
Abstract
Map-based cloning of mutant genes is straightforward if the genome sequence and sufficient molecular markers are available. When a mutated gene in Arabidopsis causes a clear phenotype and is located in a genomic region where sufficient meiotic recombination takes place, the gene can be identified within 6–12 months. However, mutated genes that cause weak phenotypes are difficult to map to small genomic intervals due to faulty selection of F2 plants. Here, we describe a method that allows for rapid identification of roughly mapped genes by using a massive parallel sequencing strategy. A genomic region of 150 kb was PCR amplified in 7–17 kb pieces from an EMS Arabidopsis onset of leaf death (old) mutant and its wild-type accession Landsberg erecta (Ler-0). Massive parallel sequencing and subsequent de novo assembly of the short sequences reliably identified 253 polymorphisms in a 110-kb region between the reference Col-0 and Ler-0 sequence. The analysis further revealed potential mutations in the old mutant of which one was confirmed to be present in the mutant. Thus the described method can be used for accelerating the map-based cloning of genes that cause weak phenotypes. An accompanying advantage is that the amplified fragments can be cloned and used to complement the mutant.
Similar content being viewed by others


Introduction
Forward genetics is a widely proven approach in plant molecular biology which aims to identify the sequence change that underlies a specific mutant phenotype. Mutants can be the result of deliberate mutagenesis or come from natural variation (Peters et al. 2003). Chemically or radiation-induced mutagenesis approaches have the broadest spectrum of mutation that can be artificially generated (Robles and Micol 2001; Greene et al. 2003). However, the identification of such a mutation can only reliably be done by a map-based cloning approach (Peters et al. 2003). Map-based cloning, also called positional cloning, is the process of identifying the location of a gene or mutation by measuring genetic linkage to markers whose physical position in the genome is known. All map-based cloning strategies rely on the decrease of the physical distance between the gene of interest and molecular markers. A succession of progressively closer flanking markers on either side of a mutation are identified and, once the physical distance between two flanking markers is small enough, the intervening DNA is sequenced to determine the genetic basis of the mutant phenotype (Jander 2006). Major advances—sequencing of the genomes, the availability of 80,127 SNP sites in the rice genome and more than 50,000 markers in the Cereon Arabidopsis Polymorphism Collection, and improvements in the methods used for detecting DNA polymorphisms (Jander et al. 2002; Peters et al. 2003)—have made map-based cloning in model plants, for example rice and Arabidopsis thaliana, fairly routine. Recently, more than one million non-redundant SNPs were identified by high-density array sequencing of 20 different Arabidopsis accessions (Clark et al. 2007).
Map-based cloning critically relies on the successful identification of the recessive phenotype in a segregating population. In cases where this cannot be reliably done, genotypes could erroneously be selected that do not carry the two recessive alleles. Such incorrect recombinant plants can greatly hamper the map-based cloning process and can put a limit on the distance one can map a phenotype to a marker. For example, if only one percent of all selected plants are incorrect recombinant plants, then one can never map closer than to ~1 cM of the gene causing the phenotype.
Selection of incorrect recombinant plants can occur if a mutation causes a weak phenotype. Propagation of apparent recessive phenotypes and F3 phenotype examination can often resolve weak phenotype issues, albeit slowly. Even if the phenotype is strong in the parent background, it may become less obvious, or may disappear altogether after pollination with other accessions. This phenomenon can arise when small phenotypic variations contribute to a phenotype as a result of differences in quantitative traits loci (QTL) between the respective accessions. Thus a certain combination of QTL can give the same phenotype as the mutant without the need for the mutation, or it can abolish the phenotype, even if two mutant alleles are present. Here, inspection of the F3 population of selected individuals may not exclude incorrect recombinants, as the combination of QTL may provide a stable phenotype in the following generation. In cases where an approximate chromosomal position of the mutation is known and where the mutation is in the Arabidopsis Landsberg erecta accession, the mutant can be crossed to a near-isogenic-line (Keurentjes et al. 2007). In this way the QTL affecting the phenotype can be separated from the mutation, which enabled for the map-based cloning of the RDO4 gene (Liu et al. 2007). Furthermore, the progress of a map-based cloning experiment can be hampered if the gene of interest is located in an area where reduced crossing-over events take place during meiosis, e.g., in centromeric regions.
