
Abstract
In the present work, a hit molecule obtained from zinc ‘clean drug-like database’ by systematically performed computational studies was modified chemically to obtain different derivatives (VS1–VS25). Structures of synthesized derivatives were confirmed by IR, 1H-NMR, 13C-NMR, 13C-DEPT, MS, and elemental analysis. All the synthesized compounds were biologically evaluated for their antidepressant activity by using tail suspension test and forced swimming test in albino mice. All these derivatives showed moderate to good antidepressant activity. The most potent compound (VS25) among the synthesized compounds showed better antidepressant potential than the standard drugs moclobemide, imipramine, and fluoxetine. To understand the time-dependent interactions of this most active compound with MAO-A molecular dynamics was carried out and reported here. Additionally, acute oral toxicity was performed for the most active compound as per OECD guidelines.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Depression is a common chronic recurrent syndrome, often refractive to drug treatment, affecting the quality of life and overall productivity. The total estimated number of people living with depression worldwide from 2005 to 2015 increased by 18.4% to 322 million, according to the World Health Organization (WHO 2017) report issued in the year 2017. Epidemiological studies in the Middle East and North Africa regions have demonstrated depression rates ranging from 13 to 18%. The rate of depression in women is double than that in men [1]. Research has shown that psychological disorders during the COVID-19 pandemic have affected individuals with symptoms of mental trauma such as emotional distress, depression, stress, mood swings, irritability, insomnia, attention-deficit/hyperactivity disorder, post-traumatic stress, and age. The etiology of depressive disorders is too complex to be understood by a single social, developmental, or biological theory. Several factors appear to work together to cause or precipitate depressive disorders. The symptoms reported by patients with major depression consistently reflect changes in brain monoamine neurotransmitters, specifically norepinephrine (NE), serotonin (5-HT), and dopamine (DA) [2,3,4]. Reduction in brain serotonin level [5] has been reported to be one of the most important etiological factors for genesis of depression. The most widely used class of antidepressants, namely serotonin reuptake inhibitors (SSRIs), increases the extracellular availability of serotonin. Further, noradrenergic and dopaminergic systems are also reported to be involved in the pathophysiology of depression acting in tandem with the serotonergic system [6,7,8]. Though patients with depression are treated adequately, the social stigma appears to be the major roadblock in seeking treatment and continuing with the treatment in the long run. As per the available data, people with depression and anxiety disorder report that the stigma and discrimination they experience may be worse than their mental health condition [9].
Monoamine oxidase (MAO) inhibitors block the enzyme catalytic activities and halt the oxidative deamination of these neurotransmitters. Inhibition of MAOs can lead to an increase in the concentrations of neurotransmitters stored in the nerve terminals (e.g., serotonin and dopamine). Thus, MAO inhibitors can be developed as therapeutic agents, particularly for disease states where MAO enzyme is over-expressed. The metabolism of monoamine neurotransmitters by MAO may generate by-products which are potentially injurious species (e.g., hydrogen peroxide and aldehydes), and lead to neuronal damage. MAO inhibitors can act as potential neuroprotective agents by reducing the production of the reactive oxygen species (ROS) by halting the MAO-catalyzed oxidation process and hence prevent their neurotoxic effect [10].
The co-crystal composition of MAO-A through X-rays with considerable MAO inhibitors resulted in novel understanding of these enzyme-ligand systems and also stimulated research in the field of MAO blockade as a possible therapeutic regime in neurodegenerative disorders. The reversible MAO-A inhibitors have been proved to be specifically potent in the treatment of depression in aged patients. Selective MAO-A and non-selective MAO inhibitors came across as therapeutically active moieties in the treatment of phobia and atypical depressions, like those including bulimia, hysterical traits, hypersomnia, tiredness for which they are better than amine-uptake inhibitors [11].
Designing strategy
One of the important studies in the past revealed that levels of MAO-A are elevated in patients suffering from major depressive disorder [12]. Elevated level of MAO-A would lead to increased metabolism of its substrates NE and 5-HT, thereby causing a decrease in the levels of these neurotransmitter monoamines in the brain leading to depression. Consequently, inhibition of MAO-A was thought to be an effective strategy, as this would treat the root cause of depression. In one of our previous work, we have reported 11 hits as potential MAO-A inhibitors by using various systematic computational techniques, and we are in process of derivatizing individual hit into series of compounds. In our previous report, 82 structurally diverse MAO-A inhibitors were selected and a robust pharmacophore model was developed, where we found DHHR (one donor, two hydrophobic, and one aromatic) features as best pharmacophoric model. This model was further validated by performing 3D-QSAR by dividing dataset in to training and test subsets. Further to validate the pharmacophoric features, the molecules under study were docked within the active site of the MAO-A receptor which showed that active inhibitors interacted with the receptor active site by H-bonding, π–π stacking with aromatic ring, and hydrophobic interactions. This observation strongly validated the generated pharmacophore model. Then to identify new potential MAO-A inhibitors this developed pharmacophore model was used as starting point for zinc clean drug-like database (140,000 molecules) screening. Along with the pharmacophoric feature mapping, Lipinski rule of five was also applied as one of the filters. Primary screen resulted into 30,052 molecules which were further filtered on the basis of fitness (>60%) with pharmacophoric features and obtained 1572 molecules. Further, the activity of these molecules was predicted using developed 3DQSAR model, and molecules with pKi ≥ 1 were retained which resulted into 960 hits. Further, these hits were screened on the basis of various levels of precision in docking, and finally, structurally diverse 11 hits were considered as promising MAO-A inhibitors [13]. Pharmacophoric feature diagram and workflow of this computational study are given in supporting information (Figure S1 and S2). We are in process of developing individual scaffold into a series, and in the present work, we have selected the hit zinc 09307121 out of the 11 hits and modified its structure chemically to obtain some novel compounds. Modeling studies and biological activity of these compounds have been reported in this paper.
