
Abstract
Drug designing and development is an important area of research for pharmaceutical companies and chemical scientists. However, low efficacy, off-target delivery, time consumption, and high cost impose a hurdle and challenges that impact drug design and discovery. Further, complex and big data from genomics, proteomics, microarray data, and clinical trials also impose an obstacle in the drug discovery pipeline. Artificial intelligence and machine learning technology play a crucial role in drug discovery and development. In other words, artificial neural networks and deep learning algorithms have modernized the area. Machine learning and deep learning algorithms have been implemented in several drug discovery processes such as peptide synthesis, structure-based virtual screening, ligand-based virtual screening, toxicity prediction, drug monitoring and release, pharmacophore modeling, quantitative structure–activity relationship, drug repositioning, polypharmacology, and physiochemical activity. Evidence from the past strengthens the implementation of artificial intelligence and deep learning in this field. Moreover, novel data mining, curation, and management techniques provided critical support to recently developed modeling algorithms. In summary, artificial intelligence and deep learning advancements provide an excellent opportunity for rational drug design and discovery process, which will eventually impact mankind.
Graphic abstract
The primary concern associated with drug design and development is time consumption and production cost. Further, inefficiency, inaccurate target delivery, and inappropriate dosage are other hurdles that inhibit the process of drug delivery and development. With advancements in technology, computer-aided drug design integrating artificial intelligence algorithms can eliminate the challenges and hurdles of traditional drug design and development. Artificial intelligence is referred to as superset comprising machine learning, whereas machine learning comprises supervised learning, unsupervised learning, and reinforcement learning. Further, deep learning, a subset of machine learning, has been extensively implemented in drug design and development. The artificial neural network, deep neural network, support vector machines, classification and regression, generative adversarial networks, symbolic learning, and meta-learning are examples of the algorithms applied to the drug design and discovery process. Artificial intelligence has been applied to different areas of drug design and development process, such as from peptide synthesis to molecule design, virtual screening to molecular docking, quantitative structure–activity relationship to drug repositioning, protein misfolding to protein–protein interactions, and molecular pathway identification to polypharmacology. Artificial intelligence principles have been applied to the classification of active and inactive, monitoring drug release, pre-clinical and clinical development, primary and secondary drug screening, biomarker development, pharmaceutical manufacturing, bioactivity identification and physiochemical properties, prediction of toxicity, and identification of mode of action.

Similar content being viewed by others

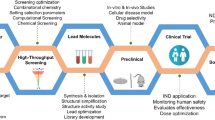

Introduction
From the past two decades, the development of efficient and advanced systems for the targeted delivery of therapeutic agents with maximum efficiency and minimum risks has imposed a great challenge among chemical and biological scientists [1]. Further, the cost of development and time consumption in developing novel therapeutic agents was another setback in the drug design and development process [2]. To minimize these challenges and hurdles, researchers around the globe moved toward computational approaches such as virtual screening (VS) and molecular docking, which are also known as traditional approaches. However, these techniques also impose challenges such as inaccuracy and inefficiency [3]. Thus, there is a surge in the implementation of novel techniques, which are self-sufficient to eliminate the challenges encountered in traditional computational approaches. Artificial intelligence (AI), including deep learning (DL) and machine learning (ML) algorithms, has emerged as a possible solution, which can overcome problems and hurdles in the drug design and discovery process [4]. Additionally, drug discovery and designing comprise long and complex steps such as target selection and validation, therapeutic screening and lead compound optimization, pre-clinical and clinical trials, and manufacturing practices. These all steps impose another massive challenge in the identification of effective medication against a disease. Thus, the biggest question that arises in front of pharmaceutical companies is managing the cost and speed of the process [5]. AI has answered all these questions in a simple and scientific manner, which reduced the time consumption and cost of the process. Moreover, the increase in data digitization in the pharmaceutical companies and healthcare sector motivates the implementation of AI to overcome the problems of scrutinizing the complex data [6].
AI, which is also referred to as machine intelligence, means the ability of computer systems to learn from input or past data. The term AI is commonly used when a machine mimics cognitive behavior associated with the human brain during learning and problem solving [7]. Nowadays, biological and chemical scientists extensively incorporate AI algorithms in drug designing and discovery process [8]. Computational modeling based on AI and ML principles provides a great avenue for identification and validation of chemical compounds, target identification, peptide synthesis, evaluation of drug toxicity and physiochemical properties, drug monitoring, drug efficacy and effectiveness, and drug repositioning [9]. With the advent of AI principles along with ML and DL algorithms, VS of compounds from chemical libraries, which comprises more than 106 million compounds, become easy and time-effective. Further, AI models eliminate the toxicity problems, which arise due to off-target interactions [10]. Herein, we briefly discuss the evolution of AI from ML to DL and big data involvement in revolutionizing the drug discovery process. Later on, we presented an overview on the congregation of AI and conventional chemistry in the improvement of the drug discovery process and the application of AI in the improvement of the traditional drug discovery process. Afterward, we discuss the numerous AI applications throughout the drug design and discovery processes such as primary and secondary screening, drug toxicity, drug release and monitoring, drug dosage effectiveness and efficacy, drug repositioning, and polypharmacology, and drug-target interactions.
Evolution of artificial intelligence: machine learning to deep learning
In September 2015, the Google search trend showed that after the introduction of ML, AI was the most searched term. Some describe ML as the primary AI application, while others describe it as a subset of AI [11, 12]. AI is an umbrella term where computer programs are able to think and behave as humans do, whereas ML is beyond that where data are inputted in the machine along with an algorithm like Naïve Bayes, decision tree (DT), hidden Markov models (HMM) and others, which helps the machine to learn without being explicitly programmed. Later, with the development of neural networks, machines could classify and organize inputted data that mimics like a human brain, which further shows advancement in AI. Around twentieth century, Igor Aizenberg and his colleagues, while talking about the artificial neural network (ANN), brought up the term “deep learning” for the first time. DL is a subset of ML, which itself is a subset of AI, and thus, the evolution goes like AI > ML > DL [13, 14]. ML either uses supervised learning, where the model is trained to use labeled data, which means that the input has been tagged with corresponding preferred output labels or uses unsupervised learning, where the model is trained to use unlabeled data but looks for recurring patterns from the input data [15]. Others are semi-supervised learning that uses the combination of both supervised and unsupervised learnings; self-supervised learning, which is a special case, uses a two-step process where unsupervised learning generates labels for unlabeled data and its ultimate goal is to make supervised learning model; reinforcement learning is a type of ML which improves its algorithm over time with the help of a constant feedback loop and lastly DL where there are many layers of ML algorithms which is called as a brain-inspired family of algorithms which mimics human brain but requires high computational power for training and big data to succeed [16, 17]. The origin of ML dates back to 1943 when McCulloch and Pitts published an article named “A logical calculus of the idea immanent in nervous activity,” where they gave the first-ever mathematical model of a neural network [18]. Alan M. Turing theorized the concept of ML in his seminal paper published in 1950 [19]. In 1952, Arthur L. Samuel popularized the term “machine learning” by writing a checker-playing program for IBM [20]. In 1957, Frank Rosenblatt developed perceptron, which was built for image recognition [21]. Henry J. Kelley developed the continuous backpropagation model in 1960, and a simpler version based only on-chain rule was developed by Stuart Dreyfus in 1962 [22, 23]. In 1965, Ivakhnenko and Lapa developed the first working DL networks. Around 1980, Kunihiko Fukushima developed an ANN called neocognitron that had a multilayered design that could help the computer learn how to recognize visual patterns [24]. He also developed the first convolutional neural network (CNN) which was based on the visual cortex organization found in animals [25] [Fig. 1].

