
Abstract
Spot blotch, caused by Cochliobolus sativus, is an important foliar disease of barley. The disease has been controlled for over 40 years through the deployment of cultivars with durable resistance derived from the line NDB112. Pathotypes of C. sativus with virulence for the NDB112 resistance have been detected in Canada; thus, many commercial cultivars are vulnerable to spot blotch epidemics. To increase the diversity of spot blotch resistance in cultivated barley, we evaluated 318 diverse wild barley accessions comprising the Wild Barley Diversity Collection (WBDC) for reaction to C. sativus at the seedling stage and utilized an association mapping (AM) approach to identify and map resistance loci. A high frequency of resistance was found in the WBDC as 95% (302/318) of the accessions exhibited low infection responses. The WBDC was genotyped with 558 Diversity Array Technology (DArT®) and 2,878 single nucleotide polymorphism (SNP) markers and subjected to structure analysis before running the AM procedure. Thirteen QTL for spot blotch resistance were identified with DArT and SNP markers. These QTL were found on chromosomes 1H, 2H, 3H, 5H, and 7H and explained from 2.3 to 3.9% of the phenotypic variance. Nearly half of the identified QTL mapped to chromosome bins where spot blotch resistance loci were previously reported, offering some validation for the AM approach. The other QTL mapped to unique genomic regions and may represent new spot blotch resistance loci. This study demonstrates that AM is an effective technique for identifying and mapping QTL for disease resistance in a wild crop progenitor.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Spot blotch, caused by Cochliobolus sativus (Ito and Kurib.) Drechsl. ex Dastur [anamorph: Bipolaris sorokiniana (Sacc. in Sorok.) Shoem.], is an important foliar disease of barley in the Upper Midwest region of the United States and eastern prairies of Canada, the largest production area in North America. Under favorable conditions, this disease can cause significant reductions in both the yield (Clark 1979) and quality of the crop (Nutter et al. 1985). Spot blotch can be controlled by applying fungicides, but the most effective and environmentally sound means of reducing the impact of this disease is through the use of resistant cultivars. Through a concerted effort of breeding and selection, the first spot blotch resistant cultivar (Dickson) was released for the Upper Midwest region in 1964. The resistance in Dickson was derived from NDB112 (Wilcoxson et al. 1990), a breeding line derived from the composite cross CC-XIII selection CIho 7117-77. In fact, all subsequent six-rowed malting cultivars released in the Upper Midwest since 1964 carry the NDB112 resistance and have remained highly resistant to spot blotch. The durability of this resistance is remarkable considering the large area (up to 2.3 million ha) over which various cultivars carrying the resistance have been grown, the ubiquity of C. sativus inoculum, and the wide range of environmental conditions under which the pathogen can cause epidemics (Fetch et al. 2008).
Two-rowed cultivars bred for resistance in the same region have not been broadly effective against populations of C. sativus. A case in point is cultivar Bowman, a two-rowed feed barley classified as moderately resistant to spot blotch when it was first released in 1984. Six years after Bowman was released, high levels of infection were observed on it and derived breeding lines in the field (Fetch and Steffenson 1994). Analysis of pathogen isolates revealed that Bowman had succumbed to a new pathotype of C. sativus (Fetch and Steffenson 1994; Valjavec-Gratian and Steffenson 1997). Recent research indicates the presence of additional C. sativus pathotypes in Canada, some of which possess virulence for the durable NDB112 resistance present in six-rowed malting cultivars (Ghazvini and Tekauz 2007). The emergence of pathotypes with virulence for the NDB112 resistance is alarming and could result in widespread epidemics given the genetic uniformity of spot blotch resistance in six-rowed malting cultivars.
The durable resistance provided by NDB112 has led to some complacency with regard to identifying new sources of spot blotch resistance in barley. Given the reports of virulent pathotypes in the region, additional research is needed to identify new sources of resistance. Recently, Fetch et al. (2008) reported on the screening of over 5,000 spring barley accessions from the USDA National Small Grains Collection for resistance to both spot blotch and net blotch. Only 5.8% (373 accessions) of the evaluated germplasm was resistant to spot blotch in the field. The geographic analysis of spot blotch resistance revealed a “center of concentration” in North America, due possibly to the wide use of the NDB112 resistance in breeding lines and cultivars that comprised the collection (Bonman et al. 2005; Fetch et al. 2008).
Another possible source of spot blotch resistance is wild barley, Hordeum vulgare subsp. spontaneum (C. Koch) Thell. This subspecies is the progenitor of cultivated barley (von Bothmer et al. 2003) and possesses a high level of genetic diversity and also novel alleles for many economically important traits (Ellis et al. 2000; Fetch et al. 2003; Shakhatreh et al. 2009; Steffenson et al. 2007). Wild barley can be found across a wide geographic range extending from North Africa in the west, throughout the Fertile Crescent, and across to Central Asia and western Pakistan in the east (von Bothmer et al. 2003). The transfer of genes from wild into cultivated barley can proceed without any difficulties because both are fully inter-fertile. Despite the rich diversity of novel alleles in H. vulgare subsp. spontaneum, this member of the primary Hordeum genepool has not been systematically characterized and exploited for genes or quantitative trait loci (QTL) conferring spot blotch resistance.
Fetch et al. (2003) evaluated 116 wild barleys from Israel and Jordan for reaction to spot blotch at the seedling stage and found 46% and 53% of accessions with resistance (i.e., infection responses [IRs] of 1–5 on a 1–9 scale) from the respective countries. However, only six of these accessions exhibited a high level of spot blotch resistance (IRs of 1–2). Based on these results, there is potential for identifying additional sources of spot blotch resistance from a wider collection of H. vulgare subsp. spontaneum germplasm. Recently, a large ecogeographically diverse collection of wild barley was assembled (the Wild Barley Diversity Collection or WBDC), comprising 318 accessions collected from 19 countries (Steffenson et al. 2007). This expanded core collection was established to catalog and exploit the genetic diversity of wild barley for a number of important agronomic traits, including spot blotch resistance.
