
Abstract
Many countries in the world have recently experienced an outbreak of COVID-19, turned out to be a pandemic which significantly affected the world economy. Among many attempts to treat/control infection or to modulate host immunity, many small molecules including steroids were prescribed based on their use against other viral infection or inflammatory conditions. A recent report established the possibility of usage of a corticosteroid against the virus through inhibiting NSP-15; an mRNA endonuclease of SARS-CoV-2 and thereby viral replication. This study aimed to identify potential anti-viral agents for the virus through computational approaches and to validate binding properties with the protein target through molecular dynamics simulation. Unlike the conventional approaches, dedicated data base of steroid like compounds was used for initial screening along with dexamethasone and cortisone, which are used in the treatment of COVID-19 affected population in some countries. Molecular docking was performed for three compounds filtered from data base in addition to dexamethasone and Cortisone followed by molecular dynamics simulation analysis to validate the dynamics of binding at the active site. In addition, analysis of ADME properties established that these compounds have favorable drug-like properties. Based on docking, molecular dynamics simulation studies and various other trajectory analyses, compounds that are identified could be suggested as therapeutics or precursors towards designing new anti-viral agents against SARS-CoV-2, to combat COVID-19. Also, this is an attempt to study the impact of steroid compounds on NSP-15 of SARS-CoV-2, since many steroid like compounds are used during the treatment of COVID-19 patients.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe Acute Respiratory Syndrome out break by SARS-CoV and Middle East Respiratory Syndrome (MERS-CoV) identified in 2002/2003 and 2012, respectively. The infections of SARS-CoV spread to 27 countries and MERS-CoV resulted in 8000 infections worldwide. Presently, SARS-CoV-2, newly identified virus is creating a havoc around the world. Infection causes a disease of Corona Virus disease -2019, a serious pneumonia where mortality is high compared with other viral infections. World Health Organization declared SARS-CoV-2 infection as pandemic. Recent emergence of many variants add complexity in understanding the disease and hence in strategies to control or manage the situation. COVID-19 has worse effect both on human life and world economy. This disease challenge the present day health care system and made the human race to think about the in-adequacy of available medical facilities to fight this invisible particles, SARS-CoV-2.
Scientific community and pharma companies have developed vaccines which are used for mass vaccination is taken up by many countries. Since no specific drug is discovered to combat COVID-19, some small molecule drugs such as remdesivir are prescribed time to time. The spike protein which is essential for viral invasion was targeted for vaccine development as well as for drug designing efforts. There are reports on different targets such as Non-Structural proteins with varying functions involved in viral replication. Also, from these reports describe the possibilities aiming to inhibit them in turn to control viral replication. Researchers employ different data bases such as drug data base, small molecule data base and natural compounds data base etc., for screening against different targets identified of SARS-CoV-2. This approach is faster and may result in effective compounds against SARS-CoV-2.
Historically, besides different approaches to design anti-virals, steroids have been used against viral diseases for treatment as immuno-suppressant. Systemic use of corticosteroids were practiced against SARS-CoV and MERS-CoV for the symptom of pneumonia resulted in increased viral replication [1]. Recent study of revisiting the usage of corticosteroids divulged the effect on viral replication as well as inflammation of the host. It was shown that the steroid compounds, Ciclesonide and mometasone had concentration dependent effect on viral growth and cytotoxicity [2]. The report suggested that the Ciclesonide interacts with NSP-15 directly or indirectly.
NSP-15 is a nidoviral RNA uridylate-specific endoribonuclease (NendoU). It carries C-terminal catalytic domain belonging to EndoU family [3]. Previously, NSP-15 was thought to be directly involving in viral replication but later found that it’s role in viral replication is indirect. Also, the immunomodulatory property of NSP-15 by interfering innate immune response is recently reported [4]. In addition, it was also suggested that NSP-15 is involving in RNA degradation which help to hide from the host defenses. Considering various roles of NSP-15, it is reported as essential in coronavirus biology.
By considering the use of steroids and the importance of NSP-15 enzyme, here we have used dedicated steroid compounds data base for screening against the target NSP-15. Also, binding of different steroid compounds with NSP-15 have been analyzed in-detail through MD simulation and tragterory analysis. This study elucidates structure based interactions of various steroid compounds with NSP-15 (endoribonuclease) which will be useful to expand research efforts on the use of steroid compounds against SARS-CoV-2.
Computational methods
Library preparation
A total of 533 steroid like compounds were retrieved from ChemDiv database and eight steroid compounds with anti-viral activity as reported [2] were chosen for the structure based virtual screening against NSP-15 protein target.