One possible solution is to analyse a substantial number of F2 plants displaying the recessive phenotype. Statistical analysis of more data points helps with the mapping of the gene of interest. False recombinant plants can be excluded by statistical analysis, if the gene of interest is mapped to a region with sufficient confidence. However, reaching a high mapping resolution suitable for cloning the gene is time costly. In fact, transformation of the recessive phenotype with clones containing multiple genes from the mapped region might be less time-consuming. The amplification of large genomic fragments up to 20 kb is feasible for Arabidopsis and thus a fairly straightforward approach for complementing the mutated gene. However, reliable complementation of the recessive phenotype often requires phenotypic analysis of the T2 population. Therefore sequence identification of a mutation before complementation is preferred, because it greatly enhances the work-flow for subsequent follow-up experiments.
Recent advances in DNA sequencing technologies have the potential for up to three orders of magnitude more sequence throughput than conventional Sanger sequencing. The invention of the Genome Sequence 20 DNA Sequencing System (454 parallel sequencing platform from Roche) has enabled the rapid and high-volume production of sequence data (Binladen et al. 2007). Recently, Applied Biosystems announced the ABI SOLiD system, which uses a ligation-based chemistry. Another instrument that utilizes a parallel sequencing strategy is available from Illumina. The Illumina genome analyser produces millions of short sequences of up to 36 bp reads (Bennett 2004). These high-throughput DNA sequencing instruments are mainly applied for re-sequencing of genomes. Here we present the use of the Illumina genome analyser as a cost-effective method for fast identification of a point mutation within a 150 kb mapped region of Arabidopsis.
Experimental procedures
Plant materials and growth conditions
Arabidopsis thaliana ecotype Landsberg erecta (Ler-0) was used in this study. The old mutants were obtained from an EMS mutagenized collection (Jing et al. 2002, 2005). All plants were sown directly on soil (Lentse potgrond, www.hortimea.nl, Elst, The Netherlands) and transferred to a climate room at 21°C and 65% humidity with 16 h light (60 μm m−2 s−1)/8 h dark cycles after three days of cold treatment at 4°C.
Genomic DNA isolation
Total DNA was extracted from 25 day-old frozen plants using a method as described by (Dellaporta et al. 1983) and (Kater et al. 1991), with minor modifications. Genomic plant DNA was isolated by grinding 5 g leaf tissue in liquid nitrogen and mixing with 45 ml cold extraction buffer (0.35 M sorbitol, 0.1 M Tris–HCl, 5 mM EDTA pH 7.5). The suspension was centrifuged at 16g for 10 min at 4°C and the supernatant discarded. The pellets were resuspended in 1.25 ml extraction buffer/1.75 ml nuclei lysis buffer (2% w/v CTAB, 0.2 M Tris–HCl pH 8.0, 2 M NaCl, 50 mM EDTA)/0.6 ml 5% w/v sarkosyl and incubated at 65°C for 1 h. The mixture was extracted with 8 ml chloroform:iso-amyl alcohol (24:1 v/v), the DNA was precipitated from the aqueous phase with an equal volume of 2-propanol, dissolved in T10E1 (10 mM Tris–HCl, 1 mM EDTA pH 8.0) containing 5 μg Dnase-free Rnase A and incubated for 30 min at 60°C.
PCR
PCR products of genomic Arabidopsis DNA were amplified in a range 7–17 kb by Extaq DNA polymerase (TaKaRa #RR001A). Primers were designed by primer3 (http://frodo.wi.mit.edu/) and run with PCR program (4′94°C/1′94°C, 1′62°C, 7′–15′68°C, 35× cycles/5′68°C). PCR products were isolated from 1% agarose gels using the Quiquick (Qiagen #28706) gel extraction kit. DNA concentrations of the individual PCR products were measured by using a Nanodrop spectrophotometer (Nanodrop ND-1000; Isogen, NL). The products were pooled in equimolar amounts at a final concentration of 0.1 μg/μl.