To effectively utilize new spot blotch resistance genes from wild barley, genetic characterization is required. The standard approach is to construct bi-parental crosses between resistant and susceptible parents and then phenotype and genotype (with molecular markers) progeny populations to determine the number and chromosomal location of resistance loci (Lander and Botstein 1986). An alternative approach for mapping resistance loci that does not require development of bi-parental crosses or screening generations of progeny is association mapping (AM) or linkage disequilibrium (LD) mapping. With AM, statistical assessments are made for associations between genotypes based on molecular markers and phenotypes of various traits in reference germplasm sets (Buntjer et al. 2005). Theoretically, this technique can be applied to any set of germplasm (including wild accessions of a crop species) and detect QTL for as many traits that show variation. Compared to traditional QTL mapping, AM has three main advantages: increased resolution power for mapping QTL, greater capacity for detecting more alleles, and faster completion time (Yu and Buckler 2006; Zhu et al. 2008). AM was pioneered in human genetics (Hardy and Singleton 2009). Since its first use with plants in 2001 (Thornsberry et al. 2001), AM has gained wide application in many important crop plants because of advances in high throughput genotyping technologies, increased interest in identifying novel alleles, and improvements in statistical methods (Gupta et al. 2005; Zhu et al. 2008).
Our long-term goal is to catalog and characterize economically important genes in wild barley for cultivated barley improvement. The objective of this study was to identify and map loci conferring spot blotch resistance in the WBDC using an AM approach.
Materials and methods
Plant materials
The WBDC consists of 318 accessions selected on the basis of various eco-geographic parameters (e.g., longitude/latitude, elevation, high/low temperature, rainfall, soil type, etc.). Most of the accessions were from the Fertile Crescent (246/318 or 77.4%), with fewer numbers from Central Asia (50/318 or 15.7%), North Africa (12/318 or 3.8%), and the Caucasus region (10/318 or 3.1%) (Steffenson et al. 2007). Single plant selections were initially made from each accession in the WBDC. These selections were then selfed several times to achieve greater homozygosity. Seeds from the first selfed (S1) or third selfed (S3) generation were used in this study. Cultivar Bowman and line NDB112 were the resistant controls and line ND5883 was the susceptible control in all spot blotch phenotyping experiments.
Sowing and plant growing conditions
Four seeds from each WBDC accession were placed on moistened filter paper in petri plates and incubated in a cold room (4°C) for 1 week to break dormancy. Then, the seeds were moved to room temperature (25 ± 2°C) for one day before being sown in pots. Seeds were planted in the corners of square plastic pots (10.2 × 10.2 cm) filled with 50% soil and 50% MetroMix 200 (a vermiculite, peat moss, perlite, and sand mix). Plants were grown in a greenhouse at 16–24°C with 14 h of supplemental lighting provided by 400 W sodium vapor lamps (~300 μmol m−2 s−1 photon flux). At planting, the seeds were fertilized with a water-soluble (Peters Dark Weather 15–0–15, N–P–K, 150 g/gal concentrate at 1/16 dilution [Scott’s Company, Marysville, OH]) and slow release (Osmocote 14–14–14 N–P–K, 3 g/pot [Scott’s Company]) fertilizer. Thereafter, plants were fertilized bi-weekly with Peters 20–10–20, N–P–K (145 g/gal concentrate at 1/16 dilution [Scott’s Company]).
Pathogen isolate, inoculation, and incubation conditions
Isolate ND85F was used in all experiments conducted in this study and represents one (i.e., pathotype 1) of the three major virulence types identified in the Upper Midwest region of the USA (Valjavec-Gratian and Steffenson 1997). This isolate is routinely used to screen breeding materials for spot blotch resistance and its virulence pattern is well characterized. The isolate was originally derived from a single spore and was maintained as conidia stored on silica gel crystals in glass vials in a refrigerator at 4°C. Detailed protocols for inoculum production are described in Bilgic et al. (2006).
When the second leaves of plants were fully expanded (usually 14 days after planting), seedlings were inoculated with a conidial suspension (8,000 conidia/ml) of isolate ND85F using a Paasche H single action, external mix airbrush (Paasche Airbrush Company, Harwood Heights, IL) pressurized by an air pump (138 kPa). To facilitate even distribution and adherence of conidia on the barley leaves, a surfactant (Tween-20: polyoxyethylene-20-sorbitan monolaurate) was added (100 μl/l) to the conidial suspension. The volume of inoculum suspension applied to each plant was approximately 0.4 ml. After inoculation, plants were transferred into a mist chamber and misted with ultrasonic humidifiers for 30–40 min to establish a layer of free moisture on the leaf surfaces. Thereafter, the humidifiers were set to come on for 2 minutes every hour to maintain leaf wetness. During this time, the plants were kept at 18–22°C and near 100% RH in the dark. After 16–18 h, the misters were turned off and the chamber doors opened in order to facilitate the slow drying of moisture from the plant surfaces. Plants were then moved to the greenhouse under the same conditions described above.
Disease phenotyping
Between 9–12 days post-inoculation, the spot blotch IRs were assessed on the second leaves of seedlings using the nine-class (1–9) rating scale of Fetch and Steffenson (1999). This scale is based on the type (presence of necrosis and/or chlorosis) and relative size of lesions. The experiment was conducted in a completely randomized design and repeated once. Accessions giving variable reactions between experiments were repeated again to confirm the phenotypes. The first two disease evaluation experiments were conducted on S1 generation plants of the WBDC as this was the only generation available at the start of the study. Subsequent phenotyping tests were done on S3 generation plants. All experiments were conducted at the Plant Growth Facility on St. Paul campus of University of Minnesota.