Protein and ligand preparation
Protein preparation
The three-dimensional structure of NSP-15 was retrieved from the Protein Data Bank (PDB ID: 6VWW) [5]. The structure of NSP-15 and the active site are shown in Fig. 1. To perform the docking studies, the protein structure was prepared using protein preparation wizard available in Schrödinger suite [6]. There are two steps involved in protein preparation. First one is the preparation; in this step hydrogens were added and side-chain atoms were neutralized [7]. The second step is refinement where the water molecules were removed and hydrogen atoms were geometry minimized until the average root mean square deviation of the non-hydrogen atoms reached 0.3 Å [8].

Crystal structure of NSP-15 of SARS-CoV-2 and the active site details (PDB ID: 6VWW)
Ligand preparation
Ligprep module included in PHASE was used to convert 2D to 3D structures, and energy minimization was performed using OPLS 2005 force field [9], with implicit GB/SA solvent model. Using the rapid torsion angle approach, the conformers of all the compounds were generated and structures with high estimated energies were eliminated. Using preprocesses, minimization of 100 steps and post-process minimization of 50 steps, maximum of 1000 conformers for each structure were generated. All the minimized conformers were filtered using a relative energy window of 10 kcal mol−1 and a minimum atom deviation of 1.0 Å [10]. Energy threshold value (10 kcal mol−1) was set as to the lowest energy conformer. Conformers having higher energy were discarded.
Structure based virtual screening
Structure based virtual screening is the simple and best approach to find suitable lead molecules against the target protein. To identify a suitable steroid like ligand against NSP-15, ChemDiv DB chemical database was used and SBVS protocol was employed. Initially, a receptor grid was generated based on the non-structural protein -15 (NSP-15) structure around the active site. The prepared database compounds were docked with structure based docking parameter of extra precision (XP) procedures with default settings [11]. The best compounds were selected based on their glide score and energy. SBVS has been carried out with 541 compounds. Based on key interaction profile and glide energy, 16 compounds were short listed. Finally, by employing induced fit docking (IFD) protocols, five lead compounds were shortlisted based on their binding energetics. The overall lead identification work flow is shown in Fig. S1.
Molecular docking (induced fit docking)
From the Schrodinger’s Glide extra precision XP docking results, best hit compounds were selected and subjected to induced fit docking (IFD) using Glide Schrödinger, LLC, New York, NY, 2015 [12]13. A mixed molecular docking method where the protein was flexible and ligand was rigid during docking was employed. During docking protocols, van der Waals (vdW) radii scaling of 0.5 for the proteins and ligand nonpolar atoms was used. The NSP-15 protein structure (PDB ID: 6VWW) was subjected to energy minimization, using force field (OPLS-2005) with an implicit solvation model. The Prime module was used to determine Docking score and Glide energy. Finally, the obtained binding poses were evaluated through glide empirical scoring function. The best pose of docked protein–ligand complexes were analyzed using Pymol and ligplot [14, 15].
Molecular dynamics simulation
The complexes were solvated with water molecules in a cubic box where a protein molecule is at 10 Å distance from the box edge. TIP3P water model was used for simulations. The system was neutralized by adding counter ions. Then, the system was subjected to maximum force minimization of 1000 kJ/mol/nm. The minimized system was subjected to initial NVT equilibration followed by NPT for 500 ps. The system was maintained at 300 K and 1 bar using modified Berendsen Thermostat [16] and Parinello-Rahman barostat [17] for temperature and pressure, respectively. Subsequently, the ligand-bound protein was subjected to 50 ns molecular dynamics. The MD simulations were carried out using Gromacs 5.1 version [18].
PCA analysis
PCA analysis was carried out based on the Cartesian coordinate deviants from the reference crystal structure (6VWW) through diagonalization of covariance matrix. PCA tends to reduce the dimension of the data. In MD, the principal components refers to the eigen vector determining the direction of motion and the eigen value, the extent of residual motion. The low dimensional displacement subspace covered by first few eigen vector representing the PC’s is termed as essential dynamics [19].
The PCA is defined by the coordinates of the trajectories \(x\)(t). The correlational atomic motions is expressed in terms of covariance matrix Cα equation below:
where \(\langle x\rangle\) represents the positional average over time. The matrix Cα can be normalized by orthogonal transformation T given by:
The transformation leads to the determination of eigen value from the diagonal matrix \(\Delta =\langle q{q}^{T}\rangle \, \text{of }{\lambda }_{i}\). The overall solution of the matrix can be given by:
All the calculations were chosen for Cα atoms to find maximum variance in implicit solvent system by stripping the solvent and ions from the trajectories. CPPTRAJ [20] implemented in AMBER was used for computing the PCA. The first three PCA with corresponding eigen values were utilized to understand the conformations.