Hit Zinc 09307121 was modified as shown in Fig. 1. The pyridine ring was replaced with benzene, which is not as deactivated as the pyridine ring in terms of the availability of electrons is concerned. This change could help with more efficient π–π staking. It was also intended to study the effect of hydrophobic interactions of cyclohexyl and benzyl groups on antidepressant activity in comparison with the substituted phenyl ring. The interaction of ligand binding residues with protein is shown in Fig. 2. The designed molecules were synthesized and evaluated for antidepressant activity.

Representation of the structural modifications in the hit molecule zinc 09,307,121

Interaction of ligand binding residues with protein
Results and discussion
Chemistry
The modified Zinc 09307121 led to a series of twenty-five 2-((1H-benzimidazol-2-yl)thio)-N-substituted-acetamide derivatives (VS1–VS25) and which were synthesized. The starting compounds 2-chloro-N-substituted-acetamides (1a–1y) were synthesized by reacting anilines/amines with chloroacetyl chloride in the presence of glacial acetic acid and sodium acetate for 2 h. The title compounds were prepared by reacting 2-chloro-N-substituted-acetamides with 2-mercaptobenzimidazole in ethanol and triethylamine (Scheme 1). The yields of the title compounds after recrystallization were in the range of 62–74%. Their structure confirmation was accomplished by infrared radiation (IR), nuclear magnetic resonance (1H-NMR), 13C NMR, distortionless enhancement of polarization transfer (13C DEPT), mass spectroscopy (MS), and elemental analysis.

Syntheses of benzimidazol-2-ylthio-N-substituted-acetamide derivatives
Biological evaluation
In vivo evaluation of antidepressant activity: tail suspension test (TST) and forced swim test (FST)
In TST and FST compound’s ability to decrease the duration of immobility of the animals is taken as a reflection of their antidepressant potential. Thus, in this test, a compound’s antidepressant potential is expressed in terms of percent decrease in immobility duration (% DID) brought about by it with respect to the control. The test compounds were administered at three different doses for evaluation of their antidepressant activity—15 mg/kg, 30 mg/kg, and 60 mg/kg in adult Swiss Albino mice (22 ± 2 g). It was noted that at the dose of 15 mg/kg all the compounds exhibited very weak antidepressant activity in both the tests. However, when the dose of the test compounds was doubled to 30 mg/kg, it was seen that most of the compounds of the present series displayed significant antidepressant activity. The most active compound in this series was VS25, which exhibited the highest % DID value of 82.23%. However, when the dose was stepped up to 60 mg/kg, it was found that no increase in activity was achieved. On the contrary, antidepressant activity of many of the active compounds decreased on increasing the dose. Thus, for both the tests, a dose of 30 mg/kg was considered as the optimum one for the test compounds. Results of the TST and FST are tabulated in Table 1. Graphical representation of the results of TST and FST at the dose of 30 mg/kg is given in Fig. 3a, b, respectively.


Graphical representation of % DID of the standard drugs and test compounds (VS1-VS25) in a TST, b FST at optimum dose of 30 mg/kg
During the biological evaluation, it was noted that most of the synthesized compounds exhibited significant antidepressant activity with meaningful statistical results. Some test compounds caused a considerable decrease in immobility duration as compared to the standard drug moclobemide, a MAO-A inhibitor. From %DID values, it is observed that electron-releasing groups like methyl and ethyl at ortho and para positions of the phenyl ring had a favorable influence on the activity as seen in VS8, VS9, VS21, and VS22 as compared to unsubstituted analog VS1, whereas from the % DID value of VS5, VS6, VS7, VS10, VS11, VS15, VS16, and VS18, it can be said that electron-withdrawing groups like chloro, fluoro, bromo, and nitro on the phenyl ring at ortho, meta, or para positions do not have significant influence on antidepressant activity of its unsubstituted analog VS1. Replacement of the phenyl ring with benzyl ring showed a significant increase in antidepressant activity as seen in VS25, while its replacement with cyclohexyl ring in VS24 resulted in a moderate increase in antidepressant activity. In general, it was noted that non-polar substituents in the phenyl ring were better tolerated than polar substituents. Further, replacement of the phenyl ring by cyclohexyl and benzyl ring led to an increase in antidepressant activity.
Acute oral toxicity
The most active compound was evaluated for acute oral toxicity in mice. It was seen that VS25 was safe in mice up to a dose of 300 mg/kg, but proved toxic, causing mortality at a dose of 2000 mg/kg. Following Organization for Economic Co-operation and Development (OECD) 423 guidelines, VS25 falls under globally harmonized classification system (GHS) category 4 (>300–2000) and has LD50 (cutoff) of 1000 mg/kg body weight. The results of this study are summarized in Table 2.
Computational studies
Docking studies
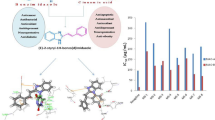
To understand the ligand receptor interactions, the most active compound (VS25) was docked within the receptor active site of MAO-A structure (PDB code: 2Z5X). Here the benzyl moiety of the scaffold was found to be oriented toward the FAD cofactor. This ring exhibited very strong pi–pi interactions with Phe208, promising pi–sulfur interaction with Cys323, and pi–alkyl interactions with Ile180, Ile335, and Leu337. Further, the complex was stabilized by strong hydrogen bond interactions between NH of acetamide with Phe208 and C=O with Val210. The benzimidazole ring further imparted stability to the complex through hydrophobic interactions within the receptor active site. This includes pi–sigma interaction with Thr211, and hydrophobic interactions with Gly110, Ala111, Ser209, and Val210 (Fig. 4).