Genotyping using diversity arrays technology markers
Genomic DNA was extracted from 2-week-old seedlings of WBDC accessions using the FASTprep kit (Q-BIOgene, Irvine, CA) and sent to Diversity Arrays Technology Pty. Ltd (Yarralumla, Australia) for genotyping. Diversity Arrays Technology (DArT®) genotyping was done first on the WBDC; thus, only the S1 generation was available at the time. A total of 558 non-redundant DArT® (a dominant marker type) markers, previously developed (Jaccoud et al. 2001) and mapped onto a barley consensus map (Wenzl et al. 2004, 2006), were used to genotype the WBDC accessions.
Genotyping using single nucleotide polymorphism markers
DNA for single nucleotide polymorphism (SNP) (a co-dominant marker type) genotyping was extracted from S3 generation plants of the WBDC. Leaf segments (5–8 cm lengths) from 2-week-old seedlings were cut and freeze-dried in 2.0 ml Eppendorf tubes. DNA was then extracted following a modified sodium dodecyl sulphate (SDS) method (Pallotta et al. 2003). The general quality and quantity of DNA was verified on a 0.8% agarose gel. Two barley oligonucleotide pool assays (BOPAs), BOPA1 and BOPA2 (Close et al. 2009), containing allele-specific oligos for a set of 3,072 SNPs (1,536 SNPs on each BOPA) were used to genotype the WBDC accessions following the protocols of Illumina’s GoldenGate Bead Array Technology (Illumina, San Diego, CA, USA) (Fan et al. 2003, 2006; Oliphant et al. 2002). SNP genotyping was done at the USDA-ARS Biosciences Research Laboratory in Fargo, ND. The 3,072 SNP markers of BOPA1 and BOPA2 were developed by a consortium of research groups led by Timothy Close at the University of California-Riverside (Close et al. 2009). Of the 3,072 SNP markers developed, 2,878 provided good quality genotype calls and were used for subsequent analyses (Supplemental Table S1). More than 95% of the 3,072 SNP markers have been mapped in a barley consensus map consisting of four populations (Close et al. 2009).
Population structure analysis
To estimate the number of subpopulations in the WBDC, the DArT and SNP marker datasets were analyzed separately for population structure using the STRUCTURE program, ver. 2.0/2.1 (Falush et al. 2003, 2007; Pritchard et al. 2000). Analyses for LD (Steffenson et al. 2007; Roy and Steffenson unpublished), population structure, and AM were conducted separately for several reasons: (a) the DArT and SNP marker maps are based on different consensus maps where marker intervals may not have the same degree of precision; (b) the WBDC accessions were assigned to different subpopulations depending on the marker type used; and (c) we were interested in comparing the results obtained for population structure and AM from the two marker types readily available within the barley research community. A burn-in period of 100,000 and Monte Carlo Markov Chain replications of 200,000 were used for the analyses. The “Admixture” and “Correlated” options were used for the ancestry and allele frequency models, respectively. STRUCTURE was run to test the hypothesis of two to fifteen (K = 2–15) subpopulations. A Q matrix, i.e., the n × p population structure incidence matrix where n is the number of individuals assayed and p is the number of populations defined, from the structure analysis was used as a covariate in AM (discussed below). The 558 DArT and 2,878 SNP marker datasets both estimated ten subpopulations in the WBDC. These population structure parameters were accounted for in the AM analyses.
Association mapping
As with structure analysis, genome-wide scans for AM of loci governing spot blotch resistance were conducted separately for DArT and SNP markers. Before analysis, the phenotype data were transformed with the Box-Cox transformation to make them conform to a more normal distribution (Box and Cox 1964). The mean spot blotch IR from all experiments was used as the response factor, with the Q matrix from population structure analysis as a covariate in the analysis. AM analysis was conducted in the TASSEL standalone program, ver. 2.1 (www.maizegenetics.net). The default settings of the program were used for filtering marker data for minimum genotype count and minor allele frequency (MAF). Marker data were filtered for a minimum count of 235 (75% of 314 accessions) and 238 (75% of 318 accessions) for the DArT and SNP datasets, respectively. Markers having a MAF less than 10% were removed from the datasets. Preliminary analyses of data revealed very little kinship among accessions in the WBDC; thus, a general linear model (GLM) was used instead of a mixed linear model. In the program, a GLM was fitted for each single marker and trait association (Yu et al. 2005). A permutation test using 10,000 permutations was conducted to correct the P-value for multiple comparisons. The experiment-wise P-value provides a test of significance (adjusted P-value) that corresponds to the experiment-wise error and was used to make decisions about the significance of marker effects.
Results
Disease phenotypes
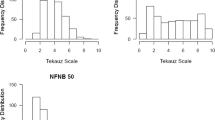
Spot blotch infection on plants was uniform across all experiments, allowing for the clear and unambiguous classification of IRs. The controls reacted as expected to C. sativus isolate ND85F: the two resistant controls NDB112 and Bowman exhibited low IRs (mean of 2.3 and 3.0, respectively), whereas the susceptible control ND5883 exhibited a high IR (mean of 7.7). Most WBDC accessions (302/318 or 95%) were resistant to spot blotch, exhibiting mean IRs of 5.0 or lower (Fig. 1). Within this resistant group, 204 accessions exhibited very low IRs of 3.0 or lower. The remaining WBDC accessions were susceptible (16/318 or 5%), exhibiting mean IRs greater than 5.0.