Free energy analysis
Protein energy was calculated with molecular mechanics with generalized born surface approximation (MM/GBSA)[21].
The protein energy was estimated using the equation having energy contribution terms given by Eq. 4:
In the above equation, first three terms represent molecular mechanics energy such as internal, electrostatics and van der Waals energy. \({G}_{\text{solvation}}\) and \({G}_{\text{non-polar solvation}}\) represents solvation energies, T represents absolute temperature, SMM represent the entropy calculated from molecular mechanics from estimated harmonic frequencies of vibrations.
ADME prediction
To evaluate pharmacological properties of ligands, QikProp [22] module in Schrödinger 2015–2 was used. The following properties were predicted such as Molecular weight (MW), Hydrogen bond donor (HBA), hydrogen bond acceptor (HBD), water partition (QPlogPo/w), blood–brain barrier (BBB) penetration (QPLogBB) and central nervous system (CNS) activity of the molecules were predicted.
Results and discussion
The structure based virtual screening was performed to identify new lead(s) of steroid like compounds with therapeutic effectiveness against COVID-19 targeting NSP-15 protein. Molecular docking through induced fit strategies followed by MD simulation revealed the binding mode, strength and stability of protein–ligand interactions.
Virtual screening
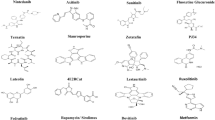
The library of 533 steroids compounds from ChemDIV database was screened against NSP-15 through structure based docking using glide XP module. Compounds were ranked using docking score and glide energy (Table S1a and S1b) and the top 8 hit compounds were shortlisted. In addition, docking studies were carried out for eight steroid compounds having anti-viral activities (tested compounds) as reported [2]. From these analysis, three compounds from ChemDIV (0449-0045, N001-0004, N006-0008) and two compounds (Dexamethasone and Cortisone) from tested compounds were identified as potential compounds based on glide scoring function and ligand–protein interaction profiles. The 2D chemical schemes are shown in Fig. 2.

2D structure of known steroids and best hit compounds
Molecular docking (induced fit docking)
Induced Fit Docking (IFD) has been carried out for the tested compounds (from earlier reports) and steroid compounds from ChemDIV against NSP-15 of SARS-CoV2. All the compounds are docked in to the active site having interactions with catalytic residues with meaningful binding energetics. The compound 0449-0045 make four hydrogen bond interactions with SER294, VAL292, GLN245 and GLU340 at a distance of 2.83 Å, 3.19 Å, 2.81 Å and 2.75 Å respectively as the best pose with glide score and energy of − 9.74, − 65.61 kcal/mol, respectively. Compound N001-0004 shows binding with docking score of − 7.87 and glide energy of − 45.99 kcal/mol by interactions with residues, HIS235, VAL292, SER292 and GLY248 at a distance of 2.97 Å, 3.30 Å, 2.98 Å and 2.99 Å, respectively. Similarly, N006-0008 compound has five hydrogen bond interactions with SER294, LYS290, HIS250, GLY248 and GLN245 and at a distance of 3.14 Å, 2.79 Å, 3.06 Å, 3.25 Å and 2.86 Å respectively with glide score of − 9.42 and glide energy of − 55.68 kcal/mol. Both dexamethasone and cortisone make two hydrogen interaction with active sites of SER290 and LYS292 at a distance of 3.33 Å and 3.21 Å and 3.14 Å and 3.06 Å, respectively. Also energetics found similar for the protein–ligand binding [− 4.07 and − 31.03 kcal/mol for dexamethasone and − 4.63 and − 28.18 kcal/mol for cortisone]. Interestingly, all the compounds show hydrogen bond interaction with SER294 which actually involving in catalysis by initiating nucleophilic attack on the substrate at the active site of NSP-15. Interactions of the compounds with the target at the active site are shown in Fig. 3 and the hydrogen bonding and hydrophobic interactions are listed in Table 1. All the shortlisted compounds found to have energetically favorable binding. Superposition of the docked compounds at the active site with NSP-15 are shown in Fig. 4.