Interactions of VS25 with human MAO-A; left image: 3D interactions, cyan color ball and stick model is ligand; right side: suggested 2D interactions
Molecular dynamic simulation
The most active compound showed promising interactions with MAO-A receptor. To understand the time-dependent stability of the ligand receptor complex, molecular dynamics study was carried out for 50-ns duration using the Desmond tool. In order to understand the interactions over the period of time, parameters related to ligand and receptor such as root-mean-square fluctuation (RMSD), root-mean-square deviation (RMSF), radius of gyration (ROG), solvent-accessible surface area (SASA), and hydrogen bonding were analyzed. Here, the docked complex of VS25 with the receptor was considered as the reference frame for the calculation of these properties over the period of time. The major fluctuation in RMSD-P, which explains the protein RMSD while having a ligand in the active site, was observed up to an initial 8-ns time; then, it was found to be stabilized for the remaining period of time in the range of 5 to 7 Ǻ. The compound VS25 showed initial fluctuation in RMSD-L, while remaining in the active site of the receptor, at around 4 Ǻ and 8 Ǻ, whereas for the remaining period of time under study, it was stable and observed in the range of 3 Ǻ to 5 Ǻ (Fig. 5a). The major initial fluctuation in the protein was mainly due to C-terminal fluctuations, which was confirmed by the RMSF plot for the protein, where except for C-terminal, all other residues showed RMSF less than 3 Ǻ. Over the period of analysis C-terminal exhibited RMSF up to 15 Ǻ (Fig. 5b). Also, RMSD of the ligand with the protein and the ligand with initial conformation of the ligand was very stable and was between 3 to 5 Ǻ and 1 to 2 Ǻ, respectively (Fig. 5c). This suggests that there is no major deviation of the ligand from the active site as well as no major change in orientation of the ligand within the receptor active site. The RMSF of the ligand with respect to the protein as well as the ligand itself also supported the stability of the ligand with the receptor. Hydrogen bond analysis suggested that for a 52% period of time this interaction was there with Val210 (Fig. 5d). Further, the ligand properties like ROG, SASA, MolSA, and PSA were found to be in acceptable limits and supported the stability of the ligand–receptor complex under study (Fig. 5e).

a RMSD of Calpha, lig_fit_prot, and lig_fit_lig for 2Z5X and VS25 complex; b RMSF–Protein for 2Z5X and VS25 complex; c RMSF–Ligand for 2Z5X and VS25 complex; d VS25 interaction with 2Z5X residues; e rGyr, MolSA, SASA, and PSA for VS25 while in contact with 2Z5X
Conclusion
In the present work, a series of twenty-five compounds (VS1–VS25) 2-(1H-benzimidazol-2-ylthio)-N-substituted acetamides were synthesized and structurally confirmed by IR, 1H-NMR, 13C-NMR, 13C DEPT, MS, and elemental analysis. All the synthesized final compounds were evaluated for antidepressant activity using two behavior models: tail suspension test (TST) and forced swim test (FST). The receptor molecular interactions and time-dependent stability were evaluated by molecular docking and molecular dynamics studies. The phenyl and substituted phenyl rings on acetamide offered poor to moderate anti-depressant activity, whereas the benzyl substituent exhibited the highest activity among the compounds of the series of the synthesized derivatives. This also suggested that the presence of the methylene bridge between aromatic ring and acetamide nitrogen contributed to high activity. Further substitution on benzyl ring and the effect of length of spacer/linker would be explored in future studies. However, the current study revealed some compounds with higher antidepressant activity than the standard drug moclobemide.
Experimental
Chemistry
General methods
Completion of reaction was monitored by thin-layer chromatography on Merck precoated silica gel F-254 plates. Melting points of the synthesized compounds were determined on Veego VMP-D digital melting point apparatus by open capillary method and are uncorrected. The IR spectra of the synthesized compounds were recorded on Jasco FT-IR 4100 in potassium bromide. The 1H-NMR, 13C-NMR, and 13C-DEPT-135 spectra were recorded in deuterated chloroform (CDCl3) and dimethylsulfoxide (DMSO) using NMR Varian Mercury plus 300 MHz using tetramethylsilane (TMS) as internal standard. The mass spectra were recorded on 410 Prostar Binary LC with a 500 MS IT PDA detector. Elemental analysis was performed on FLASH EA 1112, Thermo-Finnigan, and indicated with the element symbol. The elemental analyses were within ± 0.4% of the theoretical values.
Synthesis of substituted phenylacetamides (1a–1y)
The respective amines (0.02 mol) were dissolved in 60 mL of glacial acetic acid (GAA) and saturated solution of sodium acetate. To the reaction mixture, chloroacetyl chloride (0.02 mol) was then added dropwise in a fuming cupboard and the reaction was stirred for 1 h. The precipitate obtained was poured into ice-cold water. The crude product was recovered by filtration. The product was washed with very dilute solution of glacial acetic acid and recrystallized from a mixture of ethanol and water.
2-Chloro-N-phenylacetamide (1a)
Yield: 79%, mp: 136–137 °C; Rf 0.56 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3303, 3209, 3145, 2945, 1671, 1605, 1559, 1250, 762. 1H NMR (δ ppm: DMSO-d6): 4.6 (s, 2H, CH2), 7.18 (d, 2H, ArH), 7.85 (d, 2H, ArH), 9.95 (s, 1H, NH).
2-Chloro-N-(2-methylphenyl)acetamide (1b)
Yield: 72%, mp: 105–106 °C; Rf 0.69 (3:2 Toluene:EtOAc; UV),IR vmax(cm−1) (KBr): 3435, 3258, 1663, 1542, 1252, 747. 1H NMR (δ ppm: DMSO-d6) 1.53 (s 3H, CH3), 4.46 (s, 2H, CH2), 7.86 (d, 2H, ArH), 7.98 (d, 2H, ArH), 9.90 (s, 1H, NH).
2-Chloro-N-(3-methylphenyl)acetamide (1c)
Yield: 69%; mp: 91–93 °C; Rf 0.52 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3309, 3141, 1682, 1614, 1552, 780.
2-Chloro-N-(4-methylphenyl)acetamide (1d)
Yield: 68%, mp: 165–167 °C; Rf 0.59 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3325, 3152, 1653, 1618, 1567, 782.
2-Chloro-N-(2-chlorophenyl)acetamide (1e)
Yield: 67%, mp: 71–72 °C; Rf 0.6 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3271, 3096, 1641, 1567, 1287, 752.
2-Chloro-N-(3-chlorophenyl)acetamide (1f)
Yield: 71%, mp: 93–94 °C; Rf 0.54 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3465, 3348, 3151, 1683, 1598, 1212, 776.