a History of artificial intelligence in healthcare: the first breakthrough of artificial intelligence in healthcare comes in 1950 with the development of turning tests. Later on, in 1975, the first research resource on computers in medicines was developed, followed by NIH's first central AIM workshop marked the importance of artificial intelligence in healthcare. With the development of deep learning in the 2000s and the introduction of DeepQA in 2007, the scope of artificial intelligence in healthcare has increased. Further, in 2010 CAD was applied to endoscopy for the first time, whereas, in 2015, the first Pharmbot was developed. In 2017, the first FDA-approved cloud-based DL application was introduced, which also marked the implementation of artificial intelligence in healthcare. From 2018 to 2020 several AI trials in gastroenterology were performed. b Classification of artificial intelligence: there are seven classifications of artificial intelligence, which are reasoning and problem solving, knowledge representation, planning and social intelligence, perception, machine learning, robotics: motion and manipulation, and natural language processing, as discussed by Russel and Norvig in their book “Artificial Intelligence: A Modern Approach.” Machine learning is further divided into three significant subsets: supervised learning, unsupervised learning, and deep learning, whereas vision is divided into two subsets, such as image recognition and machine vision. Similarly, speech is divided into two subsets: speech to text and text to speech, whereas natural language processing is classified into five main subsets, including classification, machine translation, question answering, text generation, and content extraction. c Artificial intelligence in the healthcare and pharmaceutical industry has five significant applications, which change the entire scenario. These applications include research and discovery, clinical development, manufacturing and supply chain, patient surveillance, and post-market surveillance
David Rumelhart, Geoffrey Hinton, and Ronald J. Williams published a paper entitled “Learning Representations by Back-propagating Errors” in 1986, which demonstrated that backpropagation could provide an improvement in shape recognition and word prediction [26]. After the initial success, there were some setbacks, but Hinton kept working during the second AI Winter to achieve new heights. Thus, he is considered as the Godfather of DL. Soon, in 1989, Yann LeCun gave the first practical demonstration of backpropagation at Bell Labs [27]. The same year, Christopher Watkins published his thesis entitled “Learning from Delayed Rewards,” which introduced the concept of Q-learning, which further improved reinforcement learning in computer programs [28]. In 1995, Corinna Cortes and Vladimir Vapnik developed support vector machines (SVM) to map and recognize similar data [29]. After two years, in 1997, Jürgen Schmidhuber and Sepp Hochreiter developed long short-term memory (LSTM) for recurrent neural networks [30].
In 1999, a graphic processing unit (GPU) was launched as a microprocessor circuit, which was developed initially to accelerate 3D graphics processing for computer gaming. Later on, GPUs became popular in the field of technology and research as well because of their ability of parallel computing. A research report presented by META Group in 2001 stated that volume, speed, source and types of data were increasing, which was a call to prepare for the attack of Big Data. In 2007 Nvidia introduced compute unified device architecture (CUDA), a framework that allowed programmers and researchers to use GPU for general purpose computing [31]. Since then, with the help of CUDA, researchers started using GPUs for DL-driven operations, as high memory bandwidth of GPUs allowed easy handling of massive data involved in DL algorithms, and thousands of cores in GPUs allowed simultaneous parallel processing of neural networks. In 2009, Fei-Fei Li launched ImageNet, which is a free database containing millions of labeled images that can be used for research purposes [32]. AlexNet, a convolutional neural network, was created by Alex Krizhevsky around 2012, which helped in strengthening the speed and dropout using rectified linear units [33]. In the same year, “the cat experiment” conducted by Google Brain concluded that the network correctly recognizes less than 16% of the presented objects [34]. In 2014 Nvidia introduced CUDA deep neural network (cuDNN), a CUDA-based DL library, which accelerated DL-based operations [35]. Similarly, “Deep Face” was developed and released in 2014 to identify faces with 97.5% accuracy [36]. In the same year, generative adversarial networks (GANs) were introduced, using two competing neural networks to check whether the data are genuine or generated [37]. In 2016, Cray Inc. used Microsoft’s neural network software on its XC50 supercomputer with 1000 Nvidia Tesla P100 GPUs that could perform the task and gave output in a fraction of seconds. In 2017 Nvidia introduced Tesla V100 GPU, which had tensor cores that accelerated AI-based operations. However, DL is still in its growth phase, and creative ideas are required for further advancement in this field.
Revolutionizing drug discovery process: role of big data and artificial intelligence
Big data can be defined as data sets that are too gigantic and intricate to be analyzed with the conventional data analyzing software, tools, and techniques. The three main characteristic features of big data are volume, velocity, and variety, where volume represents the huge amount and mass of data generated, velocity represents the rate at which these data are being reproduced, and variety represents heterogenicity present in the data sets [38]. With the advent of microarray, RNA-seq, and high-throughput sequencing (HTS) technologies, a plethora of biomedical data is being engendered every day, due to which contemporary drug discovery has made a transition into the big data era. In drug discovery, the first and foremost step is the identification of appropriate targets (e.g., genes, proteins) involved in disease pathophysiology, followed by finding suitable drugs or drug-like molecules which can meddle with these targets, and now we have access to a constellation of biomedical data repositories which can help us in this regard [39]. Moreover, the evolution of AI has made big data analytics a lot easier as there is a myriad of ML techniques available now, which can help in extracting useful features, patterns, and structures present in these big biomedical data sets [40]. For target identification, a feature like a gene expression is widely used to understand disease mechanisms and find genes responsible for the disease. Microarray and RNA-seq technologies have generated a large amount of gene expression data for various disorders. NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) [41], The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) [42], Arrayexpress (https://www.ebi.ac.uk/arrayexpress/) [43], are some of the big repositories which contain gene expression data. By analyzing gene expression signatures, we can find out target genes responsible for different disorders. For example, using the ML approach and gene expression data, van IJzendoorn et al. 2019 found out novel biomarkers and potential drug targets for rare soft tissue sarcoma [44].
Further, genome-wide association studies (GWAS) can determine the interrelation of genomic variants with particular complex disorders [45]. GWAS central (https://www.gwascentral.org/) [46], NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/home) [47] are some of the repositories which contain GWAS data. Further, with the help of GWAS, we can ascertain the disease-associated genetic loci, and it has been observed that genes linked with these loci are potential therapeutic targets. For instance, Li et al. [48] used the GWAS catalog, gene expression, epigenomics, and methylation data to determine target genes associated with juvenile idiopathic arthritis loci through ML analysis . In addition, specific genes whose mutations can lead to different threatening diseases are also promising therapeutic targets. These risk genes can be identified by analyzing the various genome and exome sequencing data. For sequencing data, we have public repositories like Sequence read archive (https://www.ncbi.nlm.nih.gov/sra) [49], which contains sequencing data obtained from next-gen sequencing technology. The National Cancer Institute Genomic Data Commons (NCIGDC) (https://gdc.cancer.gov/) [50] and TCGA are data repositories that contain sequencing data related to cancer. Moreover, taking advantage of big data and AI, Han et al. 2019 have developed DriverML (https://github.com/HelloYiHan/DriverML), a supervised ML-based tool that can point out driver genes related to cancer [51] [Fig. 2].