Ligand interactions of experimental tested steroids and identified best hit steroid compounds with NSP-15. A Dexamethasone, B Cortisone, C 0449-0045, D N001-0004, E N006-0008

Superposition of experimental tested steroids and identified best hit steroid like compounds with NSP-15. a Surface representation of superimposed docked complexes, b binding interaction of identified steroids at the catalytic sites of NSP-15
Molecular dynamics simulations
Molecular dynamics simulations for 50 ns time scale were performed for analyzing the dynamic behaviors of the NSP-15-ligand complexes. Detailed trajectory analyses were performed to understand the dynamics of inter molecular interactions. The root mean square deviation (RMSD) profile measures the average distance between superimposed biomolecules (Cα), indicating the degree of closeness with reference to the initial structure. The radius of gyration (Rg) represents the mass-weighted RMS deviation of group of atoms from their center of mass, denoting global measurement of protein dimension. All the ligand-bound complexes were stable over 50 ns. The RMSD is minimal for N006-0008 ligand-bound structure in comparison with that of N001-0004 (Fig. 5). The RGYR (Rg) values of the NSP-15 complexes were constrained, which ranges from 23 to 24 Å, for all the cases. Hydrogen bond interactions around the active of the complexes are uniformly present, though N006-0008 compound has a larger contribution favoring binding in comparison with other compounds (Fig. 6). The fluctuation of amino acids during MDS is an important parameter to evaluate internal residual dynamics. The RMSF profiles of the complexes are similar in the overall pattern while certain active site residues are involved in the interactions behave differently. In general, complexes have linear and correlated RMSF profiles (Fig. 7).

RMSD of C-alpha atoms of best hits and known steroid compound complexes from the MD trajectories

RGYR of best hits and known steroid compound complexes from the MD trajectories

RMSF of C-alpha atoms of best hits and known steroid compound complexes from the MD trajectories
All five docked complexes were observed to be stable throughout the MD simulation of 50 ns. Hydrogen bonding interactions with residues Lys290, Val292, His250, Ser294, Leu346, Gln347, Cys293 and Gln245 at the active site vicinity found to be stable for all the complexes through out the duration of simulation. Hydrophobic interactions with the residues Trp333 and Tyr343 are also intact during simulation (Fig. 8).


Protein and ligand contact analysis from the simulated MD trajectory
Principal component analysis
NSP-15 complexes (0449-0045, N001-0004, N006-0008, cortisone and dexamethasone) were analyzed for dominant motion using principal components analysis (PCA), for which the first few eigenvectors were captured and their combined motions amplitude were identified to evaluate local fluctuations. The first three PCs accounted for the variance of motion observed for the complexes and two-dimensional plot between eigenvectors was drawn to compare the correlated motion. (Fig. S2). The graph represents the variance in conformational distribution, denoting periodic change between the conformations. All the complexes have a similar distribution profile, the subspace of N001-0004 and 0449-0045 has PC 1/2 and 1/3 distinct distribution centered on the origin, making it confined to one local energy basin. The first PCs accounted for 50.9%, 18.6%, 31.2%, 36.4% and 25.8% of conformational variance observed during the trajectories of complexes (N001-0004, N006-0008, cortisone, 0449-0045 and dexamethasone) respectively.
Assesment of binding free energy
Docked NSP-15 complexes with best hit compounds (0449-0045, N001-0004, N006-0008) from virtual screening and reported steroids cortisone, dexamethasone were analyzed to evaluate binding free energy using the MM/GBSA protocol. The total binding free energy (ΔGbind) for dexamethasone complex is − 14.87 kcal/mol which is the least when compared with other compounds 0449-0045 (− 26.01), N001-0004 (− 15.93), N006-0008 (− 28.12) and cortisone (− 28.78 kcal/mol). Table 2 describes individual contributions of polar (ΔEele) and non-polar (ΔEvdw) components. Both non polar and polar contributions are higher for shortlisted compounds than tested compounds. As a result, based on total binding energy, it can be established that 0449-0045 and N006-0008 are better lead compounds to inhibit NSP-15.