2-Chloro-N-(4-chlorophenyl)acetamide (1g)
Yield: 73%, mp: 152–154 °C; Rf 0.53 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3308, 3259, 3151, 1688, 1578, 1432, 1281, 784.
2-Chloro-N-(2-ethylphenyl)acetamide(1h)
Yield: 75%, mp: 118 °C, Rf 0.61 (3:2 Toluene:EtOAc; UV),IR vmax(cm−1) (KBr): 3274, 3202, 2964, 1894, 1668, 1614, 1407, 1253, 961, 828, 786, 1H NMR (δ ppm: DMSO-d6) 1.12 (t, 3H, CH3), 2.59 (q, 2H, CH2), 4.29 (s, 2H, CH2), 7.17 (d, 2H, ArH), 7.34 (d, 2H, ArH), 9.65 (s, 1H, NH).
2-Chloro-N-(4-ethylphenyl)acetamide (1i)
Yield: 74%, mp: 163 °C, Rf 0.59 (3:2 Toluene:EtOAc; UV),IR vmax(cm−1) (KBr): 3110, 3055, 1662, 1417, 785.
2-Chloro-N-(2-fluorophenyl)acetamide (1j)
Yield: 68%, mp: 112 °C, Rf 0.55 (3:2 Toluene:EtOAc; UV),IR vmax(cm−1) (KBr): 3319, 3174, 1659, 1469, 788.
2-Chloro-N-(4-fluorophenyl)acetamide (1k)
Yield: 65%, mp: 109 °C, Rf 0.53 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3323, 3187, 1674, 1463.1240, 783.
2-Chloro-N-(3-methoxyphenyl)acetamide (1l)
Yield: 71%, mp: 83–85 °C, Rf 0.58 (3:2 Toluene-EtOAc; UV),IR vmax(cm−1) (KBr): 3257, 3163, 1642, 1423, 784.
2-Chloro-N-(4-methoxyphenyl)acetamide (1m)
Yield: 72%, mp: 105–107 °C; Rf 0.56 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3314,3262, 3084, 1661, 1447, 784.
2-Chloro-N-(2-nitrophenyl)acetamide (1n)
Yield: 76%, mp: 85–87 °C; Rf 0.52 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3308, 3223, 1668, 1633, 1412, 779.
2-Chloro-N-(3-nitrophenyl)acetamide (1o)
Yield: 80%, mp: 91–92 °C; Rf 0.57 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344, 1254, 788.
2-Chloro-N-(4-nitrophenyl)acetamide (1p)
Yield: 78%, mp: 120–121 °C; Rf 0.61 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3374, 3262, 1670, 1298, 758.
2-Chloro-N-(2-trifluoromethylphenyl)acetamide (1q)
Yield: 76%, mp: 213 °C, Rf 0.51 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3347, 3267, 1669, 1327, 778.
2-Chloro-N-(4-bromophenyl)acetamide (1r)
Yield: 70%, mp: 170–172 °C; Rf 0.62 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3365, 3258, 1687, 1305, 782.
2-Chloro-N-(2,4-dinitrophenyl)acetamide (1s)
Yield: 67%, mp: 213 °C, Rf 0.54 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3402, 3227, 1663, 1348, 1289, 803.
2-Chloro-N-(2,3-dimethylphenyl)acetamide (1t)
Yield: 66%, mp: 213 °C, Rf 0.52 (3:2 Toluene-EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344.
2-Chloro-N-(2,5-dimethylphenyl)acetamide (1u)
Yield: 66%, mp: 213 °C, Rf 0.5 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344.
2-Chloro-N-(2,6-dimethylphenyl)acetamide (1v)
Yield: 68%, mp: 213 °C, Rf 0.57 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344.
2-Chloro-N-(2,3-dichlorophenyl)acetamide (1w)
Yield: 72%, mp: 213 °C, Rf 0.55 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344.
2-Chloro-N-cyclohexylacetamide (1x)
Yield: 74%, mp: 102–103 °C; Rf 0.6 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344.
2-Chloro-N-benzylacetamide (1y)
Yield: 70%, mp: 90–92 °C; Rf 0.56 (3:2 Toluene:EtOAc; UV), IR vmax(cm−1) (KBr): 3393, 3292, 1649, 1344.