Application of big data for drug designing and discovery: with the increase in biological and chemical data from the literature, in vitro, in vivo, clinical studies, genomics studies, proteomics studies, metabolomics studies, gene ontology studies, and molecular pathway data, different data repositories have been developed. For instance, ChemSpider, ChEMBL, ZINC, BindingDB, and PubChem are the essential databases for compound synthesis and screening in the drug designing and discovery process. The data stored in the above-said databases were curated and screened out for pharmacological and physicochemical properties of compound necessary for the drug discovery process instead of quantum mechanical calculations such as solvation energy and proton affinity the wave function, atomic forces, and transition state. The high-throughput screened data were subject to filtration based on drug-likeness, PAINS calculation, ADMET analysis, and toxicity. The filtered compounds were subject to artificial intelligence models such as deep learning, random forest, classification and regression, and neural networks for further analysis. These compounds were then subjected to quantitative-structure activity relationship and pharmacophore models followed by molecular docking and molecular dynamics simulations studies. Afterward, the final predicted compounds were visualized for binding energy calculations and active site identification. Thus, the final compound was identified and underwent in vitro and in vivo experimental studies for validation. However, quantum mechanical properties play a crucial role in the process of drug discovery and designing, but these properties cannot directly hamper the process of drug designing. QM methods include ab initio density functional theory and semi-empirical calculations, where accurate calculations use electron correlation methods. QM will become a more prominent tool in the repertoire of the computational medicinal chemist. Therefore, modern QM approaches will play a more direct role in informing and streamlining the drug-discovery process
Moreover, sometimes even published literature can be used for target identification, and PubMed (https://pubmed.ncbi.nlm.nih.gov/) [52] is a major repository of the various published biomedical literature, whose data mining can help in identifying targets for different disorders. After an appropriate target has been identified and validated, the next step is to find suitable drugs and/or drug-like molecules that can interact with the target and elicit the desired response [53]. In the age of big data, the multitude of big chemical databases is at our disposal, which can help in finding perfect drugs for a specific target. Likewise, PubChem (https://pubchem.ncbi.nlm.nih.gov/) [54] is a freely accessible chemical database that contains data of various chemical structures, including their biological, physical, chemical, and toxic properties [55]. Further, the ChEMBL database (https://www.ebi.ac.uk/chembl/) [56] is an open access big database containing data of numerous bioactive compounds exhibiting drug-like properties [57]. The ChEMBL database also contains information on absorption, distribution, metabolism, and excretion (ADME), toxicity properties of these compounds, and even their target interactions. Further, DrugBank (https://go.drugbank.com/) [58] is another open access pharmaceutical data repository which contains data of various drugs, their targets, and mechanism [59]. Additionally, the library of integrated network-based cellular signature (LINCS) L1000 (https://lincsproject.org/LINCS/) [60] is another repository that contains information on the change in gene expression signatures of human cell lines when treated with different chemical compounds. LINCS L1000 data-driven search engine, known as L1000CDS2, is an open-access search engine that contains data of drugs that can revert the expression of differentially expressed genes; hence, they too can be used for drug discovery [61]. Further, the protein data bank (PDB) (https://www.rcsb.org/) [62] is another freely accessible online repository that contains data of three-dimensional structures of proteins, DNA, RNA [63]. PDB data are also widely used to assess protein–ligand interactions and then find appropriate inhibitors of a target protein. Xu et al. [64] combined ML and molecular docking to find inhibitors of COVID 3CL proteinase; here, the crystal structure of COVID 3CL proteinase was obtained from PDB.
Congregation of artificial intelligence and conventional chemistry: improves drug discovery
In the pharmaceutical industry, AI has emerged as a possible solution to the problems raised due to classical chemistry or chemical space, which hampers drug discovery and development. With the advancements in technologies and the development of high-performance computers, AI algorithms such as ML to DL have been increased in computer-aided drug design (CADD). AI is not a new technique for scientists in drug discovery and development; neither chemists' desire to accurately forecast chemical activity-structure relationships. For example, Hammett relates equilibrium constants with reaction rates, whereas Hansch performed computer-assisted prediction of drug compounds' physicochemical properties and biological activity. The success of Hansch provides an avenue for research that will focus on (a) detailed identification and prediction of the chemical structure along with the characterization of properties such as pharmacophores and three-dimensional structure and (b) hypothesize complex mathematical equations that will relate to chemical representation and biological activity of the predicted compound. However, scientists' main aim in the current era is to improve the drug discovery and development process with high accuracy and confidence scores through ML algorithms based on classical chemistry activities. This will encourage chemists to identify the potential of AI techniques for answering two crucial questions of medical chemistry, such as "what should be the next compound?” and "what is the process of making a compound?”. Thus, the last two decades developed many techniques and tools for computational drug discovery, quantitative-structure activity relationship (QSAR) methods, and free-energy minimization techniques. For example, [65] distinguish compound cell activity using machine intelligence methods such as DT, random forest (RF) method, CNN, SVM, LSTM network, and gradient boosting machine. Among the mentioned models, in some models, the compounds were expressed as a string by the simplified molecular input line entry system and directly used as input data instead of any chemical descriptor and act as natural language processing. They have used two different cutoffs for the single data set (Z-score = 3) and the whole data set (Z-score = 5 or 6). Later on, they incorporated nine different metrics used to evaluate the model's precision, accuracy, the area under the curve, and Cohen's K value. The results demonstrated that the gradient boosting machine is competent at balanced data distribution. The experiment's outcomes also concluded that classical ML methods and DL methods could classify compound cell activity [65]. Similarly, [66] predicted the PAMPA effective permeability using a two-QSAR approach, where the authors developed a classical QSAR model and an ML-based QSAR model using a partial least square (PLS) scheme and hierarchical SVM (HSVR) scheme. The authors concluded that the HSVR scheme executed better than the PLS scheme in the training set, test set, and statistical analysis [66]. Further, for the synthesis of new compounds, chemical scientists readily depended on published literature. With advancements in automated drug discovery methods involving AI and ML, it is relatively simple to distinguish between existing drugs and novel chemical structures. For example, [67] applied a computational approach to screen the hepatotoxic ingredients in traditional Chinese medicines, whereas [68] demonstrated the phylogenetic relationship, structure–toxicity relationship, and herb-ingredient network using computational technique. Recently, Zhang et al. implemented computational analysis against a novel coronavirus, where the authors screened different compounds that were biologically active against severe acute respiratory syndrome (SARS). Later on, the compounds were subjected to ADME and docking analysis. The results concluded that 13 existing Chinese traditional medicines were effective against novel coronavirus [69]. Thus, conventional chemistry-oriented drug discovery and development concepts combined with computational drug designing provide a great future research platform. Moreover, system biology and chemical scientists worldwide, in coordination with computational scientists, develop modern ML algorithms and principles to enhance drug discovery and development.
Transforming traditional computational drug design through artificial intelligence and machine learning techniques
For many years computational methods have played an essential role in drug design and discovery, which transformed the whole process of drug design. However, many issues like time cost, computational cost, and reliability, are still associated with traditional computational methods [70, 71]. AI has the potential to remove all these bottlenecks in the area of computational drug design, and it also can enhance the role of computational methods in drug development. Moreover, with the advent of ML-based tools, it has become relatively easier to determine the three-dimensional structure of a target protein, which is a critical step in drug discovery, as novel drugs are designed based on the three-dimensional ligand biding environment of a protein [72, 73]. Recently, Google’s DeepMind (https://github.com/deepmind) has devised an AI-based tool trained on PDB structural data, referred to as AlphaFold, which can predict the 3D structure of proteins from their amino acid sequences [74]. AlphaFold predicts 3D structures of proteins in two steps: (i) firstly, using a CNN it transforms an amino acid sequence of a protein to distance matrix as well as a torsion angle matrix, (ii) secondly, using a gradient optimization technique it translates these two matrices into the three-dimensional structure of a protein [75]. Likewise, Mohammed AlQuraishi from Harvard Medical school has also designed a DL-based tool that takes protein’s amino acid sequence as input and generates its three-dimensional structure. This model, referred as Recurrent Geometric Network (https://github.com/aqlaboratory/rgn), uses a single neural network to figure out bond angles and angle of rotation of chemical bonds connecting different amino acids in order to predict the three-dimensional structure of a given protein [76].