Frequency distribution of the mean spot blotch infection response for 318 accessions of the Wild Barley Diversity Collection (WBDC). The x-axis includes nine mean infection response intervals of 1 = 0.0–1.0; 2 = 1.1–2.0; 3 = 2.1–3.0; 4 = 3.1–4.0; 5 = 4.1–5.0; 6 = 5.1–6.0; 7 = 6.1–7.0; 8 = 7.1–8.0, and 9 = 8.1–9.0. The y-axis represents the number of WBDC accessions with mean infection response in the respective intervals
AM of QTL for spot blotch resistance based on DArT markers
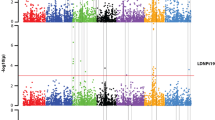
From the AM analysis with DArT markers after correction for multiple comparisons, five QTL conferring resistance to spot blotch were identified at a significance level of P ≤ 0.05 (Table 1; Fig. 2). Two QTL (Rcs-qtl-1H-bPb-2813 and Rcs-qtl-1H-bPb-3089) were identified on chromosome 1H and explained 2.7 and 3.9% of the phenotypic variation as assessed by R 2, respectively. The remaining three QTL were identified on chromosomes 3H (Rcs-qtl-3H-bPb-1068), 5H (Rcs-qtl-5H-bPb-2378), and 7H (Rcs-qtl-7H-bPb-4584) and explained 3.6, 3.6, and 3.1% of the phenotypic variation, respectively. Of the five QTL identified in this analysis, three mapped to bins where spot blotch resistance loci were previously reported and two mapped to novel locations (Table 1; Fig. 2). The MAF of DArT markers showing associations with spot blotch resistance ranged from 0.16 (bPb-2378 on chromosome 5H) to 0.40 (bPb-1068 on chromosome 3H).


Two consensus maps of arbitrarily selected DArT® (left) and SNP markers (right) for the seven barley chromosomes: 1H–7H. Positions of DArT markers and associated bins were according to Wenzl et al. (2006), and those for SNP markers and associated bins were according to Close et al. (2009). Between the representative DArT and SNP chromosomal maps, the bin numbers of Kleinhofs and Graner (2001) are indicated by the prefix ‘B’. The bolded black marker names indicate loci associated with spot blotch resistance at P = 0.05 after correction for multiple comparisons in the Wild Barley Diversity Collection (WBDC). Vertical solid black bars along the chromosomes indicate the regions or bins where previously reported spot blotch resistance genes or QTLs were identified in biparental mapping populations that did not involve a wild barley parent. Vertical open bars along the chromosomes indicate the regions or bins where previously reported spot blotch resistance genes or QTLs were identified in biparental mapping populations that included a wild barley parent. A centiMorgan scale is given at the left side
AM of QTL for spot blotch resistance based on SNP markers
From the AM analysis with SNP markers after correction for multiple comparisons, eight QTL conferring resistance to spot blotch were identified at a significance level of P ≤ 0.05 (Table 1, Fig. 2). Four QTL (Rcs-qtl-5H-11_21121, Rcs-qtl-5H-11_10641, Rcs-qtl-5H-11_20189, and Rcs-qtl-5H-11_11364) were identified on chromosome 5H and explained from 2.3 to 3.8% of the phenotypic variation. The other QTL were identified on chromosome 2H (Rcs-qtl-2H-12_10970 and Rcs-qtl-2H-12_11316 with R 2 of 2.6 and 2.3%, respectively) and chromosome 7H (Rcs-qtl-7H-12_30004 with R 2 of 2.6). The location of the QTL identified by SNP marker 11_11361 (with R 2 of 3.7) could not be assigned to a chromosome because the mapping location has not been determined. Of the seven QTL identified and mapped in this analysis, two mapped to bins where spot blotch resistance loci were previously reported and five mapped to novel locations (Table 1; Fig. 2). The MAF of SNP markers showing associations with spot blotch resistance ranged from 0.10 (11_20189 on chromosome 5H) to 0.48 (12_11316 on chromosome 2H).
LD pattern at QTL for spot blotch resistance
To determine the extent of LD at QTL detected in this study, we identified all of the markers in LD (R 2 > 0.2) with the marker associated with spot blotch resistance. We then characterized the size of this region in cM and determined whether the other markers in LD were significantly associated with spot blotch resistance. Of the 12 QTL identified and mapped in this study, three had at least one adjacent marker in LD with the most significant marker associated with the trait. Strong LD (R 2 ≥ 0.96) was found for the QTL-identifying marker bPb-2813 on chromosome 1H at 59.7 cM. Three other flanking markers (bPb-0468, bPb-7325, and bPb-7859) at the same location were in strong LD (R 2 range of 0.96–1.0) with bPb-2813 (Fig. 3). These three markers also showed highly significant associations with spot blotch resistance (P = 0.003–0.006). Additionally, the above-mentioned four markers also were in strong LD with bPb-2967, a marker significantly associated (P = 0.000; R 2 range of 0.96–1.0) with spot blotch resistance on chromosome 1H at 60.7 cM. Although the QTL detected by markers bPb-2813 and bPb-2967 were 1.0 cM apart, we considered these five adjacent markers to be detecting the same QTL because they have strong LD with each other and are all significantly associated with spot blotch resistance. On chromosome 5H, the QTL-identifying marker at 276.8 cM (11_20189) exhibited moderate LD (R 2 = 0.39) with one flanking marker (data not shown); however, this flanking marker was not significantly associated with spot blotch resistance. The QTL-identifying marker on chromosome 7H at 107.4 cM (12_30004) had moderate to strong LD (R 2 range of 0.11–1.0) with seven other markers at the same position (data not shown). Two of these markers (12_31215 and 12_30563) showed a highly significant association with spot blotch resistance (P = 0.003 and 0.008, respectively).