Synthesis of 2-(1H-benzimidazol-2-ylthio)-N-aryl/acyl acetamides (VS1-VS25)
A mixture of appropriate 2-chloro-N-substituted arylacetamide (0.1 mol), 2-mercaptobenzimidazole (0.1 mol), and triethylamine (0.12 mol) was refluxed for 2 to 3 h in ethanol (30 mL). Completion of the reaction was monitored by TLC. The reaction mixture was quenched with cold water. The product was recovered and was crystallized using acetone/water. For synthesizing 2-[(substituted-N-arylacetamido)mercapto]benzimidazoles, the reaction depicted in Scheme 1 was followed.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-phenylacetamide(VS1)
Yield: 69%, mp: 166–168 °C; Rf 0.57 (1:1 n-Hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3244–2926, 1672, 1602, 1443, 1178, 736; 1H NMR (δ ppm: CDCl3) 11.07 (s, 1H, NH), 7.13–7.40 (m, 9H, ArH), 3.97 (s, 2H, CH2); 13C NMR (δ ppm: CDCl3): 166.47, 137.86, 128.04, 123.04, 121.72, 118.65, 35.44; DEPT-135, (δ ppm: CDCl3): positive peaks-128.05, 123.13, 121.73, 118.66, negative peaks- 35.44. MS m/z:284.3 [M + 1]. Anal. Calcd for C15H13N3SO: C, 67.10; H, 3.97; N, 18.41; O, 10.52, Found: C, 67.02; H, 4.00; N, 18.36.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2-methylphenylacetamide(VS2)
Yield: 67%, mp: 157–159 °C; Rf 0.55 (1:1 n-Hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3246–2829, 2605, 1676, 1618, 1590, 1560, 1414, 1227, 1137, 974, 739; 1H NMR (δ ppm: CDCl3) 10.68 (s, 1H, NH), 10.24 (s, 1H, NH), 7.04–7.83 (m, 8H, ArH), 4.06 (s, 2H, CH2), 2.24 (s, 3H, CH3); 13C NMR (δ ppm: CDCl3): 168.96, 150.69, 135.88, 130.53, 126.50, 125.33, 123.18, 35.93, 18.24; DEPT-135, (δppm: CDCl3): positive peaks-130.62, 126.60, 125.42, 123.28, 18.34, negative peaks- 36.02. MS m/z:297 [M + 1]; Anal. Calcd for C16H15N3SO: C, 64.62; H, 5.08; N, 14.13; O, 5.38; S, 10.78 Found: C, 64.49; H, 5.11; N, 14.09.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-3-methylphenylacetamide(VS3)
Yield: 72%, mp: 185–186 °C; Rf 0.52 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3339, 3071, 2946, 1721,1663, 1610, 1467; 1H NMR (δ ppm:CDCl3) 7.02–7.88 (m, 8H, ArH), 4.06 (s, 2H, CH2), 2.24 (s, 3H, CH3).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-methylphenylacetamide(VS4)
Yield: 76%, mp: 188–190 °C, Rf 0.54 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3041–3011, 2945, 1730, 1662, 1605, 1457, 1H NMR (δ ppm:CDCl3) 7.11–7.89 (m, 8H, ArH), 4.29 (s, 2H, CH2), 2.19 (s, 3H, CH3).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2-chlorophenylacetamide(VS5)
Yield: 70%, mp: 154–155 °C; Rf 0.57 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3493, 3051- 2923, 1733, 1668, 1604, 1268, 1H NMR (δ ppm: CDCl3) 12.75 (s, 1H, NH), 9.84 (s, 1H, NH), 7.16–7.48 (m, 8H, ArH), 4.23 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-3-chlorophenylacetamide(VS6)
Yield: 72%, mp: 156–158 °C; Rf 0.58 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3062- 2940, 1726, 1666, 1601, 1468, 1H NMR (δ ppm:CDCl3) 9.89 (s, 1H, NH), 7.05–7.54 (m, 8H, ArH), 4.24 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-chlorophenylacetamide(VS7)
Yield: 74%, mp: 177–178 °C; Rf 0.56 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3043–2893, 1724, 1676, 1615, 1571, 1453, 1254, 765, 1H NMR (δ ppm: CDCl3) 12.68 (s, 1H, NH), 10.51 (s, 1H, NH), 7.14–7.45 (m, 8H, ArH), 4.25 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2-ethylphenylacetamide(VS8)
Yield: 67%, mp: 174–177 °C; Rf 0.50 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3223, 3062, 2969, 1661, 1538, 1396, 1269, 983, 742; 1H NMR (δ ppm: CDCl3) 10.45 (s, 1H, NH), 10.06 (s, 1H, NH), 7.09–7.82 (m, 8H, ArH), 4.07 (s, 2H, CH2), 2.57 (s, 2H, CH2), 1.02 (s, 3H, CH3);13C NMR: 168.72, 150.68, 136.77, 135.18, 128.58, 126.42, 125.60, 123.81, 122.41, 117.78, 110.52, 35.68, 24.48, 13.95; DEPT-135: positive peaks- 128.59, 126.40, 125.62, 123.82, 122.90, 122.42, 117.78, 110.53, 13.96, negative peaks- 36.89, 24.48; MS m/z: 312.1 [M + 1]; Anal. Calcd for C17H17N3SO; C, 65.57; H, 5.50; N, 13.49; O, 5.14; S, 10.30. Found: C, 65.43; H, 5.52; N, 13.45.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-ethylphenylacetamide(VS9)
Yield: 64%, mp: 182–184 °C, (dec.): (296.5), Rf 0.55 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3032–2847, 1728, 1673, 1614, 1475, 758, 638, 1H NMR (δ ppm: CDCl3) 10.34 (s, 1H, NH), 10.12 (s, 1H, NH), 7.08–7.84 (m, 8H, ArH), 4.07 (s, 2H, CH2), 2.56 (s, 2H, CH2), 1.03 (s, 3H, CH3).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2-fluorophenylacetamide(VS10)
Yield: 69%, mp: 169–170 °C; Rf 0.51 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3073- 2936, 1726, 1657,1607, 1482, 1466; 1H NMR (δ ppm: CDCl3): 10.33 (s, 1H, NH), 10.03 (s, 1H, NH),77.04–8.34 (m, 8H, ArH), 4.06 (s, 2H, CH2);MS m/z: 301 [M + 1]; Anal. Calcd for C15H12N3SOF: C, 64.66; H, 4.22; N, 16.76; O, 14.36 Found: C, 64.57; H, 4.26; N, 16.69.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-fluorophenylacetamide(VS11)
Yield: 65%, mp: 170–172 °C; Rf 0.53 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3484, 3035–2934, 1725, 1671, 1604, 1485, 1H NMR (δ ppm: CDCl3): 10.71 (s, 1H, NH), 6.97–7.66 (m, 8H, ArH), 3.32 (s, 3H, OCH3), 4.71 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-3-methoxyphenylacetamide(VS12)
Yield: 70%, mp: 155–157 °C; Rf 0.51 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3090–2882, 1666, 1614,1551, 1490, 1403, 1371, 1349, 1093, 1009, 981, 827, 767; 1H NMR (δ ppm: CDCl3) 12.10 (s, 1H, NH), 11.01 (s, 1H, NH), 6.38–7.25 (m, 8H, ArH), 3.76 (s, 2H, CH2), 3.53 (s, 3H, OCH3); 13C NMR: 167.18, 159.42, 150.30, 139.34, 129.02, 111.13, 109.10, 104.60, 54.64, 35.63; DEPT-135: positive peaks- 129.03, 121.81, 121.59, 111.14, 109.10, 104.60, 54.64, negative peak- 36.63; MS m/z:314.0 [M + 1]. Anal. Calcd for C16H15N3SO2: C, 61.32; H, 4.82; N, 13.41; O, 10.21; S, 10.23. Found: C, 61.24; H, 4.90; N, 13.36.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-methoxyphenylacetamide(VS13)