Further, quantum mechanics is used to determine the properties of molecules at a subatomic level, which is used to estimate protein–ligand interactions during drug development. However, sometimes with conventional computational techniques, quantum mechanics can be computationally very expensive and demanding, which can affect its accuracy [77]. However, with AI, quantum mechanics can get more user-friendly and efficacious. Schtutt et al. 2019 have recently developed a DL-driven tool, referred to as SchNOrb (https://github.com/atomistic-machine-learning/SchNOrb), which can predict molecular orbitals and wave functions of organic molecules accurately. With these data, we can determine the electronic properties of molecules, the arrangement of chemical bonds around a molecule, and the location of reactive sites [78]. Thus, SchNOrb can help researchers in designing new pharmaceutical drugs. Moreover, molecular dynamics (MD) simulation analyzes how molecules behave and interact at an atomistic level [79]. In drug discovery, MD simulation is used to evaluate protein–ligand interactions and binding stability. One major issue with MD simulation is that it can be very arduous and time-consuming. AI has the capacity to accelerate the process of MD simulation [80]. In this regard, Drew Bennett et al. performed MD simulations to calculate free energies for transferring 15,000 small molecules from water to cyclohexane to train a 3D convolutional network and spatial graph CNN using these free energies and some other atomistic features. The researchers found that the trained neural networks predicted free energies of transfer with almost similar accuracy compared to MD simulation calculations [81]. This study shows that ML techniques can improvize and expedite MD simulations. However, a large amount of training data is required to achieve this.
Moreover, de novo drug design has also taken advantage of AI in recent years. For example, Q.Bai et al. 2020 have devised MolAIcal (https://molaical.github.io/), a tool that can design three-dimensional drugs in three-dimensional protein pockets [82]. MolAICal designs 3D drugs by action of two components: (i) first component uses DL and genetic algorithm trained on the US food and drug administration (FDA)-approved drugs, for de novo drug design, (ii) second component combines molecular docking and DL model trained on ZINC database (https://zinc.docking.org/) [83]. Likewise, Popova et al. 2018 designed a deep reinforcement learning-based algorithm, referred to as ReLeaSE (https://github.com/isayev/ReLeaSE), for de novo drug design. ReLeaSE achieves its desired outcome by integrating two deep neural networks (DNN), known as generative and predictive, where the generative model is used to produce new compounds, and the predictive model is used to predict the properties of the compound [84]. Further, in recent times, AI has been used to upgrade the process of synthesis planning as well, a process that is used to determine an optimal synthesis pathway for a molecule of interest. Recently, Grzybowski et al. [85] developed a DT-based program, referred to as chematica, to design novel synthesis pathways for desired molecules. Similarly, Genheden et al. have implemented AiZynthFinder (https://github.com/MolecularAI/aizynthfinder), an open-source tool for retrosynthesis planning built on Monte Carlo tree search, which is regulated by a neural network [86]. Likewise, Segler et al. [87] used the integration of three distinct neural networks in conjugation with the Monte Carlo tree search to discover novel retrosynthesis routes. ICSYNTH (https://www.deepmatter.io/products/icsynth/) is another tool that can produce novel chemical synthesis pathways by using a collection of chemical rules which are generated via ML models [88].
Additionally, various text mining-based tools have also been developed, which can aid the process of traditional drug discovery. Text mining uses methods like natural language processing (NLP) to transform unstructured texts in various literature and databases into structured data, which can be analyzed appropriately to gain new insights. NLP is a branch of AI, which allows computers to process and analyze human languages like speech and text through AI-based algorithms. Taking advantage of this AI driven techniques, various text mining-based tools have been developed. For instance, Jang et al. 2018 developed PISTON (http://databio.gachon.ac.kr/tools/PISTON/), a tool that can predict drug side effects and drug indications, using NLP and topic modeling [89]. Likewise, DisGeNET (https://www.disgenet.org/) is a text mining-driven database that contains a plethora of information on gene-disease and variants-disease relationships [90]. Data in DisGeNET can analyze various biological processes like adverse drug reactions, molecular pathways involved in disease, drug action on targets. Further, STRING (https://string-db.org/) is another text mining-driven database containing a myriad of information on protein–protein interactions for various organisms [91]. In addition, STITCH (http://stitch.embl.de/) is another text mining-driven database, which contains information on interactions between proteins and chemicals/small molecules [92]. Information in STICH can also be used to ascertain binding affinities of drugs and drug-target association.
Artificial intelligence in primary and secondary drug screening
Today AI has come out as a very successful and demanding technology because it saves time and is cost-efficient [93]. In general, cell classification, cell sorting, calculating properties of small molecules, synthesizing organic compounds with the help of computer programs, designing new compounds, developing assays, and predicting the 3D structure of target molecules are some time-consuming and tiresome tasks which with the help of AI can be reduced and can speed up the process of drug discovery [94, 95]. The primary drug screening includes the classification and sorting of cells by image analysis through AI technology. Many ML models using different algorithms recognize images with great accuracy but become incompetent when analyzing big data. To classify the target cell, firstly, the ML model needs to be trained so that it can identify the cell and its features, which is basically done by contrasting the image of the targeted cells, which separates it from the background [96]. Images with varying textured features like wavelet-based texture features and Tamura texture features are extracted, which is further reduced in dimensions through principal component analysis (PCA). A study suggests that least-square SVM (LS-SVM) showed the highest classification accuracy of 95.34% [97, 98]. Regarding cell sorting, the machine needs to be fast to separate out the targeted cell type from the given sample. Evidence suggests that image-activated cell sorting (IACS) is the most advanced device that could measure the optical, electrical, and mechanical properties of the cell [99] [Fig. 3].

Artificial intelligence in primary and secondary drug screening: in drug discovery and designing pipeline, screening of potential lead is crucial, and artificial intelligence plays a great role in identifying novel and potential lead compounds. There are approximately 106 million chemical structure presents in chemical space from different studies such as OMIC studies, clinical and pre-clinical studies, in vivo assays, and microarray analysis. With machine learning models such as reinforcement models, logistic models, regression models, and generative models, these chemical structures are screened out based on active sites, structure, and target binding ability. The complete drug discovery process through artificial intelligence will take about 14–18 years, which is comparatively less than the traditional drug discovery process. The first step in the drug discovery process is lead identification, in which disease-modifying target protein is identified through reverse docking, bioinformatics analysis, and computational chemical biology. In the second step, primary screening of compounds is done to select potential lead compounds, which can inhibit target protein. This can be done through virtual screening and de novo designing. The next step in the drug discovery process includes lead optimization and lead compound identification through focused library design, drug-like analysis, drug-target reproducibility, and computational biology. Afterward, secondary screening of compounds is performed, followed by pre-clinical trials. The drug discovery process's final step is clinical development through cell-culture analysis, animal model experimentation, and patient analysis
The secondary drug screening includes analyzing the physical properties, bioactivity, and toxicity of the compound. Melting point and partition coefficient are some of the physical properties that govern the compound's bioavailability and are also essential to design new compounds [100], while designing a drug, molecular representation can be done using different methods like molecular fingerprinting, simplified molecular-input line-entry system (SMILES), and Coulomb matrices [101]. These data can be used in DNN, which comprises two different stages, namely generative and predictive stage. Though both the stages are trained separately through supervised learning, when they are trained jointly, bias can be applied to the output, where it is either rewarded or penalized for a specific property. This whole procedure can be used for reinforcement learning [84]. Matched molecular pair (MMP) has been extensively used for QSAR studies. MMP is associated with a single change in a drug candidate, which further influences the bioactivity of the compound [102]. Along with MMP, other ML methods are used like DNN, RF, and gradient boosting machines (GBM) to get modifications. It has been observed that DNN can predict better than RF and GBM [103]. With the increase in databases, which are publicly available like ChEMBL, PubChem, and ZINC, we have access to millions of compounds annotating information like their structure, known targets and purchasability; MMP plus ML can predict bioactivity like oral exposure, intrinsic clearance, ADMET, and method of action [98, 104, 105]. Optimizing the toxicity of a compound is the most time-consuming and expensive task in drug discovery and is a crucial parameter as it adds significant value to the drug development process.