Triangular linkage disequilibrium (LD) matrix plot of nine DArT® markers on chromosome 1H in the Wild Barley Diversity Collection (WBDC). The plots were produced using pairwise R 2 estimates of LD with the computer program, Haploview (Barrett et al. 2005). In the figure, the pairwise R 2 values are given inside the boxes. The solid back boxes represent a pairwise R 2 value of 1.0
Discussion
Six-rowed malting barley cultivars grown in the Upper Midwest region of the United States and the eastern prairie provinces of Canada are genetically uniform for spot blotch resistance as nearly all carry the NDB112 resistance. Given the recent reports of isolates carrying virulence for this resistance, it is readily apparent that the barley crop is vulnerable to spot blotch epidemics. A recent evaluation of cultivated barley germplasm revealed a low percentage of adult plant spot blotch resistance and that many of the resistant accessions were likely derived from NDB112 (Fetch et al. 2008). To identify new sources of spot blotch resistance and to map the underlying QTL, we evaluated the WBDC for reaction to C. sativus at the seedling stage in the greenhouse. A high frequency of seedling resistance was found in the WBDC as 95% (302/318) of the accessions exhibited mean IRs of 5 or lower. This frequency of resistance is much higher than that reported by Fetch et al. (2003) for wild barley accessions from Israel (46%) and Jordan (53%) using the same C. sativus isolate and methodology. Considering only the accessions from Israel and Jordan in the WBDC, the frequency of resistance within the respective countries was still higher at 92.9% (39/42) and 86.9% (53/61). Thus, seedling resistance to spot blotch in wild barley is extremely common and likely governed by many different loci given the diversity of genotypes in the WBDC (see below).
Evaluations of the WBDC revealed many potential sources of spot blotch resistance that may be useful for cultivated barley improvement. An important question to consider is what scheme will most effectively identify spot blotch resistance QTL in these accessions and their utilization in breeding programs? The standard method would be to select diverse accessions (i.e., based on ecogeographic origin and molecular marker diversity) exhibiting resistance and then develop individual bi-parental mapping populations. In previous research, three bi-parental mapping populations were developed from wild barley accessions that are included in the WBDC. QTL analyses for spot blotch resistance and other traits have been completed in these wild × cultivated barley populations (i.e., OUH602/Harrington [Yun et al. 2005, 2006], Damon/Harrington [Alsop 2009], and Shechem/Harrington [Alsop 2009]), revealing from one to three QTL for seedling spot blotch resistance in the respective populations. In total, these three mapping populations carried only four QTL (Rcs-3H-1-2, Rcs-5H-11, Rcs-7H-3-4/Rcs-QTL-7H-2-4, and Rcs-7H-7), all of which mapped to regions coincident with previously identified QTL for seedling spot blotch resistance. Despite the development of cost-effective, high-throughput marker systems such as DArT, QTL mapping efforts in individual bi-parental populations (e.g., Alsop 2009) will not reveal, in the most efficient way, the diverse alleles present in large germplasm collections and their chromosomal locations.
AM is an alternative strategy for mapping QTL in germplasm collections that does not require development of bi-parental crosses or screening generations of progeny. Since AM relies on statistical assessments of associations between genotypes and phenotypes, one can apply the technique to any set of germplasm and detect QTL for as many traits that show variation. As a prerequisite for conducting these AM studies, we initially genotyped the WBDC with 558 non-redundant DArT® markers (Steffenson et al. 2007) and then later with >3,000 SNP markers (Roy and Steffenson unpublished). Marker data for the WBDC revealed a low level and rapid decay of LD to within 1.0 cM. This LD profile can facilitate high resolution mapping of QTL that exceeds the level possible through standard bi-parental mapping (Rafalski 2002). However, rapid LD decay, as in the case of the WBDC, necessitates a high density of markers in order to identify QTL through AM. With the LD decay and current marker density in the WBDC, the chances for identifying significant associations are moderately low; yet, if ones are detected, they likely represent close linkages between the markers and respective QTL. In this study using 558 DArT markers (average inter-marker distance of 2.3 cM) and 2,878 SNP markers (average inter-marker distance of 0.4 cM), we identified 13 QTL for seedling spot blotch resistance in 318 wild barley accessions (Table 1). Although a number of QTL were likely not detected due to rapid LD decay and low marker density, AM still represents a marked improvement in the efficiency of identifying QTL for spot blotch resistance over that obtained through bi-parental mapping. Moreover, the significantly associated markers identified in this study are probably tightly linked to the respective QTL given the level of LD found across the genome for the WBDC accessions (Roy and Steffenson unpublished). Such markers will be valuable in breeding programs using marker-assisted selection to introgress spot blotch resistance genes.