Yield: 74%, mp: 202–204 °C; Rf 0.54 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3349, 3051- 2946, 1720, 1667, 1612, 1465, 1H NMR (δ ppm: CDCl3) 9.65 (s, 1H, NH), 6.91–7.34 (m, 8H, ArH), 4.29 (s, 2H, CH2), 2.59 (s, 3H, OCH3).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2-nitrophenylacetamide(VS14)
Yield: 67%, mp: 162–165 °C; Rf 0.56 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3041, 1720, 1697, 1601, 1485, 1268, 1H NMR (δ ppm: CDCl3) 7.05–7.55 (m, 8H, ArH), 4.31 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-3-nitrophenylacetamide(VS15)
Yield: 65%, mp: 168–170 °C; Rf 0.55 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3043–2895, 1729, 1599, 1485, 1270, 1H NMR (δ ppm: CDCl3) 7.07–8.68 (m, 8H, ArH), 4.55 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-nitrophenylacetamide(VS16)
Yield: 69%, mp: 165–166 °C; Rf 0.52 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3054, 2955, 1705, 1537, 1439, 1224, 1H NMR (δ ppm: CDCl3) 9.89 (s, 1H, NH), 7.16–8.19 (m, 8H, ArH), 3.87 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2-trifluoromethylphenylacetamide(VS17)
Yield: 63%, mp: 167–169 °C; Rf 0.57 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3234, 3045, 2847, 1737, 1595, 1441, 1227, 1H NMR (δ ppm: CDCl3): 7.14–7.68 (m, 8H, ArH), 2.89 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-4-bromolphenylacetamide(VS18)
Yield: 68%, mp: 222–225 °C; Rf 0.52 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3233, 3128, 2847, 1764, 1537, 1467, 1235, 1H NMR (δ ppm: CDCl3): 11.90 (s, 1H, NH), 6.96–7.65 (m, 8H, ArH), 4.22 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2,4-dibromolphenylacetamide(VS19)
Yield: 72%, mp: 175–178 °C; Rf 0.55 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3044, 2951, 1738, 1564, 1475, 1239, 1H NMR (δ ppm: CDCl3): 7.06–8.08 (m, 7H, ArH), 4.12 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2,3-dimethyllphenylacetamide(VS20)
Yield: 66%, mp: 144–146 °C; Rf 0.58 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3067, 2945, 1754, 1537, 1448, 1267, 1H NMR (δ ppm: CDCl3): 12.36 (s, 1H, NH), 10.23 (s, 1H, NH), 6.66–7.45 (m, 7H, ArH), 4.20 (s, 2H, CH2), 3.70 (s, 6H, (CH3)2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2,5-dimethyllphenylacetamide(VS21)
Yield: 68%, mp: 143–145 °C; Rf 0.51 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3048, 2887, 1733, 1557, 1439, 1218, 1H NMR (δ ppm: CDCl3): 6.89–7.59 (m, 7H, ArH), 4.14 (s, 2H, CH2), 2.37 (s, 3H, CH3), 2.20 (s, 3H, CH3).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2,6-dimethyllphenylacetamide(VS22)
Yield: 71%, mp: 220–222 °C; Rf 0.50 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3065, 2874, 1769, 1557, 1451, 1243, 1H NMR (δ ppm: CDCl3): 6.94–8.16 (m, 7H, ArH), 4.22 (s, 2H, CH2), 2.32 (s, 3H, CH3), 2.25 (s, 3H, CH3).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-2,3-dichlorophenylacetamide(VS23)
Yield: 63%, mp: 240 °C; Rf 0.52 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3064, 2867, 1764, 1587, 1422, 1250, 1H NMR (δ ppm: CDCl3): 10.74 (s, 1H, NH), 7.37–7.65 (m, 7H, ArH), 4.51 (s, 2H, CH2).
2-(1H-Benzimidazol-2-ylsulphanyl)–N-cyclohexylacetamide(VS24)
Yield: 62%, mp: 210–212 °C; Rf 0.49 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3067, 2995, 1734, 1635, 1587, 1437, 1266.
2-(1H-Benzimidazol-2-ylsulphanyl)–N-benzylacetamide(VS25)
Yield: 69%, mp: 185–189 °C; Rf 0.54 (1:1 n-hexane:EtOAc; UV), IR vmax(cm−1) (KBr): 3069, 2856, 1787, 1534, 1481, 1246, 1H NMR (δ ppm: CDCl3): 7.66–8.68 (m, 9H, ArH), 4.55 (s, 2H, CH2), 4.31 (s, 2H, CH2).
Biological
For the biological evaluation of the synthesized compounds, adult male Swiss Albino mice (22 ± 2 g) were used. Animals were maintained in humidity- and temperature-controlled rooms with a day/night cycle. Animals were allowed to acclimatize with the environment for one week before commencement of the experiments and had free access to food and water. Standard drug moclobemide was procured from Sigma-Aldrich, USA. Marketed formulations of standard drugs imipramine and fluoxetine were used.
Solutions of the synthesized compounds and standard drugs (moclobemide, imipramine, and fluoxetine) were prepared in DMSO and were administered i.p. Test compounds were administered at three doses: 15 mg/kg, 30 mg/kg, and 60 mg/kg, whereas the standard drugs (moclobemide, imipramine, and fluoxetine) were administered at a fixed dose of 30 mg/kg. The synthesized compounds were administered thrice in 24-h duration (subchronic) at t = 0, 18, and 24 h. A similar dosing regimen was followed for the standard drugs. The test was performed 1 h after administration of the last dose [14]. Antidepressant activity was expressed in terms of percent decrease in the duration immobility, calculated as:
where X is duration of immobility in control group (s) and Y is duration of immobility in test group (s).
In vivo pharmacological evaluation of the synthesized compounds was carried out by employing the following well-established behavior models:
Tail suspension test (TST)
In TST, mouse is suspended by its tail, which induces hemodynamic stress of being hung in an uncontrollable position. After an initial period of struggle, mouse becomes immobile. Administration of antidepressant drugs decreases the duration of immobility, and mouse remains actively engaged in escape-directed behavior for longer period of time [15, 16].
