to MADS, MYB, bHLH, AP2-EREBP, NAC, zf-HD, GRAS, C2H2, and C2C2-CO-like family (Supplemental Table 4). Most of them displayed upregulation in dt mutant, which were supposed to adapt to the vigorous development of SAM.
In order to understand the biological pathways of genes, KEGG database (Kanehisa et al. 2008) was used to perform pathway enrichment analysis of DEGs between dt and the wild type. Based on the results, 667 DEGs were mapped to 106 biological pathways, in which 21 of them were significantly enriched. “Metabolic pathways” is the most enriched pathway, followed by “biosynthesis of secondary metabolites” and “ribosome” (Supplemental Fig. 3). Plant hormones played essential roles during SAM development. As for “tryptophan metabolism” and “plant hormone signal transduction” pathways, some genes displayed significant expression, in which Aldehyde dehydrogenase (ALDH), MYROSINASE and CYP83B1 participants in auxin biosynthesis, AUXIN RESPONSE FACTOR (ARF), small auxin-up RNA (SAUR), AHK4/CYTOKININ RESPONSE1 (CRE1), GIBBERELLIN INSENSITIVE DWARF1 (GID1), DELLA, Transcription Factor (TF), Brassinosteroid-insensitive1 (BRI1), and Xyloglucan Endo-transglycosylase (TCH4) involve in auxin, cytokinin, gibberellin, and brassinosteroid signal transduction (Supplemental Table 5).
Based on the annotation of differently expressed genes, we found genes participating in SAM activity maintenance and cytokinin signal transduction were significantly altered in dt SAM. Therefore, 22 genes were selected for qPCR validation and further analysis (Fig. 4, Supplemental Fig. 4). These genes displayed the same variation trend in expression using both techniques. The results of the transcriptome and qPCR showed that the expressions of WUS, CUC2 and CUC3 were up-regulated, and the expressions of CLV3 and STM were down-regulated (Fig. 4a), which activated the SAM. LONELY GUYs (LOGs) act at the bioactive step of cytokinin (Kurakawa et al. 2007). Three LOGs (LOG3, LOG5, and LOG6) were upregulated in dt, especially LOG5, for which the expression had enlarged to 84-fold. ISOPENTENYLTRANSFERASEs (IPTs) are key genes to participate in the biosynthesis of cytokinin (Kieber and Schaller 2014). In this study, only IPT1 displayed upregulation; however, IPT5 and IPT7 were downregulated in the mutant (Fig. 4b). As for cytokinin signal transduction, most of the selected type A ARABIDOPSIS RESPONSE REGULATORs (ARRs) (ARR8, ARR9, and ARR17) were downregulated in the dt mutant. Conversely, some type B ARRs (ARR14 and ARR18) showed substantial upregulation in the dt mutant (Fig. 4b). Other genes involved in CK signal transduction exhibited significant variation in dt. Therefore, the dramatic upregulation of CK bioactive genes in dt might lead to a significant variation of CK content and signal transduction, followed by the activation or suppression of downstream genes, leading to abnormal SAM formation.

Comparison of results between RNA-Seq and qPCR. a Genes that participated in SAM activity maintenance and bioactivation were chosen for qPCR validation. b Genes participated in CK biosynthesis and signal transduction were chosen for qPCR validation. Y-axis indicates the fold change in the dt SAM relative to the wild type
CK contents increased in the shoot apex of dt mutant
CK performs a positive role during the development of SAM (Shani et al. 2006). According to the above results, we supposed that the content of CK had changed in dt SAM. Therefore, the detection of CK component was performed to confirm this speculation (Fig. 5). Compared to the wild type, the detected CK components in dt mutant increased in different degrees. The content of active CK, such as tZ (trans-Zeatin) and IP (isopentenyladenine), increased by 101% and 35%, which may be caused by the upregulation of LOGs. The riboside status of these two kinds of CK, tZR (tZ riboside) and IPR (iP riboside), also increased by 380% and 190%. In addition, the content of DHZR (dihydrozeatin riboside) and IP7G (N6-(Δ2-isopentenyl) adenosine-7-beta-D-glucoside) increased by 87% and 36%. Taken together, the increasing of CK in dt mutant might lead to a dominant role during the development of dt SAM.