Five of the twelve mapped QTL identified in this study were found in the same bins where spot blotch resistance loci were previously reported (Fig. 2). This includes previous studies describing either qualitative or quantitative resistance loci identified at either the seedling or adult plant stage and in both cultivated and wild barley (Alsop 2009; Bilgic et al. 2005; Steffenson et al. 1996; Yun et al. 2005, 2006) (Table 1). The chromosomal location of one QTL could not be determined because the marker (11_11361) showing the association has not been mapped. The chromosome 3H QTL Rcs-qtl-3H-bPb-1068 mapped to the same bin as one found at the seedling stage in the Dicktoo/Morex population (Bilgic et al. 2005). The chromosome 7H QTL Rcs-qtl-7H-bPb-4584 mapped to the same bin as one identified at the seedling stage in the Steptoe/Morex, Dicktoo/Morex, Harrington/Morex, Harrington/TR306, and OUH602/Harrington populations (Bilgic et al. 2005; Yun et al. 2005). The second chromosome 7H QTL Rcs-qtl-7H-12_30004 mapped to the same bin as one identified at the seedling stage in the Dicktoo/Morex, Harrington/TR306, and Damon/Harrington populations (Alsop 2009; Bilgic et al. 2005). The other two QTL were found in coincident bins for adult plant resistance reported from previously investigated populations. Specifically, QTL Rcs-qtl-1H-bPb-2813 was identified in a bin coincident to one reported in the Steptoe/Morex population and QTL Rcs-qtl-2H-12_11316 in a bin coincident to one reported in the Harrington/Morex and Damon/Harrington populations (Alsop 2009; Bilgic et al. 2005). The chromosome 7H QTL Rcs-qtl-7H-bPb-4584 mapped to a bin coincident with the major effect spot blotch resistance gene Rcs5, which has been implicated in conferring resistance at the seedling and/or adult plant stages in eight previously analyzed bi-parental mapping populations (Alsop 2009; Bilgic et al. 2005; Steffenson et al. 1996; Yun et al. 2005, 2006). While spurious associations can be a problem in AM (Breseghello and Sorrells 2006), the number of previously reported QTL that we have possibly confirmed suggests false discoveries are relatively low. In this study, we identified one of three QTL for spot blotch resistance reported in three wild × cultivated barley crosses (either seedling or adult plant resistance: Alsop 2009; Yun et al. 2005, 2006) and also two additional ones reported in several cultivated × cultivated barley crosses (Bilgic et al. 2005; Steffenson et al. 1996). Although based only on general coincident bin mapping locations, these results offer some validation for the effectiveness of the AM approach. Validation of QTL identified in the WBDC through AM also was reported by Steffenson et al. (2007) for the rpg4/Rpg5 stem rust resistance gene complex in two wild × cultivated barley populations. Additionally, Cockram et al. (2008) validated the AM approach with the VRN-H1 and VRN-H2 genes for flowering time that control the vernalization requirement in barley. They also showed discrimination between intragenic VRN-H1 markers, demonstrating the feasibility of fine mapping candidate genes.
The other seven QTL identified in the WBDC appear to map at unique locations for spot blotch resistance and may therefore represent new loci (Table 1). While easy and fast to assay, spot blotch resistance at the seedling stage may not be indicative of the reaction at the adult plant stage because some genes are known to function at specific ontogenetic stages (Steffenson et al. 1996). Adult plant resistance is essential for barley cultivars in North America because spot blotch does not increase significantly until after the crop has headed. Additional evaluations of the WBDC for adult plant resistance in the field should be completed before utilizing any accession in the breeding program. Moreover, accessions carrying putative novel alleles also should be tested against a collection of different C. sativus isolates in the greenhouse to determine whether they possess broad-based resistance, as was done by Fetch et al. (2008). The genetics of spot blotch resistance in barley can be complex and dependent on genetic background. For example, Bilgic et al. (2005) identified three different major effect QTL derived from the resistant donor cultivar Morex in individual crosses with three different susceptible parents. One may therefore expect to see an even greater number of spot blotch resistance loci revealed in crosses between WBDC accessions and various breeding lines. These loci will significantly increase the diversity for spot blotch resistance in cultivated barley, thereby reducing the threat of epidemics.
LD, population structure, and AM analyses for the WBDC were conducted separately for DArT and SNP markers. This was done for several reasons: (a) the DArT and SNP marker maps are based on different consensus maps where marker intervals may not have the same degree of precision (Close et al. 2009; Wenzl et al. 2006), (b) the WBDC accessions were assigned to different subpopulations depending on the marker type used, and (c) we were interested in comparing the results obtained for the two marker types readily available to the barley research community. Of the five and eight QTL identified separately by DArT and SNP markers (one QTL identified with a SNP marker had an unknown mapping location), respectively, none mapped in the same bin for the two marker types. The fact that DArT and SNP markers detected unique sets of QTL for spot blotch resistance is not completely surprising. Given the low number of DArT and SNP markers found significantly associated with spot blotch resistance and the very rapid LD decay in the WBDC, the chances of identifying the same QTL with any two markers at distinct genomic positions regardless of type is extremely low.
As mentioned previously, LD in the WBDC is low and decays very rapidly based on both DArT and SNP markers (Steffenson et al. 2007; J. Roy and B. Steffenson, unpublished). To specifically assess the LD pattern at the identified spot blotch resistance QTL, we investigated the association of flanking markers. No LD was observed between nine QTL-identifying markers and their respective flanking markers. This result was expected because of the rapid LD decay. For the remaining three QTL-identifying markers, we observed moderate or strong LD. Moderate LD was observed between the QTL-identifying marker 11_20189 on chromosome 5H (276.8 cM) and one flanking marker. This flanking marker, however, was not significantly associated with spot blotch resistance (data not shown). In contrast, strong LD was observed between the QTL-identifying marker bPb-2813 on chromosome 1H (59.7 cM) and the three flanking markers bPb-0468, bPb-7325, and bPb-7859 (Fig. 3). These same four markers also showed strong LD with another nearby QTL-identifying marker (bPb-2967) on chromosome 1H at 60.7 cM (Fig. 3). We considered these five adjacent markers to be detecting the same QTL because they have strong LD with each other and are also significantly associated with spot blotch resistance. In addition to the chromosome 5H and 1H LD cases, the QTL-identifying marker on chromosome 7H at 107.4 cM (12_30004) had moderate to strong LD with seven other markers at the same position, two of which showed a highly significant association with spot blotch resistance. In predominantly self-pollinated species such Arabidopsis and rice, previous investigators have demonstrated strong LD surrounding genes controlling flowering time and disease resistance—in some cases extending from tens of kb up to 1 cM (Thornsberry et al. 2001; Hagenblad and Nordborg 2002; Nordborg et al. 2002; Garris et al. 2003; Olsen et al. 2004). In cultivated barley, strong LD beyond a target gene has been observed: 212 kb at the Ha locus controlling grain hardness (Caldwell et al. 2006); 5.5 cM at a gene conferring resistance to the Barley Yellow Mosaic Virus (BYMV) complex (Stracke et al. 2007); and 0.7 and 6.4 cM for the VRN-H1 and VRN-H2 loci (Cockram et al. 2008). In this study, only one of 12 identified and mapped QTL showed strong LD up to 1.0 cM. This indicates that higher resolution mapping can be done in the WBDC compared to other reported studies utilizing cultivated barley collections or populations (Cockram et al. 2008; Stracke et al. 2007). Additionally, the low LD of the WBDC can be exploited for conducting candidate gene analyses based on homology with Brachypodium and rice sequences.