ADME properties
By the encouragement of docking and dynamics results, the identified potential NSP-15 inhibitor lead compounds were analyzed for their drug likeliness. ADME analysis was performed to predict biological properties of these lead compounds. Using Qikprop module, properties were calculated and tabulated (Table 3). Action of these identified leads on central nervous system were predicted. Molecular weight < 500 Da, Hydrogen bond donor ≤ 5, hydrogen bond acceptor ≤ 10, QPlogPo/w ≤ 5, QPlogBB) range of − 3.0 to 1.2, Central nervous system (CNS) activity − 2 to + 2
Conclusion
NSP-15 encodes for an endonuclease, exhibiting EndoU activity which in turn interfere innate immune response. The importance of this target is maximized by its action on dsRNA to degrade so that host RNA sensors could not recognize which resulted effective evasion of host immune system. Binding of a compound at the active site would inhibit the function of NSP-15, and so effective against SARS-CoV-2. Since steroids are widely used to treat inflammatory conditions because of viral invasion, the dedicated steroid like compounds data base was screened and three compounds 0449-0045, N001-0004, N006-0008 were identified with potential NSP-15 binding. Also Cortisone and Dexamethasone were included for Induced-Fit-docking and the MD simulation studies. All the compounds were found to bind with NSP-15 at the active site with favorable binding energetics and conformation. The residue SER294 which was implicated in the catalytic mechanism was having interaction with all compounds and hence these compounds may stop the nucleophilic attack on the RNA substrate. MD simulation and trajectory analyses established the stability of protein–ligand interactions. As revealed by favorable glide score, glide energy, and stable interactions during simulation, the identified steroids are proposed as anti-viral agents against SARS-CoV-2.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Yang J, Fan L, Miao X et al (2015) Corticosteroids for the treatment of human infection with in fl uenza virus : a systematic review and meta-analysis. Clin Microbiol Infect 21:956–963. https://doi.org/10.1016/j.cmi.2015.06.022
Matsuyama S, Kawase M, Nao N et al (2020) The inhaled steroid ciclesonide blocks SARS-CoV-2 RNA replication by targeting the viral replication-transcription complex in cultured cells. J Virol. https://doi.org/10.1128/JVI.01648-20
Kim Y, Jedrzejczak R, Maltseva NI et al (2020) Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci 29:1596–1605. https://doi.org/10.1002/pro.3873
Liu X, Fang P, Fang L et al (2019) Porcine deltacoronavirus nsp15 antagonizes interferon-β production independently of its endoribonuclease activity. Mol Immunol 114:100–107. https://doi.org/10.1016/j.molimm.2019.07.003
Kim Y, Jedrzejczak R, Maltseva NI et al (2020) Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV -2. Protein Sci 29:1596–1605. https://doi.org/10.1002/pro.3873
Sastry GM, Adzhigirey M, Day T et al (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. https://doi.org/10.1007/s10822-013-9644-8
Jacobson MP, Friesner RA, Xiang Z, Honig B (2002) On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol 320:597–608. https://doi.org/10.1016/S0022-2836(02)00470-9
Shelley JC, Cholleti A, Frye LL et al (2007) Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21:681–691. https://doi.org/10.1007/s10822-007-9133-z
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–11236. https://doi.org/10.1021/ja9621760
Schrödinger Release 2014–2 (2014) LigPrep, Schrödinger, LLC, New York, NY, 2014–2
Friesner RA, Murphy RB, Repasky MP et al (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem 49:6177–6196. https://doi.org/10.1021/jm051256o
Sherman W, Beard HS, Farid R (2006) Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des 67:83–84. https://doi.org/10.1111/j.1747-0285.2005.00327.x
Farid R, Day T, Friesner RA, Pearlstein RA (2006) New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem 14:3160–3173. https://doi.org/10.1016/j.bmc.2005.12.032
Holm L, Laakso LM (2016) Dali server update. Nucleic Acids Res 44:W351–W355. https://doi.org/10.1093/nar/gkw357
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel 8:127–134. https://doi.org/10.1093/protein/8.2.127
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:014101. https://doi.org/10.1063/1.2408420
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. https://doi.org/10.1063/1.328693
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Amadei A, Linssen ABM, Berendsen HJC (1993) Essential dynamics of proteins. Proteins Struct Funct Genet 17:412–425. https://doi.org/10.1002/prot.340170408
Roe DR, Cheatham TE (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9:3084–3095. https://doi.org/10.1021/ct400341p
Bello M (2014) Binding free energy calculations between bovine β-lactoglobulin and four fatty acids using the MMGBSA method. Biopolymers 101:1010–1018. https://doi.org/10.1002/bip.22483
Ioakimidis L, Thoukydidis L, Mirza A et al (2008) Benchmarking the reliability of QikProp. correlation between experimental and predicted values. QSAR Comb Sci 27:445–456. https://doi.org/10.1002/qsar.200730051
Acknowledgements
Anantha Krishnan Dhanabalan, thank the Indian Council of Medical Research, India for Senior Research Fellowship (Grant No: ISRM/11(69)/2017). Authors thank DBT Bioinformatics Infrastructure Facility, University of Madras and CAS in Crystallography and Biophysics for computing facility.
Funding
The authors declare that no funds, Grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dhanabalan, A.K., Raghavan, S.S., Rajendran, S. et al. Evaluation of action of steroid molecules on SARS-CoV-2 by inhibiting NSP-15, an endoribonuclease. Mol Divers 27, 2715–2728 (2023). https://doi.org/10.1007/s11030-022-10576-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10576-5