Comparison of cytokinin components between dt and wild type. tZ, trans-Zeatin; tZR, trans-Zeatin riboside; IP, isopentenyladenine; IPR, isopentenyladenine riboside; DHZR, dihydrozeatin riboside; TZ7G trans-Zeatin-7-glucoside. Error bars represent ± standard deviation from three replicates. *, **, and *** indicate significant difference with the wild type at P < 0.05, 0.01, and 0.001, respectively (t test)
Discussion
The molecular mechanism underlying shoot development in rapeseed is largely unknown. In this study, we observed a natural mutant of rapeseed, which exhibited several morphological differences from the wild type, including an increased number of leaves, additional main stems, dwarfism, and partly abnormal differentiation of flowers. We speculated that the variation of the aerial part was caused by abnormal development of the SAM. Further study indicated that leaf generation rate in dt is higher than wild type, which indicates a higher SAM activity. Tissue sections of dt demonstrated an altered shape and number of SAMs. The data indicate that the abnormal development of dt is the result of an increased number and irregular development of SAM.
We used RNA-seq to explore the internal reason of abnormal development in dt. According to GO enrichment analysis, several transcription factors were identified to participate in the main stem formation process. MADS transcription factors are reported involving in reproductive development in plants (Favaro et al. 2003; Honma and Goto 2001). In this study, the enriched MADS transcription factors all displayed upregulation, which suggested a prime influence on the regulation of SAM development in dt. Meanwhile, the result has pointed out new functions of these genes beyond regulators in flower development. In addition, MYB and bHLH are two transcription factor superfamilies in the plant, which participate in regulatory networks controlling development, metabolism, and responses to biotic and abiotic stresses (Dubos et al. 2010; Pires and Dolan 2010). Eight of them showed a dramatic differential expression, which provides new ideas for SAM development of these two kinds of transcription factors.
In this study, both RNA-seq and qPCR indicated an upregulation of WUS and downregulation of CLV3 in dt (Fig. 4a). WUS is known to promote the generation of stem cells (Gallois et al. 2004). The overexpression of WUS induced the generation of new vegetative buds in Arabidopsis (Xu et al. 2005). In this study, the downregulation of CLV3 could not provide enough suppression to restrict the expression region of WUS, which indirectly enhanced the activity of stem cells. What’s more, the expression of WUS was induced by CK. Combine the results of CK detection, the increased CK level could promote the expression of WUS as well. STM and CUCs are critical genes in lateral organ differentiation (Aida et al. 1999). STM promote cell division by suppressing organ differentiation, and downregulation of STM is beneficial to the initiation of lateral organs (Heisler et al. 2005). Moreover, the high expression level of CLV3 needs STM activity (Brand et al. 2002). The expression level of STM was downregulated in this study (Fig. 4a), which supposed to remove the support to high-level CLV3 activity and promote the differentiation of leaves in dt mutant. CUC genes function in the specification of boundaries between the meristem and initiating organs (Aida et al. 1999). The expression of CUC genes suppresses cell division in order to promote lateral organ differentiation. In this study, CUC2 and CUC3 were up regulated significantly (Fig. 4a), which might be caused by the downregulation of STM. This could be the reason of the increased leaf number observed in dt mutant.
The expression of genes which participate in cytokinin biosynthesis and signal transduction was altered significantly in the dt mutant. LOG genes act at the final step of bioactive cytokinin synthesis, which catalyze the transformation of isopentenyladenine riboside 5′-monophosphate (IPRMP) to isopentenyladenine (IP) and trans-Zeatin riboside 5′-monophosphate (tZRP) to trans-Zeatin (tZ) (Kurakawa et al. 2007; Kuroha et al. 2009; Tokunaga et al. 2012). In this study, the upregulation of LOG5 and LOG6 might be the reason for high level IP and tZ in dt mutant (Fig. 5). However, the IPT genes that participate in cytokinin biosynthesis showed different expression trends (Fig. 4b), which could be caused by the downregulation of STM. The selected type A ARRs were downregulated (Fig. 4b), which were suitable for their feedback suppressor roles in CK signal transduction. Type B ARRs regulate cytokinin signal transduction positively (Kieber and Schaller 2014) and were upregulated in this study (Fig. 4b). Taken together, genes involved in regulating SAM stem cell activity, cytokinin biosynthesis, and signal transduction exhibited significant variation from the wild type in dt, which could form the transcriptional basis for the enhanced SAM development in dt mutant.