In this study, the R 2 for all of the identified QTL was low, ranging from 2.3 to 3.9% (Table 1). A low R 2 value (3.1%) also was found for the QTL identified by DArT marker bPb-4584 at the putative Rcs5 locus. Rcs5 is a major effect locus conferring seedling and/or adult plant resistance depending on the population studied. It was mapped as a Mendelian locus in the Steptoe/Morex population (Steffenson et al. 1996) and as a QTL in eight other populations explaining from 8 to 85% of the phenotypic variation (Alsop 2009; Bilgic et al. 2005; Steffenson et al. 1996; Yun et al. 2005, 2006). If DArT marker bPb-4584 has indeed identified Rcs5, the low variation explained is somewhat surprising and may be due to several reasons. First, previous studies used crosses with relatively small population sizes that are likely to overestimate the effects of QTL (Beavis 1998; Melchinger et al. 1998). Secondly, since many QTL were identified in this study, the presence of a resistance allele at more than one QTL in any one accession could affect the estimate of the QTL effects. For example, if most of the QTL identified had a relatively large effect and most accessions carried the resistance allele at more than one of these loci, then the R 2 for any one QTL would be lower.
In summary, the data accumulated from this study provides a wealth of information from which to initiate preemptive breeding efforts to reduce the vulnerability of barley to spot blotch. The first step would be to identify a small subset of resistant accessions that carry resistance alleles at different QTL. These accessions would then be used as donor parents in a marker-assisted backcrossing scheme to create sets of lines carrying resistance alleles at different loci in a cultivated target background. Further characterization of these sets of near-isogenic lines would confirm the QTL, provide more accurate estimates of allelic effects, and assess whether individual QTL are equally effective against a broad range of pathogen isolates. Finally, the closely linked markers identified as a result of low LD in the WBDC can be used to efficiently pyramid multiple QTL into cultivars, thereby providing more stable spot blotch resistance as was done for stripe rust of barley (Castro et al. 2003).
References
Alsop BP (2009) Linkage analysis and inheritance of multiple disease resistance in intra-specific wild × cultivated barley populations. PhD dissertation, University of Minnesota, St. Paul
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Beavis WB (1998) QTL analyses: power, precision, and accuracy. In: Patterson AH (ed) Molecular dissection of complex traits. CRC Press, Boca Raton
Bilgic H, Steffenson BJ, Hayes P (2005) Differential expression of seedling and adult plant resistance to spot blotch in different genetic backgrounds of barley. Theor Appl Genet 111:1238–1250
Bilgic H, Steffenson BJ, Hayes P (2006) Molecular mapping of loci conferring resistance to different pathotypes of the spot blotch pathogen in barley. Phytopathol 96:699–708
Bonman JM, Bockelman HE, Jackson LF, Steffenson BJ (2005) Disease and insect resistance in cultivated barley accessions from the USDA National Small Grains Collection. Crop Sci 45:1271–1280
Box GEP, Cox DR (1964) An analysis of transformations. J R Stat Soc Ser B 26:211–252
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Buntjer JB, Sorensen AP, Peleman JD (2005) Haplotype diversity: the link between statistical and biological association. Trends Plant Sci 10:466–471
Caldwell KS, Russell J, Langridge P, Powell W (2006) Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics 172:557–567
Castro AJ, Chen X, Hayes P, Johnston M (2003) Pyramiding quantitative trait locus (QTL) alleles determining resistance to barley stripe rust. Crop Sci 43:651–659
Clark RV (1979) Yield losses in barley cultivars caused by spot blotch. Can J Plant Pathol 1:113–117
Close TJ, Bhat PR, Lonardi S, Wu Y, Rostoks N, Ramsay L, Druka A, Stein N et al (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genomics 10:582
Cockram J, White J, Leigh FJ, Lea VJ, Chiapparino E, Laurie DA, Mackay IJ, Powell W, O’Sullivan DM (2008) Association mapping of partitioning loci in barley. BMC Genet 9:16
Ellis RP, Forster BP, Robinson D, Handley LL, Gordon DC, Russell JR, Powell W (2000) Wild barley: a source of genes for crop improvement in the 21st century? J Exp Bot 51:9–17
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F et al (2003) Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 68:69–78
Fan JB, Gunderson KL, Bibikova M, Yeakley JM, Chen J, Wickham Garcia E, Lebruska LL, Laurent M, Shen R, Barker D (2006) Illumina universal bead arrays. Methods Enzymol 410:57–73
Fetch TG Jr, Steffenson BJ (1994) Identification of Cochliobolus sativus isolates expressing differential virulence on two-row barley genotypes from North Dakota. Can J Plant Pathol 16:202–206
Fetch TG Jr, Steffenson BJ (1999) Rating scales for assessing infection responses of barley infected with Cochliobolus sativus. Plant Dis 83:213–217