Sequence data generation by Illumina genome analyser sequencing technology
Sample preparation was performed as described by the manufacturer. Briefly, following fragmentation by nebulization and ligation of the Illumina adapters to the fragments, the fragments were purified from 2% TAE agarose gel to remove unligated adapters or adaptors that have ligated to one another. Ligation products of approximately 150 bp were purified from gel, and PCR amplified using the adapter primers for 18 cycles to avoid skewing the representation of the library. Flow cell cluster generation using the Illumina cluster station and sequence data generation using the 1G analyser were performed using manufacturer’s protocols. Image analysis, base calling, and alignment to a reference sequence were performed using the Illumina data analysis pipeline (version 2.2.5). A total of 85% of the sequence tags passed the quality filter with standard parameters (Chastity ≥ 0.6).
ELAND and SSAKE sequence data analysis
Sequence tags were mapped to the reference sequence using the ELAND software (Illumina), which aligns short sequence tags to a reference with up to two mismatches. After alignment of the fragments, the identities of calls for each base of the reference sequence were counted using a shell script. Sequence coverage and ratios of wild-type/mutant alleles were calculated and putative SNPs were selected for further investigation.
For SSAKE analysis, approximately 500,000 36-bp sequence tags were trimmed at the 5 and 3′ end to 32 bp. SSAKE (http://www.bcgsc.ca/platform/bioinfo/software/ssake) assembly v2.0 (Warren et al. 2007) was subsequently performed on a 3-GHz Intel Pentium IV computer with 512 MB RAM running KNOPPIX (http://www.knoppix.org/) with default parameters (–m 16, –o 3, –r 0.7). Running parameters were: –m 25, –o 2 –s 1. Generated contigs were sorted by blasting against the TAIR7 whole genome BAC clones database. Sequence alignment was performed with the SeqMan utility of DNASTAR Lasergene (Version 6; Madison, WI, USA). Scanning of the sequence resulted in the detection of possible mutations between the wild type and the mutant line. Detected mutations were scored on the basis of the coverage ratio of the contig as presented by SSAKE. Limiting the coverage to a factor of >10 clears the data of all background mutations that might have come from PCR or Illumina read-mistakes.
Results and discussion
The old101 phenotype is masked in the F2 population of a cross between the old101 mutant and the non-parental Col-0 accession
The Arabidopsis old101 mutant was identified during screening for altered leaf senescence phenotypes (Jing et al. 2002). The EMS-induced old101 mutation segregates as a monogenic recessive trait when backcrossed to its parental wild-type accession Ler-0 (Table 1). For positional cloning a mapping population was generated by crossing the old101 mutant, which is in the Ler-0 background, to the Col-0 accession. Subsequent analysis of the F2 population revealed that the old101 phenotype did not segregate in a 3:1 (wild-type:mutant) ratio (Table 1). However, a two-gene model did not fit with the segregation data, and therefore is seems likely that QTL associated with leaf senescence affect the phenotype. Previously, senescence-associated QTL were identified in Arabidopsis (Diaz et al. 2006; Luquez et al. 2006) which have a significant effect on crosses between Ler-0 and Col-0. The presence of QTL affecting leaf senescence, is likely to hamper the unequivocal identification of the mutant old101 phenotype in the mapping population.