Method: The animal was suspended by its tail on a rod 80 cm above the floor with the help of an adhesive tape applied 1 cm from the tip of its tail. Initial escape-orientated behavior ceases after sometime, and the mouse undergoes spells of immobility. Duration of immobility was measured for a period of 6 min. Mouse was considered immobile only when it was completely motionless [17, 18].
Forced swim test (FST)
In FST the animal is forced to swim in a confined space. It becomes immobile following a phase of extensive swimming and climbing. Treatment with antidepressants reduces the immobility duration. Practice session is usually carried out 24 h before the actual test [19].
Method: The animals were submitted to a swimming stress session for 15 min, 24 h before the test session, in plastic cylinders (height 18.5 cm, diameter 12.5 cm) containing 13.5 cm of water at 25 °C. On the test day, the animals were dropped individually in to the same plastic cylinders containing water and behavioral observations were performed for up to 5 min. Duration of immobility was recorded during the last 4 min of the observation period. Animals were judged to be immobile when they ceased struggling and remained floating motionless in the water, making only those movements necessary to keep the head above water [20, 21].
Acute oral toxicity
Adult female albino mice, nulliparous and non-pregnant, in the weight range of 25 ± 2 g were used for the study. Suspensions of the test substances were prepared in water for injection containing 2% Tween 80 and were administered orally. The final administration volume did not exceed 2 mL/100 g body weight. Three animals were used for each step. The dose level used was 5, 50, 300 and 2000 mg/kg body weight as per OECD 423 [22].
Statistical analysis
The data obtained from the above studies were analyzed by one-way analysis of variance (ANOVA) to determine the significant difference between the groups. The intergroup significance or post hoc comparison was analyzed using Dunnett’s t test. P values < 0.05 were considered to be significant. All the values were expressed as percentage inhibition of the respective behavior model relative to the control group.
Computational studies
Docking studies
Molecular docking is a useful tool to understand interactions between the ligand and the receptor [23, 24]. In order to study the binding interactions, docking study was carried out between the designed structures and crystal structure of the target hMAO-A (PDB ID 2Z5X). Docking was carried out using Glide module of Schrodinger (Schrodinger-2009) [25]. The shape and properties of the receptor are represented on a grid by different sets of fields that provide progressively more accurate scoring of the ligand pose. From the poses selected by initial screening, the ligand is refined in torsional space in the field of the receptor using OPLS2005 force field.
Molecular dynamics studies
Molecular dynamics is the time-dependent analysis of ligand–receptor interactions. This analysis helps to understand the stability of interactions between the ligand and receptor complex [26, 27]. Molecular dynamics study for the ligand receptor complex (VS25–2Z5X) was carried out using Maestro-Desmond (with OPLS-2005 force field) for 10-ns duration [28,29,30]. The system was solvated with the TIP3P water model in an orthorhombic box and neutralized by Cl− ions. The smooth particle-mesh Ewald (PME) approximation and MSHAKE algorithm were used to evaluate long-range electrostatic interactions and non-bonded interactions, respectively. To relax the system, the default six-step relaxation protocol was followed, which consisted of restrained and unrestrained minimization (2 steps) followed by the equilibration processes (4 steps). The production MD was performed for 10 ns with NPT ensemble at 300 K and 1 atm pressure using a Nose–Hoover thermostat and Martyna–Tuckerman–Klein barostat. The stability of ligand protein complexes was evaluated by calculating root-mean-square deviation for the protein and ligand, root-mean-square fluctuation of the protein, molecular surface area (MolSA), solvent-accessible surface area (SASA), radius of gyration (rGyr), and polar surface area (PSA).
References
Hira AR, Alya H, Shaima A (2019) Depression: prevalence and associated risk factors in the United Arab Emirates. Oman Med J 34(4):274–283. https://doi.org/10.5001/omj.2019.56
Hirschfield RM (2000) History and the evolution of the monoamine hypothesis of depression. J Clin Psych 61(6), 4–6. https://pubmed.ncbi.nlm.nih.gov/10775017/
Delgado PL (2000) Depression: the case for a monoamine deficiency. J Clin Psych 61(Suppl 6): 7–11. https://pubmed.ncbi.nlm.nih.gov/10775018/
Stahl SM (2000) Blue genes and the mechanism of action of antidepressants. J Clin Psych 61:164–165. https://doi.org/10.4088/jcp.v61n0302
Anguelova M, Benkelfat C, Turecki G (2003) A systematic review of association studies investigating genes coding for serotonin receptors and the serotonin transporter: affective disorders. Mol Psych 8:574–591. https://doi.org/10.1038/sj.mp.4001328
Schreiber R, Brocco M, Audinot V, Gobert A, Veiga S, Millan MJ (1995) (1-(2, 5-dimethoxy-4-iodophenyl)-2-aminoropane)-induced head twitches in the rat are mediated by 5-hydroxytryptamine 5-HT2A receptors: modulation by novel 5-HT2A/2C antagonists, D1 antagonists and 5-HT1A agonists. J Pharmacol Exp Ther 273:101–12. https://pubmed.ncbi.nlm.nih.gov/7714755/
Millan MJ, Lejeune F, Gobert A (2000) Reciprocal autoreceptor and heteroreceptor control of Serotonergic, dopaminergic and noradrenergic transmission in the frontal cortex: relevance to the actions of antidepressant agents. J Psychopharmacol 14:114–138. https://doi.org/10.1177/026988110001400202
Koch S, Perry KW, Nelson DL, Conway RG, Threlkeld PG, Bymaster FP (2002) R-fluoxetine increases extracellular DA, NE, as well as 5-HT in rat prefrontal cortex and hypothalamus: an in vivo microdialysis and receptor binding study. Neuro-psychopharmacology 27:949–959. https://doi.org/10.1016/S0893-133X(02)00377-9
Sahoo S, Grover S, Malhotra R, Avasthi A (2018) Internalized stigma experienced by patients with first-episode depression: a study from a tertiary care center. Indian J Soc Psych. https://doi.org/10.4103/ijsp.ijsp11317
Shalabya R, Petzerb JP, Petzerb A, Ashrafc UM, Ataric E, Alasmarid F, Kumarasamyc S, Sarid Y, Khalil A (2019) SAR and molecular mechanism studies of monoamine oxidase inhibition by selected chalcone analogs. J Enzyme Inhib Med Chem 34(1):863–876. https://doi.org/10.1080/14756366.2019.1593158
Tapan B, Dapinder K, Aayush S, Sukhbir S, Neelam S, Gokhan Z, Felicia LA, Mirela MT, Simona B, Adrian GB (2021) Role of monoamine oxidase activity in Alzheimer’s disease: an insight into the therapeutic potential of inhibitors. Molecules 26:3724. https://doi.org/10.3390/molecules26123724
Jeffrey HM, Nathalie G, Anahita B, Sandra S, Doug H, Armando G, Trivor Y, Nicole P, Alan AW, Sylvain H (2006) Elevated monoamine oxidase A levels in the brain: An explanation for the monoamine imbalance of major depression. Arch Gen Psych 63(11):1209. https://doi.org/10.1001/archpsyc.63.11.1209
Shelke SM, Bhosale SH, Dash RC, Suryawanshi MR, Mahadik KR (2011) Exploration of new scaffolds as potential MAO: a inhibitors using pharmacophore and 3D-QSAR based in silico screening. Bioorg Med Chem Lett 21: 2419–424. https://europepmc.org/article/med/21397504.