The regulation of CK to plants is reflected in many aspects. In addition to the function of promoting meristem development, high level of CK content delay the senescence of leaves and repress the mature of siliques, which results in the reduction of production (Gan and Amasino 1995; Riefler et al. 2006). Therefore, we performed the yield-related agronomic traits analysis of dt (Supplemental Fig. 5). Comparing to the wild type, the thousand seed weight and silique number per plant displayed a significant decrease, which resulted in the decreased yield of dt mutant. At the mature stage, we found the maturity of siliques of dt is much slower than the wild type. In addition, we have detected the oil content and oil component of the seed. Comparing to the wild type, the content of oleic acid has decreased in dt and the linoleic acid displayed a significant increase (Supplemental Fig. 6). However, the total oil content did not display significant change. What’s more, the entire plant body exhibited delayed senescence phenomenon, and we suggested that the continuous high level of cytokinin might retard the senescence of dt mutant and finally reduce the grain yield.
In conclusion, we suggested that the formation of more main stems in rapeseed is closely related to abnormal development of SAM. The changes of gene expression levels promoted the CK biosynthesis in dt SAM, influenced the expression levels of downstream genes and thereby caused the abnormal development of aerial parts. Our study offers clues for further studies of the dt mutant and provides useful material for the study of SAM development in B. napus.
Change history
25 April 2019
The correct article title is presented above. Page 2, last paragraph, line 8: the term "duo tou" has been corrected in the last paragraph of Introduction section.
References
Aichinger E, Kornet N, Friedrich T, Laux T (2012) Plant stem cell niches. Annu Rev Plant Biol 63:615–636. https://doi.org/10.1146/annurev-arplant-042811-105555
Aida M, Ishida T, Tasaka M (1999) Shoot apical meristem and cotyledon formation during Arabidopsis embryogenesis: interaction among the CUP-SHAPED COTYLEDON and SHOOT MERISTEMLESS genes. Development (Cambridge, England) 126:1563–1570
Aida M, Vernoux T, Furutani M, Traas J, Tasaka M (2002) Roles of PIN-FORMED1 and MONOPTEROS in pattern formation of the apical region of the Arabidopsis embryo. Development (Cambridge, England) 129:3965–3974
Barton MK (2010) Twenty years on: the inner workings of the shoot apical meristem, a developmental dynamo. Dev Biol 341:95–113. https://doi.org/10.1016/j.ydbio.2009.11.029
Brand U, Fletcher JC, Hobe M, Meyerowitz EM, Simon R (2000) Dependence of stem cell fate in Arabidopsis on a feedback loop regulated by CLV3 activity. Science (New York, NY) 289:617–619
Brand U, Grunewald M, Hobe M, Simon R (2002) Regulation of CLV3 expression by two homeobox genes in Arabidopsis. Plant Physiol 129:565–575. https://doi.org/10.1104/pp.001867
Burian A, Raczynska-Szajgin M, Borowska-Wykret D, Piatek A, Aida M, Kwiatkowska D (2015) The CUP-SHAPED COTYLEDON2 and 3 genes have a post-meristematic effect on Arabidopsis thaliana phyllotaxis. Ann Bot 115:807–820. https://doi.org/10.1093/aob/mcv013
Chen W, Zhang Y, Liu X, Chen B, Tu J, Tingdong F (2007) Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F2 populations. Theor Appl Genet 115:849–858. https://doi.org/10.1007/s00122-007-0613-2
Chickarmane VS, Gordon SP, Tarr PT, Heisler MG, Meyerowitz EM (2012) Cytokinin signaling as a positional cue for patterning the apical-basal axis of the growing Arabidopsis shoot meristem. Proc Natl Acad Sci U S A 109:4002–4007. https://doi.org/10.1073/pnas.1200636109
Clark SE, Williams RW, Meyerowitz EM (1997) The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell 89:575–585
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L (2010) MYB transcription factors in Arabidopsis. Trends Plant Sci 15:573–581