Mapping the old101 mutation
Approximately 500 F2 plants displaying the old101 phenotype were selected and genotyped with two molecular markers for each chromosome. In general, analysis of 500 F2 plants should allow for the gene causing the phenotype to be mapped within a 1 cM region. However, the mapping of old101 mutation resulted in a linkage of ~30 cM with a marker on chromosome 2 and another of ~20 cM on chromosome 5. Unable to narrow down the map position through F2 analysis, we selected 400 new F2 plants with a senescence phenotype, the seeds were collected, and the F3 population was screened for segregation of the phenotype. Molecular analysis of the F3 population resulted in the identification of a single linkage within a genomic region of ~500 kb on chromosome V between markers on the BACs MJH22 and MUB3 (data not shown). Subsequently, 1,000 random F2 plants were selected and checked for possible recombination events between the markers for the BAC clones MJH22 and MUB3. Plants showing a recombination between the markers were selected and propagated. The F3 plants were screened for segregation of the senescence phenotype and the position of the recombination event was estimated by molecular analysis of the mapped region. This approach allows for using F2 plants that are heterozygous or homozygous for the old101 mutation and plants that show a wild-type phenotype. The chromosomal position of the old101 mutation could be defined within a ~150 kb region (Fig. 1). However, some of the identified recombinant plants gave contradicting results, indicating the mutation was in different areas within the mapped region. Taken together, the influence of QTL on the phenotype could not be excluded and this hindered the fine mapping of the old101 mutation.

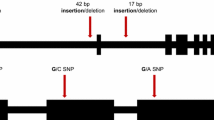
Genomic region of the old101 mutation. A schematic representation of a section of Arabidopsis thaliana chromosome 5 between coordinates 25,400 and 25,800 kb covering five BAC clones is shown. The BAC number is indicated above each of the five thick horizontal lines (a). Within the ~400-kb region recombinant plants were genotyped (c) as indicated by arrows. Symbols above the arrows represent the number of recombinants followed by the BAC number and the approximate position of the marker on the indicated BAC (e.g. 2/MBK5/5000 indicates two recombinants on BAC MBK5 at approximate position 5,000). About 150 kb of genomic DNA from the Ler-0 accession and from the old101 mutant was amplified by PCR (b) Fourteen PCR amplicons of 7–17 kb were sequenced using the Illumina genome analyser (small horizontal lines)
Sequencing of large genomic regions of the mutant and parent accession using massive parallel sequencing
The Illumina genome analyser is a high-throughput DNA sequencing instrument capable of determining tens of millions of short (~35 bp) sequence reads (Bennett 2004). The sequencing technology uses templates that are immobilized on a flow cell surface consisting of eight lanes. A single lane can provide up to five million short sequence tags of 36 bp.
We decided to sequence the ~150 kb region encompassing the old101 mutation using the genome analyser. The genome of the Arabidopsis Col-0 accession has been determined and the old101 mutation is in the Ler-0 background. Since many genomic polymorphisms exist between Col-0 and Ler-0 we expect that identification of the mutation is only feasible when the natural variation between the ecotypes has been determined. Therefore, we needed to obtain the Ler-0 sequence also. To reduce the cost of the sequencing, both the Ler-0 and the mutant were multiplexed on to a single lane. Thus, after alignment of the sequence tags to the reference sequence we would expect to see a continuous sequence alignment, as the sequence from the Ler-0 accession is essentially identical to that from the mutant. However, at the position of the mutation, tags originating from the old101 mutant will have a single base change compared with those originating from the wild type. Thus, at the position of the old101 mutation, half the sequence tags were expected to be different from the other half. To enable straightforward identification of the mutation it is crucial to have high redundancy in the amount of sequence reads. Preferentially the redundancy is more than 30-fold, however, we aimed for redundancy of ~50-fold. This would be exceeded by a factor of more than two in the case of the minimum expected one million tags and a sequence region of 150 kb. We therefore used the overcapacity to sequence other areas of the Ler-0 genome.
The total length of the genomic DNA that was being sequenced was ~700 kb, of which 150 kb was from the old101 mutant and 150 kb of equivalent area of the wild-type Ler-0 accession. Primers were designed to cover the target area and 14 amplicons of sizes ranging from 7 to 17 kb were PCR amplified, using a high-fidelity enzyme as described in Experimental procedures (Fig. 1). PCR fragments were separated and isolated from agarose gel. Subsequently, fragments were pooled in such a way that the final sample for sequencing contained a total of 700 kb PCR products at a concentration of 0.1 μg/μl.