Applications of artificial intelligence in drug development process
The most arduous and desponding step in the drug discovery and development process is identifying suitable and bioactive drug molecules present in the vast size of chemical space, which is in the order of 1060 molecules. Further, the drug discovery and development process are considered a time- and cost-consuming process. The most infuriating point is that nine out of ten drug molecules usually fail to pass phase II clinical trials and other regulatory approvals [106,107,108]. The above-said limitations of drug discovery and development can be addressed by implementing AI-based tools and techniques. AI is involved in every stage of the drug development process such as small molecules design, identification of drug dosage and associated effectiveness, prediction of bioactive agents, protein–protein interactions, identification of protein folding and misfolding, structure and ligand-based VS, QSAR modeling, drug repurposing, prediction of toxicity and bioactive properties, and identification of mode of action of drug compounds as discussed below.
Peptide synthesis and small molecule design
Peptides are a biologically active small chain of around 2–50 amino acids, which are increasingly being explored for therapeutic purposes as they have the ability to cross the cellular barrier and can reach the desired target site [109]. In recent years, researchers have taken advantage of AI and used it to discover novel peptides. For instance, Yan et al. 2020 developed Deep-AmPEP30, a DL-based platform for the identification of short anti-microbial peptides (AMPs) [110]. Deep-AmPEP30 (https://cbbio.online/AxPEP/) is a CNN-driven tool that predicts short AMPs from DNA sequence data. Using Deep-AmPEP30, Yan et al. identified novel AMPs from the genome sequence of C. glabrate, a fungal pathogen present in the GI tract. Likewise, Plisson et al. 2020 combined the ML algorithm with an outlier detection technique to discover AMPs with non-hemolytic profiles [111]. In addition, Kavousi et al. developed IAMPE (http://cbb1.ut.ac.ir/), a web server for the identification of anti-microbial peptides, which integrates 13CNMR-based features and physicochemical features of peptides as input to ML algorithms, in order to identify novel AMPs [112]. Similarly, Yi et al. 2019 devised ACP-DL (https://github.com/haichengyi/ACP-DL), a DL-based tool for the discovery of novel anti-cancer peptides [113]. ACP-DL uses the LSTM algorithm, which is an improved version of the recursive neural network (RNN), for differentiating anti-cancer peptides from non-anti-cancer peptides. Moreover, Yu et al. [114] proposed DeepACP, a deep recurrent neural network-based model for identifying anti-cancer peptides. Likewise, Tyagi et al. 2013 developed an SVM-based platform for identifying new anti-cancer peptides [115]. In addition, Rao et al. 2020 combined a graphical convolutional network and one-hot encoding to design ACP-GCN for the discovery of anti-cancer peptides [116]. Moreover, Grisoni et al. used an ensemble of four counter propagation ANN for identifying new anti-cancer peptides. Likewise, Wu et al. [117] proposed PTPD, a tool based on CNN and word2vec, for the discovery of novel peptides for therapeutics.
Moreover, small molecules are molecules that have very low molecular weight, and like peptides, small molecules are too being explored for therapeutic purposes using AI-based tools. For instance, Zhavoronkov et al. [118] devised generative tensorial reinforcement learning (GENTRL), a generative reinforcement learning-based tool for the de novo design of small molecules. With the help of GENTRL (https://github.com/insilicomedicine/GENTRL), Zhavoronkov et al. discovered novel inhibitors of an enzyme, DDR1 kinase [118]. Likewise, McCloskey et al. [119] combined DNA-encoded small molecule libraries (DEL) data with ML models like Graph CNN and RF to discover novel small drug-like molecules. Similarly, Xing et al. [120] integrated XGBoost, SVM, and DNN to find small molecules for targets implicated in rheumatoid arthritis.
Identification of drug dosage and drug delivery effectiveness
Administering an improper dose of any drug to a patient can lead to undesirable and lethal side effects; hence, it is crucial to determine a safe drug dose for treatment purposes. Over the years, it has been challenging to ascertain the optimum dose of a drug that can achieve the desired efficacy with minimum toxic side effects [121]. With the emergence of AI, lots of researchers are taking the help of ML and DL algorithms to determine appropriate drug dosage. For instance, Shen et al. [122] developed an AI-based platform, referred to as AI-PRS, to determine the optimum dose and combinations of drugs to be used for HIV treatment through antiretroviral therapy. AI-PRS is a neural network-driven approach, which relates drug combinations and dosage to efficacy through a parabolic response curve (PRS). In their study, Shen et al. administered a combination of tenofovir, efavirenz, and lamivudine to 10 HIV patients, and in due course, using the PRS method, they found out the dose of tenofovir could be reduced by 33% of the starting dose without causing virus relapse. Hence, using AI-PRS optimum drug dosage can be found out for other diseases as well. Further, Pantuck et al. [123] developed CURATE.AI, to determine adequate drug dose, which uses a patient’s personal data and transforms it to CURATE.AI profile in order to ascertain optimum dose. The study was performed, where a combination of cancer drug enzalutamide and investigation drug ZEN-3694 was given to a patient with metastatic castration-resistant prostate cancer. Using CURATE.AI, in the course of time, they found a 50% lower than starting dose of ZEN-3694, which can achieve desired results and arrest the cancer growth.
Further, Julkunen et al. [124] devised comboFM (https://github.com/aalto-ics-kepaco/comboFM), a novel ML-driven tool, which ascertain appropriate drug combinations and dose in pre-clinical studies like cancer cell lines. comboFM determines appropriate drug combinations and dose by using factorization machines (https://github.com/geffy/tffm), an ML framework for high-dimensional data analysis. In their study, using comboFM, Julkunen et al. identified a novel combination of anti-cancer drugs crizotinib and bortezomib, showing promising efficacy in lymphoma cell lines. Similarly, Sharabiani et al. used the ML approach to determine the optimum initial dose of anticoagulant drug warfarin. They used relevance vector machines to classify different patients based on their dose demands, and then, regression models were used to predict appropriate doses for the patients [125]. Likewise, Nemati et al. [126] developed a deep reinforcement learning model trained on multiparameter intelligent monitoring in intensive care II database (MIMIC II) to find an ideal dose of another anticoagulant drug, heparin. Likewise, Tang et al. [127] used ML techniques like ANN, Bayesian additive regression trees, boosted regression trees, multivariate adaptive regression splines to determine the optimum dose of immunosuppressive drug Tacrolimus. Moreover, Hu et al. [128] performed ML analysis with techniques like classification and regression trees, multilayer perceptron network, k-nearest neighbor to find out the safe initial dose of cardiac drug digoxin. In addition, Imai et al. [129] developed a DT model to find a safe starting dose of antibiotic drug vancomycin.
Predicting bioactive agents and monitoring of drug release
Designing and monitoring of drug-likeness is a tedious and time-consuming process. Lately, multiple online tools have been developed to analyze drug release and check accountability of selected bioactive compounds as a carrier. Benchmark data sets are later used to validate the computational analysis. For such evaluation’s pharmacophore based on the chemical feature suits the best. These models construct large 3D data sets developed via in silico experiments or in house compound collection [130]. To study ligand-based chemical features, various successful experiments have been established using the CATALYST program (www.accelrys.com), and a group of researchers was successful in predicting 11β-hydroxysteroid dehydrogenase type 1 inhibitors using the VS experiments [131].
Determining bioactive ligands is a crucial step for selecting a potent drug for a specific target. Now, researchers are taking advantage of artificial intelligence in determining bioactive compounds that can be used for specific targets associated with a disease. For instance, Wu et al. integrated DL and RF methods to devise WDL-RF (https://zhanglab.ccmb.med.umich.edu/WDL-RF/) for determining bioactivity of G protein-coupled receptors (GPCRs) targeting ligands. Likewise, Cichonska et al. [132] developed pairwiseMKL (https://github.com/aalto-ics-kepaco), a multiple kernel learning-based method, for determining the bioactivity of compounds [133]. To test their model's efficiency, they used to predict the anti-cancerous potency of compounds. Further, Mustapha et al. [134] developed an Xgboost model to determine bioactive chemical molecules. In addition, Merget et al. [135] created machine learning models like DNN, RF to determine the bioactivity of more than 280 different kinases. Furthermore, Arshadi et al. [136] have devised DeepMalaria, a DL-based model for identifying compounds having Plasmodium falciparum inhibitory activity. Likewise, Sugaya et al. [137] created a ligand-efficiency-driven support vector regression model to ascertain the biological activity of various chemical compounds. Moreover, Afolabi et al. [138] used data from the MLD drug data report (MDDR) repository and applied it to a combination of boosting algorithms to identify novel bioactive compounds. Additionally, Petinrin et al. [139] used the majority voting technique with an ensemble of different machine learning models to determine biologically active molecules.