Favaro R, Pinyopich A, Battaglia R, Kooiker M, Borghi L, Ditta G, Yanofsky MF, Kater MM, Colombo L (2003) MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell 15:2603–2611. https://doi.org/10.1105/tpc.015123
Fletcher JC, Brand U, Running MP, Simon R, Meyerowitz EM (1999) Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science (New York, NY) 283:1911–1914
Friml J, Yang X, Michniewicz M, Weijers D, Quint A, Tietz O, Benjamins R, Ouwerkerk PB, Ljung K, Sandberg G, Hooykaas PJ, Palme K, Offringa R (2004) A PINOID-dependent binary switch in apical-basal PIN polar targeting directs auxin efflux. Science (New York, NY) 306:862–865. https://doi.org/10.1126/science.1100618
Furutani M, Vernoux T, Traas J, Kato T, Tasaka M, Aida M (2004) PIN-FORMED1 and PINOID regulate boundary formation and cotyledon development in Arabidopsis embryogenesis. Development (Cambridge, England) 131:5021–5030. https://doi.org/10.1242/dev.01388
Gallois JL, Nora FR, Mizukami Y, Sablowski R (2004) WUSCHEL induces shoot stem cell activity and developmental plasticity in the root meristem. Genes Dev 18:375–380. https://doi.org/10.1101/gad.291204
Gan S, Amasino RM (1995) Inhibition of leaf senescence by autoregulated production of cytokinin. Science 270:1986–1988
Goncalves B et al (2015) A conserved role for CUP-SHAPED COTYLEDON genes during ovule development. Plant J 83:732–742. https://doi.org/10.1111/tpj.12923
Ha CM, Jun JH, Fletcher JC (2010) Shoot apical meristem form and function. Curr Top Dev Biol 91:103–140. https://doi.org/10.1016/S0070-2153(10)91004-1
Heisler MG, Ohno C, Das P, Sieber P, Reddy GV, Long JA, Meyerowitz EM (2005) Patterns of auxin transport and gene expression during primordium development revealed by live imaging of the Arabidopsis inflorescence meristem. Curr Biol 15:1899–1911. https://doi.org/10.1016/j.cub.2005.09.052
Hibara K, Karim MR, Takada S, Taoka K, Furutani M, Aida M, Tasaka M (2006) Arabidopsis CUP-SHAPED COTYLEDON3 regulates postembryonic shoot meristem and organ boundary formation. Plant Cell 18:2946–2957. https://doi.org/10.1105/tpc.106.045716
Honma T, Goto K (2001) Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409:525–529. https://doi.org/10.1038/35054083
Ikeda M, Mitsuda N, Ohme-Takagi M (2009) Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 21:3493–3505. https://doi.org/10.1105/tpc.109.069997
Jeong S, Trotochaud AE, Clark SE (1999) The Arabidopsis CLAVATA2 gene encodes a receptor-like protein required for the stability of the CLAVATA1 receptor-like kinase. Plant Cell 11:1925–1934
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484. https://doi.org/10.1093/nar/gkm882
Kieber JJ, Schaller GE (2014) Cytokinins. Arabidopsis Book 12:e0168. https://doi.org/10.1199/tab.0168
Kurakawa T, Ueda N, Maekawa M, Kobayashi K, Kojima M, Nagato Y, Sakakibara H, Kyozuka J (2007) Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 445:652–655. https://doi.org/10.1038/nature05504
Kuroha T, Tokunaga H, Kojima M, Ueda N, Ishida T, Nagawa S, Fukuda H, Sugimoto K, Sakakibara H (2009) Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. Plant Cell 21:3152–3169
Leibfried A, To JPC, Busch W, Stehling S, Kehle A, Demar M, Kieber JJ, Lohmann JU (2005) WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 438:1172–1175. https://doi.org/10.1038/nature04270
Lenhard M, Jurgens G, Laux T (2002) The WUSCHEL and SHOOTMERISTEMLESS genes fulfil complementary roles in Arabidopsis shoot meristem regulation. Development (Cambridge, England) 129:3195–3206
Long JA, Moan EI, Medford JI, Barton MK (1996) A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 379:66–69. https://doi.org/10.1038/379066a0
Lu K, Peng L, Zhang C, Lu J, Yang B, Xiao Z, Liang Y, Xu X, Qu C, Zhang K, Liu L, Zhu Q, Fu M, Yuan X, Li J (2017) Genome-wide association and transcriptome analyses reveal candidate genes underlying yield-determining traits in Brassica napus. Front Plant Sci 8:206. https://doi.org/10.3389/fpls.2017.00206
Lyndon RF (1999) The shoot apical meristem: its growth and development. New Phytol 144:51–54