Analysis of the sequence data using ELAND and SSAKE identifies potential mutations
The single lane on the Illumina genome analyser produced ~0.6 million sequence tags of 36 nucleotides. The number of sequence tags was less than the expected 1–2 million tags, because of suboptimum cluster formation on the flow cell after sample application. However, the resulting amount of tags was sufficient for comprehensive analysis.
To determine the mutated sequence within the mapped region two approaches were used to analyse the data. The first approach makes use of the efficient local alignment of nucleotide data (ELAND) software, provided by the manufacturer, and performs a direct alignment against the Col-0 reference sequence. Approximately 300,000 short sequence tags were aligned with the reference sequence of the old101-containing region. We assumed that ~150,000 were to be sequence reads from the old101 mutant while the other half would originate from the wild-type Ler-0. The alignment of ~300,000 tags from the old101 area demonstrated that the ELAND software can efficiently align short sequence reads since sequence tags belonging to other areas of the Ler-0 genome were separately aligned. In-silico analysis of the alignment revealed 81 potential SNPs between the mutant and wild-type sequences. The large number of potential mutations can be in part explained by the fact that Col-0 has been used as reference. A sequence read is only added to the alignment when no more than two mismatches occur. However, because of natural variation between Col-0 and Ler-0, misalignments can occur, especially within repetitive sequences of, for example, similar protein domains. Figure 2 shows a snapshot of the alignment of sequence tags against the reference sequence. For each base the number of times it was called identically and differently from the reference was determined. Since wild-type and mutant DNAs were mixed in a 1:1 ratio, putative SNPs/polymorphisms could be identified by selecting nucleotide ratios close to 1:1. However, ratios in which one allele is under-represented (in our case less than 20%), due to either PCR amplification mistakes or sequencing errors were excluded from further analysis. Because EMS induces G/C to A/T transitions in >99% of the cases, we anticipated a single EMS-induced mutation in the mapped region (Greene et al. 2003). By screening the potential mutations for a G/C to A/T transition, which would result in an amino acid change, only three potential mutations remained. Subsequently the genomic intervals containing the three potential mutations were cloned and analysed by Sanger sequencing. Of the three possible mutations, one was confirmed to be present in a coding region for the old101 mutant region as a G to A change (Fig. 2). Complementation of the mutant phenotype with the wild-type gene is needed to confirm that the discovered mutation is the cause for the mutant phenotype.

Snapshot of the alignment of the old101 sequence tags against the Col-0 reference sequence (a). The quality filtered sequence tags from wild-type and mutant were aligned to the reference using the ELAND software. Mutations indicated by circles, can be due PCR amplification mistakes or sequencing errors. A putative SNP between the wild type and mutant that shows an allele frequency ratio close to 1:1 is indicated, and subsequently confirmed by Sanger sequencing. (b) The trace of old101 mutant versus the Ler-0 parental sequence
The analysis showed, furthermore, that direct ELAND alignment of the short sequence reads of the Illumina against the Col-0 reference sequence is inaccurate when the Ler-0 sequence becomes highly polymorphic, because only two mismatches between sequence tags and the reference sequence are allowed. Therefore, a second method was used which includes a de-novo assembly of the short-sequence reads with a novel algorithm called SSAKE (Warren et al. 2007). After the assembly, the fragments were aligned to the Col-0 reference sequence. SSAKE assembly of 500,000 short-sequence reads was performed and resulted in contigs varying in size from 36 up to ~1,500 base pairs. Assembled contigs were aligned with the Col-0 DNA reference sequence spanning the mapped region. This resulted in an almost complete alignment of the sequenced region with the reference except for 18 gaps with an average size of 34 