Further, adverse drug reactions (ADRs) are unexpected, pernicious, fatal side effects caused by drug administration. ADRs are a major challenge in drug development, and it has become essential to identify possible ADRs during the nascent stage of drug development to make the drug development process more robust and efficacious. Lately, researchers have used AI to determine possible ADRs associated with different drugs before they are launched in the market for public use. For instance, Dey et al. [140] used DL-based model, which can predict ADRs associated with a drug and even identify chemical substructures responsible for those ADRs. In addition, Liu et al. [141] integrated chemical, biological, phenotypic properties of drugs to predict ADR associated with it via machine learning analysis. Likewise, Jamal et al. [142] combined biological, chemical, and phenotypic properties to predict nervous system ADRs linked with drugs through machine learning analysis. The authors also used their model to find out ADRs associated with current Alzheimer's drugs. Further, Xue et al. [143] integrated biomedical network topology with a DL algorithm to predict Drug-ADR correlation. Moreover, Raja et al. [144] used machine learning analysis to predict ADRs, which are a result of drug-drug interactions. They further used their model to predict ADR related to cutaneous disease drugs. Besides screening for an effective bioactive agent, another critical area to work with is drug likeliness and its interaction post-release. Recently, a freely accessible, user-friendly graphical interface SwissADME (http://www.swissadme.ch) was developed to evaluate the compatibility of the drug and its pharmacokinetic actions [145]. Mathematical models such as Higuchi, Hixson–Crowell, Ritger–Peppas–Kormeyers, Brazel–Peppas, Baker–Lonsdale, Hopfenberg, Weibull, and Peppas–Sahlin have also been applied in drug discovery, and one of the most common practice has been the calculation of drug loading capacity of the selected or screened bioactive molecule.
Prediction of protein folding and protein–protein interactions
Analyzing protein–protein interactions (PPIs) is crucial for effective drug development and discovery. Most of the protein annotation methods use sequence homology that has limited scope. High-throughput protein–protein interaction data, with ever-increasing volume, are becoming the foundation for new biological discoveries. A great challenge to bioinformatics is to manage, analyze, and model these data. Hence, computational models were developed that predicts multiple inputs at one place simultaneously [146]. Computational methods are implied to study both PPIs and protein–protein non-interactions (PPNIs), although PPIs are considered more informative than PPNIs. PPIs prediction can be identified as direct PPI, direct PPI with indirect functional associations and PPIs for signal transduction pathways [147]. Machine and statistical learning approaches like K-nearest neighbor, Naïve Bayesian, SVM, ANN, DT, and RF are used to predict the hindrance in PPIs. Use of Bayesian network (BN) has been applied to predict PPIs essentially using gene co-expression, gene ontology (GO), and other biological process similarity. Data set integration using BN produces precise and accurate PPI networks illustrating comprehensive yeast interactome [148]. Another group also used BN to combine data sets for the yeast to study PPIs [149]. A novel hierarchical model PCA-ensemble extreme learning machine (PCA-EELM) to predict protein–protein interactions only using protein sequences information has appeared as a powerful tool that gives output with accuracy and less duration [150]. Further, DNNs PPIs prediction efficiency was improved by a novel method known as DNN for protein–protein interactions prediction (DeepPPI) (http://ailab.ahu.edu.cn:8087/DeepPPI/index.html) [151]. In mammalian cells, signal transduction is mostly controlled by PPIs between unstructured motifs and globular proteins binding domains (PBDs). To predict these PBDs across multiple protein families bespoke ML tool was developed, known as hierarchical statistical mechanical modeling (HSMM) [152]. Prediction of protein–protein interactions based on ML, domain-domain affinities and frequency tables, a novel tool referred to as PPI_SVM, was developed in 2011, which is freely accessible at (http://code.google.com/p/cmater-bioinfo/) [153]. Due to the increased number of solved complex structures, a multimeric threading approach, MULTIPROSPECTOR, has been developed. In this method, proteins with known template structures are rethreaded, and their interaction with other proteins, their interfacial energy, and Z-score are established [154]. Structure-based threading logistic regression tool Struct2Net (http://struct2net.csail.mit.edu) to evaluate the probability of interaction is the first structure-based PPI predictor apart from homology modeling [155]. Gene cluster-based methods calculate the co-occurrence probability of orthologs of query proteins encoded from the same gene clusters. This method is also named domain/gene co-occurrence. If two proteins’ genes are not close by in the genome, then this method cannot reliably predict an interaction between these two genes [156, 157].
Structure-based and ligand-based virtual screening
In drug designing and drug discovery, VS is one of the crucial methods of CADD. VS refers to the identification of a small chemical compound that binds to a drug target. VS is an efficient method to screen out the promising therapeutic compound from a pool of compounds [158]. Thus, it becomes an important tool in high-throughput screening, which incurred the problem of high-cost and low-accuracy rate. In general, there are two important types of VS that are structure-based VS (SBVS) and ligand-based VS (LBVS) [159, 160]. The LBVS depends on the chemical structure and empirical data of both active and inactive ligands, which uses the chemical and physiochemical similarities of active ligands to predict the other active ligand from a pool of compounds with high bioactivity. However, the LBVS does not depend on the 3-D structure of the target protein, and thus, this method is implemented where target structure or information is missing, and the obtained structural accuracy is low [161]. On the other hand, SBVS has been implemented in such cases where 3-D structural information of protein or target has been elucidated either through in vitro or in vivo experiments or through computational modeling [162, 163]. In general, this method is used to predict the interaction between the active ligand or its associated target and to predict the amino acid residues, which are involved in drug-target binding. In comparison with LBVS, SBVS possesses high accuracy and precision. However, SBVS is associated with the problem of an increasing number of disease-causing proteins and their complicated conformations [164]. To use ML for VS, there should be a filtered training set comprising of known active and inactive compounds. These training data are used to train a model using supervised learning techniques. The trained model is then validated, and if it is accurate enough, the model is used on new data sets to screen compounds with desired activity against a target [165]. After that, the shortlisted compounds can go for ADMET analysis, followed by various bioassays before entering clinical trials. Hence, ML has the power to speed up VS, make it more robust, and can even reduce false positives in VS. Docking is the main principle applied in SBVS, where several AI and ML-based scoring algorithms have been developed such as NNScore, CScore, SVR-Score, and ID-Score [166]. Similarly, ML and DL methods such as RFs, SVMs, CNNs, and shallow neural networks have been constructed to predict protein–ligand affinity in SBVS. Moreover, AI-based algorithms have been developed for molecular dynamic simulation assays in SBVS [167]. On the other hand, LBVS consists of several steps, and each step comes up with novel AI- and ML-based algorithms to speed up the process and increase reliability. For example, several ML- and DL-based algorithms have been constructed for the preparation of useful decoy sets such as Gaussian mixture models (GMMs), isolation forests, and artificial neural networks (ANNs).