Duarte FS, Martins P, Romeirob GA, De Limaa T (2007) Antidepressant like profile of action of two 4-amino derivatives of 10,11-dihydro-5H-dibenzo [a, d] cycloheptane in mice evaluated in the forced swimming test. Bioorg Med Chem 15:1645–1650
Cryan JF, Mombereau C, Vassout A (2005) The tail suspension test as a model for mice. Neurosci Biobehav Rev 29:571–625. https://doi.org/10.1016/j.neubiorev.2005.03.009
Heba A, Hassan HA, Mohamed A, Gamal EA (2009) 1-Malonyl-1,4-dihydropyridine as a novel carrier for specific delivery of drugs to the brain. Bioorg Med Chem 17:1681–1692. https://doi.org/10.1016/j.bmc.2008.12.051
Mohamed AA, Gamal EDA, Abuo-Rahma AAH (2009) Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Euro J Med Chem 44:3480–3487. https://doi.org/10.1016/j.ejmech.2009.01.032
John FC, Michelle EP, Irwin L (2005) Differential behavioural effects of the antidepressants reboxetine, fluoxetine and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology 182(3):335–344. https://doi.org/10.1007/s00213-005-0093-5
Zuhal OZ, Burak K, Bulent G, Unsal CA, Altan B (2007) Synthesis and studies on antidepressant and anticonvulsant activities of some 3-(2-furyl)-pyrazoline derivatives. Euro J Med Chem 42:373–379. https://doi.org/10.1002/ardp.200800068
Filipe SD, da Evilazio S, Ricardo AV, Marina U, Ricardo JN, Thereza CM (2006) Synthesis and antidepressant-like action of stereoisomers of imidobenzenesulfonylaziridines in mice evaluated in the forced swimming test. Bioorg Med Chem 14:5397–5401. https://doi.org/10.1016/j.bmc.2006.03.036
Mailard CA, Edwards DA (1991) Excitotoxin lesions of the zonaincerta/lateral tegmentum continuum: effects on male sexual behaviour in rats. Behav Brain Res 46:143–149. https://doi.org/10.1016/s0166-4328(05)80107-x
OECD Guidelines for Testing of Chemicals. Guidelines 425, Acute Oral toxicity-Acute Toxic Class Method 2001.
Bhosale SH, Kanhed AM, Dash RC, Suryawanshi MR, Mahadik KR (2014) Design, synthesis, pharmacological evaluation and computational studies of 1-(biphenyl-4-yl)-2-{4-(substituted phenyl)-piperazin-1-yl]ethanones as potential antipsychotics. Eur J Med Chem 74: 358–365. https://www.sciencedirect.com/science/article/abs/pii/S0223523414000051
Kanhed AM, Zambre VP, Pawar VA, Sharma MK, Giridhar R, Yadav MR (2014) Structural requirements for Imidazo[1,2-A]pyrazine derivatives as Aurora A kinase inhibitors and validation of the model. Med Chem Res 23:5215–5253. https://doi.org/10.1007/s00044-014-1094-x
Schrödinger, LLC, New York, NY, 2009.
Kanhed AM, Dash RC, Parmar N, Das TK, Giridhar R, Yadav MR (2015) Benzo[E]pyrimido[5,4-B]Diazepin-6(11H)-one derivatives as Aurora A kinase inhibitors: LOTA-QSAR Analysis and detailed systematic validation of the developed model. MolDiv 19:965–974. https://doi.org/10.1007/s11030-015-9618-y
Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR (2021) Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. MolDiv 25:383–401
Modi SJ, Kulkarni VM (2021) Exploration of structural requirements for the inhibition of VEGFR-2 tyrosine kinase: binding site analysis of type II, ‘DFG-out’ inhibitors. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.1872417
Bowers KJ, Chow DE, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD (2007) Scalable algorithms for molecular dynamics simulations on commodity clusters. 43. https://www.cines.fr/wp-content/uploads/2014/01/pap259.pdf
Tuckerman M, Berne BJ, Martyna GJ (1992) Reversible multiple time scale molecular dynamics. J Chem Phys 97:1990–2001. https://doi.org/10.1063/1.463137
Funding
There was no funding source pertaining to this research work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Suryawanshi, M.R., Kanhed, A.M., Kulkarni, V.M. et al. Design, synthesis, and computational studies of phenylacetamides as antidepressant agents. Mol Divers 26, 3157–3172 (2022). https://doi.org/10.1007/s11030-021-10374-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10374-5