bp. These gaps were all present in an approximately 10-kb area on BAC MHJ24 where sequence coverage was low because of poor PCR amplification. Polymorphisms between the Ler-0 genome and the Col-0 reference were found and in total 253 polymorphisms in a region of 110 kb were identified (Table 2 and S1). A great advantage of this method is that also InDels can be identified, which is impossible with the ELAND analysis. Approximately 90% of the identified polymorphisms could be verified against the Monsanto Arabidopsis polymorphism database (Jander et al. 2002) and the TAIR website (www.arabidopsis.org), demonstrating the potential of this method for novel polymorphism detection. The frequency of polymorphisms between Arabidopsis accessions has been estimated to be one SNP every 166 bp (Buckler and Gore 2007) or one SNP every 336 bp (Schmid et al. 2003). Our data of one polymorphism every 230 bp lies within those values. Next, the EMS-induced mutation was discovered by scanning the sequence for potential mutations as described in the Experimental procedures section. This resulted in identification of eight potential mutations between the old101 genomic DNA and the wild-type accession. The SSAKE log file enables identification of the number of base calls within an assembled contig. Of the eight possible mutations, only one had a read redundancy of more than 50-fold while the others were on contigs with less than 6-fold redundancy and were, therefore, regarded as unreliable. The detected mutation was consistent with the previously detected G/C to A/T transition using the ELAND analysis. Taken together, the de-novo assembly was successful in identifying a single-point mutation and was more comprehensive than ELAND analysis because more than 250 polymorphisms were detected. Thus SSAKE is a valuable tool for analysis of experimental data from the genome analyser.
Implications for the identification of mutations in large genomic regions
The genome analyser can read up to 40 million sequence tags in a single run of a flow cell consisting of eight lanes. Pooling of mutant and wild-type DNA samples proved to be an acceptable solution in order to reduce the costs of the sequencing to a single lane. Theoretically the capacity enables sequence analysis of 400-kb fragments of mutant and wild-type DNA in a single run while a 100-fold read redundancy is maintained. If a larger genomic area needs to be sequenced, the use of two lanes, of which one is used for the wild type and the other for the mutant(s), may be preferable. A single lane may allow the identification of a single mutation in a 400-kb genomic area, but equally well enables identification of eight mutations in eight different genomic areas when the affected genes are mapped to an average region of 50 kb. Therefore the parallel-sequencing strategy may provide significant advantages for the identification of genes that are mapped to small genomic regions and those that are mapped on large regions. The availability of a reference sequence is necessary for amplification of target DNA and identification of the mutation through sequence alignment. Unless de-novo sequence assembly can be improved to generate longer contigs, the method described is for now only applicable to genomic areas with good sequence coverage. The total experiment can be done in less than two months and taking into account that mutations can be identified at a current cost of 1% of the cost of conventional capillary sequencing, significant savings on time and labour can be achieved. Increased use of the system, together with increasing sequence read lengths should make this method even more affordable in the near future.
The use of the novel SSAKE algorithm greatly enhanced the accuracy of sequence alignment and enabled the discovery of a great number of polymorphisms. Therefore we expect that even without reference sequences this method will become useful in the analysis of crops and model plants for assessment of genetic diversity, detection of genotype/phenotype associations, and marker-assisted breeding. A number of whole-plant genome sequence projects are in progress, including those for economically important crops such as tomato, potato, maize, barley, and soybean. The presented method implies that identification of mutations in these crops may become routine in the near future.