Further, ML models such as PARASHIFT, HEX, USR, and ShaPE algorithms have been constructed for LBVS [168, 169]. Currently, with the rise of AI algorithms in the healthcare and pharma industry, different tools and models have been developed for both LBVS and SBVS. For example, tools such as MTiOpenScreen (http://bioserv.rpbs.univ-paris-diderot.fr/services/MTiOpenScreen/) [170], FlexX‐Scan [171], CompScore (http://bioquimio.udla.edu.ec/compscore/) [172], PlayMolecule BindScope (PlayMolecule.org) [173], GeauxDock (http://www.brylinski.org/geauxdock) [174], EasyVS (http://biosig.unimelb.edu.au/easyvs) [175], DEKOIS 2.0 [176], PL-PatchSurfer2 (http://www.kiharalab.org/plps2/) [177], SPOT-ligand 2 (http://sparks-lab.org/) [178], Gypsum-DL (https://durrantlab.pitt.edu/gypsum-dl/) [179], and ENRI [180] have been developed for SBVS. Moreover, mounting evidence validates the hypothesis that AI plays a critical role in SBVS, such as identification of non-peptide cysteine-cysteine chemokine receptor 5 receptor agonists [181], screening of partial agonists of the β2 adrenergic receptor [182], identification of bromodomain-containing protein 4 inhibitors [183], discovery of natural product-like signal transducer and activator of transcription 3 dimerization inhibitor [184], prediction of VHL and hypoxia-inducible factor 1-alpha inhibitors [185], and prediction of Kelch-like ECH-associated protein-nuclear factor erythroid 2-related factor 2 (Keap-Nrf2) small-molecule inhibitors [186]. Likewise, Liu et al. 2017 discovered low toxicity O-GlcNAc transferase inhibitors, whereas Dou et al. [187] identified novel glycogen synthase kinase 3 beta (GSK-3β) inhibitors through SBVS [188]. Different studies were conducted on cancer and leukemia through SBVS, such as the discovery of novel GSK-3β for treatment of acute myeloid leukemia [189], identification of novel protein arginine methyltransferase 5 inhibitor in non-small cell lung cancer [190], identification of vascular endothelial growth factor receptor 2 potent compounds for the treatment of renal cell carcinoma [191], identification of multi-targeted inhibitors against breast cancer [192], and discovery of Mdm2-p53 inhibitor [193]. Recently, novel corona virus became a huge problem worldwide, and thus, here also SBVS provides a great opportunity for chemical and biological scientists to identify novel drug compounds against disease-causing targets. For example, Gahlawat et al. 2020 identified that saquinavir, lithospermic acid, and 11m_32045235 were promising therapeutic compound against SARS-Cov-2 main protease, whereas Selvaraj et al. 2020 demonstrated that TCM 57,025, TCM 3495, TCM 5376, TCM 20,111, and TCM 31,007 were therapeutic compounds that interact with the substrate-binding site of N7-MTase [194, 195]. On the same trend, Cruz et al. 2018 concluded that ZINC91881108 was potent compound against RIPK2, whereas Simoben et al. 2018 demonstrated eight novel N-(2,5-dioxopyrrolidin-3-yl)-n-alkylhydroxamate derivatives as smHDAC8 inhibitors with IC50 values ranging from 4.4 to 20.3 µM against smHDAC8 [196, 197] [Fig. 4].

a Ligand-based virtual screening: in the drug design and discovery process, ligand-based virtual screening is the most crucial step, which comprises different steps as shown in the figure. The initial step consists of database screening and the 3-D structural model's prediction through the active site for a special target and X-ray structure of complexes. Later on, pharmacophore modeling of selected compounds with selected features is performed, followed by pharmacophore and docking-based virtual screening of compounds. The screened compounds are subjected to different toxicity and physiochemical properties for further analysis. Finally, the lead compounds are subjected to in vitro and in vivo bioassays for validation. b structure-based virtual screening: it is another type of virtual screening applied in the drug discovery process, where target structure preparation and chemical compound library preparation are initial steps. Afterward, structural analysis and binding site prediction are done, followed by molecular docking of compounds with the selected target. Later on, molecular dynamics simulation studies are carried out to validate the screened compounds in silico, followed by experimental validation through bioassays
Moreover, different algorithms and tools have been developed for LBVS such as SwissSimilarity (http://www.swisssimilarity.ch/) [198], METADOCK [199], Open-source platform [200], HybridSim-VS (http://www.rcidm.org/HybridSim-VS/) [201], PKRank [202], PyGOLD (http://www.agkoch.de/) [203], BRUSELAS (http://bio-hpc.eu/software/Bruselas) [204], RADER (http://rcidm.org/rader/) [205], QEX [206], IVS2vec (https://github.com/haiping1010/IVS2Vec) [207], AutoDock Bias (http://autodockbias.wordpress.com/) [208], Ligity [209], D3Similarity (https://www.d3pharma.com/D3Targets-2019-nCoV/D3Similarity/index.php) [210], and GCAC (http://ccbb.jnu.ac.in/gcac) [211]. Emerging evidence suggests the potential implementation of AI algorithms in LBVS such as identification of aurora kinase A inhibitors [212], G-quadruplex-targeting chemotypes [213], PI3Kα inhibitors [214], targeting dengue virus non-structural protein 3 helicases [215], potential selective histone deacetylase 8 inhibitors [216], and novel p-Hydroxyphenylpyruvate dioxygenase inhibitors [217]. Apart from these mentioned studies number of literature validated the possible implementation of AI in LBVS, such as identification of HIV entry inhibitors and potent inhibitors of DNA methyltransferase [218, 219]. Like SBVS, LBVS also plays a crucial role in identifying potential therapeutic compounds against novel human coronaviruses. For example, Amin et al. 2020 demonstrated the molecular docking study of some in-house molecules as papain-like protease inhibitors, whereas Hofmarcher et al. 2020 through DNN identified 30,000 compounds from the library across 3.6 M compounds as CoV-2 inhibitors [220, 221]. Similarly, Choudhary et al. 2020 identified SARS-CoV-2 cell entry inhibitors, whereas Ferraz et al. 2020 identified bedaquiline, glibenclamide, and miconazole as potential therapeutic compounds against coronavirus [222, 223]. Xiao et al. 2018 developed ligand-based big data DNN models for VS of compound libraries against six anti-cancer targets. The study integrated 0.5 M chemical compounds, and the models developed were evaluated by tenfold cross-validation [224]. With the growing size of chemical compound libraries, it is become so difficult to find a potential hit and it is like finding a “needle in a haystack.” Thus, SBVS and LBVS have huge role in minimizing the complexity in identification of potential therapeutic compounds against the disease-causing target. Further, AI-based models in SBVS and LBVS make it simpler with high accuracy and precision. Table 1 discusses the different AI- and DL-based web tools and algorithms implemented in LBVS and SBVS.