Fetch TG Jr, Steffenson BJ, Nevo E (2003) Diversity and sources of multiple disease resistance in Hordeum spontaneum. Plant Dis 87:1439–1448
Fetch TG Jr, Steffenson BJ, Bockelman HE, Wesenberg DM (2008) Spring barley accessions with dual spot blotch and net blotch resistance. Can J Plant Pathol 30:534–542
Garris AJ, McCouch SR, Kresovich S (2003) Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165:759–769
Ghazvini H, Tekauz A (2007) Virulence diversity in the population of Bipolaris sorokiniana. Plant Dis 91:814–821
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Hagenblad J, Nordborg M (2002) Sequence variation and haplotype structure surrounding the flowering time locus FRI in Arabidopsis thaliana. Genetics 161:289–298
Hardy J, Singleton A (2009) Genomewide association studies and human disease. N Engl J Med 360:1759–1768
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:e25
Kleinhofs A, Graner A (2001) An integrated map of the barley genome. In: Phillips RL, Vasil IK (eds) DNA-based markers in plants, 2nd edn. Kluwer, Dordrecht, pp 187–199
Lander ES, Botstein D (1986) Strategies for studying heterogeneous genetic traits in humans by using a linkage map of restriction fragment length polymorphisms. Proc Natl Acad Sci USA 83:7353–7357
Melchinger AE, Utz HF, Schön CC (1998) Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149:383–403
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, Stahl EA, Weigel D (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30:190–193
Nutter FW Jr, Pederson VD, Foster AE (1985) Effects of inoculations with Cochliobolus sativus at specific growth stages on grain yield and quality of malting barley. Crop Sci 25:933–938
Oliphant A, Barker DL, Stuelpnagel JR, Chee MS (2002) BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotech Suppl 5:6–58
Olsen KM, Halldorsdottir SS, Stinchcombe JR, Weinig C, Schmitt J, Purugganan MD (2004) Linkage disequilibrium mapping of Arabidopsis Cry2 flowering time alleles. Genetics 167:1361–1369
Pallotta MA, Warner P, Fox RL, Kuchel H, Jefferies SJ, Langridge P (2003) Marker assisted wheat breeding in the southern region of Australia. In: Pogna NE, Romano M, Pogna EA, Galterio Z (eds) Proceedings of the 10th International Wheat Genetics Symposium, Paestum, pp 789–791
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5:94–100
Shakhatreh Y, Haddad N, Alrababah M, Grando S, Ceccarelli S (2009) Phenotypic diversity in wild barley (Hordeum vulgare L. ssp. spontaneum (C. Koch) Thell.) accessions collected in Jordan. Genet Resour Crop Evol. doi:10.1007/s10722-009-9457-8
Steffenson BJ, Hayes PM, Kleinhofs A (1996) Genetics of seedling and adult plant resistance to net blotch (Pyrenophora teres f. teres) and spot blotch (Cochliobolus sativus) in barley. Theor Appl Genet 92:552–558
Steffenson BJ, Olivera P, Roy JK, Jin Y, Smith KP, Muehlbauer GJ (2007) A walk on the wild side: mining wild wheat and barley collections for rust resistance genes. Aust J Agric Res 58:532–544
Stracke S, Presterl T, Stein N, Perovic D, Ordon F, Graner A (2007) Effects of introgression and recombination on haplotype structure and linkage disequilibrium surrounding a locus encoding Bymovirus resistance in barley. Genetics 175:805–817
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES IV (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Valjavec-Gratian M, Steffenson BJ (1997) Pathotypes of Cochliobolus sativus on barley in North Dakota. Plant Dis 81:1275–1278
von Bothmer R, Sato K, Kamatsuda T, Yasuda S, Fischbeck G (2003) The domestication of cultivated barley. In: von Bothmer R, van Hintum T, Knupffer H, Sato K (eds) Diversity in barley (Hordeum vulgare). Elsevier, Amsterdam, pp 9–27
Wenzl P, Carling J, Kudrna D, Jaccoud D, Huttner E, Kleinhofs A, Kilian A (2004) Diversity arrays technology (DArT) for whole-genome profiling of barley. Proc Natl Acad Sci USA 101:9915–9920
Wenzl P, Li H, Carling J, Zhou M, Raman H, Paul E, Hearnden P, Maier C, Xia L, Caig V et al (2006) A high-density consensus map of barley linking DArT markers to SSR, RFLP and STS loci and agricultural traits. BMC Genomics 7:206
Wilcoxson RD, Rasmusson DC, Miles MR (1990) Development of barley resistant to spot blotch and genetics of resistance. Plant Dis 74:207–210
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Bucker ES (2005) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yun S-Y, Gyenis L, Hayes PM, Matus I, Smith KP, Steffenson BJ, Muehlbauer GJ (2005) Quantitative trait loci for multiple disease resistance in wild barley. Crop Sci 45:2563–2572
Yun SJ, Gyenis L, Bossolini E, Hayes PM, Matus I, Smith KP, Steffenson BJ, Tuberosa R, Muehlbauer GJ (2006) Validation of quantitative trait loci for multiple disease resistance in barley using advanced backcross lines developed with a wild barley. Crop Sci 46:1179–1186
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Roy, J.K., Smith, K.P., Muehlbauer, G.J. et al. Association mapping of spot blotch resistance in wild barley. Mol Breeding 26, 243–256 (2010). https://doi.org/10.1007/s11032-010-9402-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-010-9402-8