References
Bennett S (2004) Solexa Ltd. Pharmacogenomics 5:433–438. doi:10.1517/14622416.5.4.433
Binladen J, Gilbert MT, Bollback JP, Panitz F, Bendixen C, Nielsen R et al (2007) The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One 2:e197. doi:10.1371/journal.pone.0000197
Buckler E, Gore M (2007) An Arabidopsis haplotype map takes root. Nat Genet 39:1056–1057. doi:10.1038/ng0907-1056
Clark RM, Schweikert G, Toomajian C, Ossowski S, Zeller G, Shinn P et al (2007) Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317:338–342. doi:10.1126/science.1138632
Dellaporta SL, Woods J, Hicks JB (1983) A plant DNA minipreparation version II. Plant Mol Biol Rep 1:19–21. doi:10.1007/BF02712670
Diaz C, Saliba-Colombani V, Loudet O, Belluomo P, Moreau L, Daniel-Vedele F et al (2006) Leaf yellowing and anthocyanin accumulation are two genetically independent strategies in response to nitrogen limitation in Arabidopsis thaliana. Plant Cell Physiol 47:74–83. doi:10.1093/pcp/pci225
Greene EA, Christine A, Codomo CA, Taylor NE, Henikoff JG, Till BJ et al (2003) Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics 164:731–740
Jander G (2006) Gene identification and cloning by molecular marker mapping. Methods Mol Biol 323:115–126
Jander G, Norris SR, Rounsley SD, Bush DF, Levin IM, Last RL (2002) Arabidopsis map-based cloning in the post-genome era. Plant Physiol 129:440–450. doi:10.1104/pp.003533
Jing HC, Sturre MJ, Hille J, Dijkwel PP (2002) Arabidopsis onset of leaf death mutants identify a regulatory pathway controlling leaf senescence. Plant J 32:51–63. doi:10.1046/j.1365-313X.2002.01400.x
Jing HC, Schippers JH, Hille J, Dijkwel PP (2005) Ethylene-induced leaf senescence depends on age-related changes and OLD genes in Arabidopsis. J Exp Bot 56:2915–2923. doi:10.1093/jxb/eri287
Kater MM, Koningstein GM, Nijkamp HJJ, Stuitje AR (1991) cDNA cloning and expression of Brassica napus enol-acyl carrier protein reductase in Escherichia coli. Plant Mol Biol 17:895–909. doi:10.1007/BF00037070
Keurentjes JJ, Bentsink L, Alonso-Blanco C, Hanhart CJ, Blankestijn-De Vries H, Effgen S et al (2007) Development of a near-isogenic line population of Arabidopsis thaliana and comparison of mapping power with a recombinant inbred line population. Genetics 175:891–905. doi:10.1534/genetics.106.066423
Liu Y, Koornneef M, Soppea WJJ (2007) The absence of Histone H2B Monoubiquitination in the Arabidopsis hub1 (rdo4) mutant reveals a role for chromatin remodeling in seed dormancy. Plant Cell 19:433–444. doi:10.1105/tpc.106.049221
Luquez VM, Sasal Y, Medrano M, Martín MI, Mujica M, Guiamét JJ (2006) Quantitative trait loci analysis of leaf and plant longevity in Arabidopsis thaliana. J Exp Bot 57:1363–1372. doi:10.1093/jxb/erj112
Peters JL, Cnudde F, Gerats T (2003) Forward genetics and map-based cloning approaches. Trends Plant Sci 8:484–491. doi:10.1016/j.tplants.2003.09.002
Robles P, Micol JL (2001) Genome-wide linkage analysis of Arabidopsis genes required for leaf development. Mol Genet Genomics 266:12–19. doi:10.1007/s004380100535
Schmid KJ, Sorensen TR, Stracke R, Torjek O, Altmann T, Mitchell-Olds T et al (2003) Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res 13:1250–1257. doi:10.1101/gr.728603
Warren RL, Sutton GG, Jones SJ, Holt RA (2007) Assembling millions of short DNA sequences using SSAKE. Bioinformatics 23:500–501. doi:10.1093/bioinformatics/btl629
Acknowledgements
We would like to thank Rene Warren for his help with the SSAKE algorithm. This work was partially funded by a grant from the Ministry of Science, Research and Technology of Islamic Republic of Iran to Reza Shirzadian-Khorramabad.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Marcel J. G. Sturre and Reza Shirzadian-Khorramabad contributed equally to this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sturre, M.J.G., Shirzadian-Khorramabad, R., Schippers, J.H.M. et al. Method for the identification of single mutations in large genomic regions using massive parallel sequencing. Mol Breeding 23, 51–59 (2009). https://doi.org/10.1007/s11032-008-9213-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-008-9213-3