Mayer KF, Schoof H, Haecker A, Lenhard M, Jurgens G, Laux T (1998) Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95:805–815
Nishimura C, Ohashi Y, Sato S, Kato T, Tabata S, Ueguchi C (2004) Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell 16:1365–1377. https://doi.org/10.1105/tpc.021477
Ogawa M, Shinohara H, Sakagami Y, Matsubayashi Y (2008) Arabidopsis CLV3 peptide directly binds CLV1 ectodomain. Science (New York, NY) 319:294–294
Pires N, Dolan L (2010) Origin and diversification of basic-helix-loop-helix proteins in plants. Mol Biol Evol 27:862–874. https://doi.org/10.1093/molbev/msp288
Riefler M, Novak O, Strnad M, Schmulling T (2006) Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. The Plant Cell 18:40–54
Sablowski R (2007) Flowering and determinacy in Arabidopsis. J Exp Bot 58:899–907. https://doi.org/10.1093/jxb/erm002
Schoof H, Lenhard M, Haecker A, Mayer KF, Jurgens G, Laux T (2000) The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 100:635–644
Shani E, Yanai O, Ori N (2006) The role of hormones in shoot apical meristem function. Curr Opin Plant Biol 9:484–489. https://doi.org/10.1016/j.pbi.2006.07.008
Stahl Y, Simon R (2005) Plant stem cell niches. Int J Dev Biol 49:479–489. https://doi.org/10.1387/ijdb.041929ys
Tan XL, Huang Q, Tan RK, Wu L, Zhang ZY, Wang Z, Lu CM, Li XF (2015) Cloning and functional characterization of a fatty acyl-acyl carrier protein thioesterase gene (BnFatB) in Brassica napus L. J Agric Sci Technol 17:987–997
Takada S, Tasaka M (2002) Embryonic shoot apical meristem formation in higher plants. J Plant Res 115:411–417. https://doi.org/10.1007/s10265-002-0061-7
Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21:2213–2223. https://doi.org/10.1101/gr.124321.111
Tokunaga H, Kojima M, Kuroha T, Ishida T, Sugimoto K, Kiba T, Sakakibara H (2012) Arabidopsis lonely guy (LOG) multiple mutants reveal a central role of the LOG-dependent pathway in cytokinin activation. Plant J 69:355–365. https://doi.org/10.1111/j.1365-313X.2011.04795.x
Vroemen CW, Mordhorst AP, Albrecht C, Kwaaitaal MA, de Vries SC (2003) The CUP-SHAPED COTYLEDON3 gene is required for boundary and shoot meristem formation in Arabidopsis. Plant Cell 15:1563–1577
Wang B, Smith SM, Li J (2018) Genetic Regulation of Shoot Architecture. Annu Rev Plant Biol
Werner T, Motyka V, Laucou V, Smets R, Van Onckelen H, Schmulling T (2003) Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 15:2532–2550. https://doi.org/10.1105/tpc.014928
Werner T, Motyka V, Strnad M, Schmulling T (2001) Regulation of plant growth by cytokinin. Proc Natl Acad Sci U S A 98:10487–10492. https://doi.org/10.1073/pnas.171304098
Xu YY, Wang XM, Li J, Li JH, Wu JS, Walker JC, Xu ZH, Chong K (2005) Activation of the WUS gene induces ectopic initiation of floral meristems on mature stem surface in Arabidopsis thaliana. Plant Mol Biol 57:773–784
Funding
This work was supported by the National Key Research and Development Program of China (2016YFD0100305 and 2016YFD0101904), the National Natural Science Foundation of China (31671720, 31471527, 31271760 and 31671590), the Open Foundation of Key Laboratory of Biology and Genetic Improvement of Oil Crops (KF2018004), and the Distinguished Scholars Research Foundation of Jiangsu University (10JDG134).
Author information
Authors and Affiliations
Contributions
Ke-Ming Zhu and Shuo Xu performed most of the experiments and wrote the manuscript. Kai-Xia Li, Sheng Chen, Sundus Zafar, and Wei Cao helped perform the phenotype analysis. Zheng Wang, Li-Na Ding, Yan-Hua Yang, and Yao-Ming Li participated in data analysis. Xiao-Li Tan conceived the study and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhu, KM., Xu, S., Li, KX. et al. Transcriptome analysis of the irregular shape of shoot apical meristem in dt (dou tou) mutant of Brassica napus L.. Mol Breeding 39, 39 (2019). https://doi.org/10.1007/s11032-019-0943-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0943-1