QSAR modeling and drug repurposing
In drug designing and discovery, it is crucial to develop the relationship between chemical structures and their physiochemical properties with biological activities. Thus, QSAR modeling is a computational approach through which quantitative mathematical models can be created between chemical structure and biological activities. The main advantage of developing a mathematical model is identifying the diverse chemical structure from molecular databases, which can be used as therapeutic compounds against a disease target. Once the most promising compound is selected, it is subjected to laboratory synthesis and in vitro or in vivo testing. QSAR models are broadly classified into two types that are regression model and classification models. Gaussian processes (GPs) are a type of QSAR building regression model, which is a robust and powerful method of QSAR modeling. GP methods can handle a large number of descriptors and identify the crucial ones. Recently, two classification models have been demonstrated using GP that is intrinsic GP classification methods, and the other is a combination of GP regression technique and probit analysis [235, 236]. Further, the method is suitable for modeling nonlinear relationships and does not require subjective determination of the model parameters [237]. Recent advancements and increasing applications of ML algorithms such as neural networks, DL, and SVM provide a great avenue for QSAR modeling. Several web-based tools and algorithms have been developed for QSAR modeling such as VEGA platform (https://www.vega-qsar.eu/) [238], QSAR-Co (https://sites.google.com/view/qsar-co) [239], FL-QSAR (https://github.com/bm2-lab/FL-QSAR) [240], Meta-QSAR (https://github.com/meta-QSAR/simple-tree) (https://github.com/meta-QSAR/drug-target-descriptors) [241], DPubChem (www.cbrc.kaust.edu.sa/dpubchem) [242], Transformer-CNN (https://github.com/bigchem/transformer-cnn) [243], Cloud 3D-QSAR (http://chemyang.ccnu.edu.cn/ccb/server/cloud3dQSAR/) [244], MoDeSuS and Chemception (https://github.com/Abdulk084/Chemception) [245]. Karpov et al. 2020 developed a novel algorithm for QSAR modeling based on ANN called transformer-CNN. The method uses SMILES augmentation for training and interference. Similarly, Wang et al. 2020 developed QSAR modeling web-based tools by integrating the characteristics features of molecular structure generation, alignment, and molecular interaction field. Jin et al. through Cloud 3D-QSAR discovered a potent and selective monoamine oxidase B (MAO-B) inhibitor. In this study, the authors concluded that (S)-1-(4-((3-fluorobenzyl)oxy)benzyl)azetidine-2-carboxamide (C3) were more potent and selective inhibitor of MOB as compared to safinamide. Further, in vivo analysis revealed that compound C3 could inhibit cerebral MAO-B activity and rescue 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neuronal loss [246]. On the same trend, Bennett et al. 2020, through Chemception, predicted the small molecules transfer free energy by combining MD simulations and DL [81]. Moreover, the QSAR-Co tool was implemented in different studies such as the development of multi-target chemometric models for the inhibition of class I phosphoinositide 3-kinases enzyme isoforms, screening of ERK inhibitors as anti-cancer agents, prediction of K562 cells functional inhibitors, and prediction of antifungal properties of phenolic compounds [247,248,249,250]. Likewise, Kim and Cho 2018 developed a novel algorithm called PyQSAR (https://github.com/crong-k/pyqsar_tutorial) for a fast QSAR modeling platform using ML and Jupyter notebook. PyQSAR is a standalone python package that combines all QSAR modeling processes in a single workbench [251]. A. S. Geoffrey et al. 2020 conducted two different studies using PyQSAR, such as identification of potent drug candidates for novel coronavirus and development of QSAR of quercetin and its tumor necrosis factor-alpha inhibition activity [252, 253]. Further, Zuvela et al. developed ANN-based QSAR models for prediction of antioxidant activity of flavonoids. In this study, the authors integrated six methods such as PaD, PaD2, weights, stepwise, perturbation, and profile for interpretation and elucidation of ANN-based models, which calculates trolox-equivalent antioxidant properties. The results concluded that the ANN-based algorithm could eliminate the difficulties that arise due to poor interpretation of quantum mechanical parameters describing the molecular structure [254]. In parallel, Ding et al. 2020 generated a web-based tool known as VISAR (https://github.com/Svvord/visar) for dissecting chemical features through the DNN QSAR approach [255]. The mounting evidence demonstrates the implementation of QSAR modeling in drug designing and discovery process such as modeling of ToxCast assays relevant to the molecular initiating events of AOPs in Hepatic Steatosis [256], development of dipeptidyl peptidase 4 inhibitors against dipeptidyl peptidase 8 and dipeptidyl peptidase 9 enzymes [257], the applicability of QSAR model on domain analysis of HIV-1 protease inhibitors [258], and targeting HIV/HCV coinfection [259]. A well-recognized problem of ML models is data imputation for missing values in the bioassay data for SAR model generation. Basically there are three major types of missing values: (i) Missing Completely at Random (MCAR), which occurs when the probability of missing values in a variable is the same for all samples; (ii) Missing at Random (MAR), which means that probability of missing values, at random, in a variable depends only on the available information in other predictors; (iii) Missing Not at Random (MNAR), which means when probability of missing values is not random and depends on the information which is not recorded and the existing information predicts the missing values [260]. There are several ways to handle missing values like imputation using zero, mean, median or mode common value, imputation using a randomly selected value, imputing with a model or imputation using Deep Learning Library–Datawig. Every data set has missing values that need to be handled wisely in order to build a robust model [261]. Moreover, the complexity of data should be removed, and data must be curated to increase the accuracy and precision of the models generated. Moreover, initially QSAR models were implemented for predicting the toxicity and metabolism of small molecules such as molecules having molecular weight (mw) less than 1500 m.w. However, the QSAR technology applied in the early 2000s comes with some sort of constraints such as accuracy and reliability [262]. With the growing application of QSAR in drug discovery and design process such as VS, lead optimization, and target identification medicinal scientists and biologist were in constant efforts for development of more reliable and dependable approaches [263]. AI/ML algorithms-based QSAR models have potential to eliminate the constraints imposed by early methods. AI/ML-based QSAR model, namely hologram-based QSAR (HQSAR), group-based QSAR (G-QSAR), and Ensemble-based, have accelerated the drug discovery process by several folds [264, 265]. Further, apart from classical Hansch and Free-Wilson approaches, QSAR has gradually evolved over the past few years with newer refinement approaches, new methods for descriptors calculations, implementation of methodical validation tests, and involvement of receptor structural information. Similarly, apart from classical lead optimization, QSAR have been applied in different emerging areas of drug discovery and designing such as peptide QSAR, mixture toxicity QSAR, nanoparticles QSAR, QSAR of ionic liquids, cosmetic QSAR, phytochemical QSAR, and material informatics [266] [Fig. 5].

source is performed. Later on, evaluation of repositioning models through cross-validation, case analysis, and evaluation metrics is performed. Finally, validation of repurposed drugs is carried out through clinical trials, in vitro studies, and in vivo studies
a Quantitative structure–activity relationship workflow: the initial step comprises of data set compilation, where data from public database and literature database are accumulated and compiled, which further divided into different subsets for investigation. Afterward, data set processing is performed, where data pre-processing and curation followed by calculation of molecular descriptors are done. After description calculation, data set processing normalization of data and splitting of data into different sets are performed. In the third step, model construction is performed, where data sets such as internal data and external data are accumulated, and learning algorithms are applied for QSAR modeling. Finally, the statistical calculation is done to measure the model robustness. The final step in the quantitative-structure activity relationship is model evaluation, where the model is evaluated by comparison from previous benchmark models, identifying characteristics features, performance evaluation, and interpretation of essential features. b Drug repurposing or repositioning workflow: the first step is collection of data and data pre-processing followed by computational model generation. The models generated are support vector machines, logistic regression, random forest, deep learning, and matrix factorization. Afterward, the generation of proof-of-concept from a literature
Apart from QSAR modeling, the AI algorithm has also been implemented in drug repurposing or drug repositioning method. In drug designing and discovery, drug repositioning refers to the investigation of drugs that have already been developed for one diseased condition and reposition them for other diseased conditions. Repositioning drugs might be successful due to the possibility of multiple-target involvement in multiple diseases [267,268,269]. On another note, the emergence of large data sets from genomics, proteomics, and pharmacological in vivo and in vitro studies provides a great avenue for drug repositioning. Recently, the emergence of AI-based tools and algorithms in drug discovery provides a platform for future research. ML algorithms replace the chemical similarity and molecular docking-based conventional methods with new system biology methods, which can evaluate drug effects [270,271,272,273]. Thus, different AI-based algorithm and web-based tools have been developed in recent times such as DrugNet (http://genome2.ugr.es/drugnet/) [274], DRIMC (https://github.com/linwang1982/DRIMC) [275], DPDR-CPI (http://cpi.bio-x.cn/dpdr/) [276], PHARMGKB (https://www.pharmgkb.org/) [277], PROMISCUOUS 2.0 (http://bioinformatics.charite.de/promiscuous2) [278], and DRRS (http://bioinformatics.csu.edu.cn/resources/softs/DrugRepositioning/DRRS/index.html) [279]. Moreover, Yella and Jegga et al. 2020 constructed a model for drug repositioning using a multi-view graph attention approach known as MGATRx [280], whereas Yan et al. 2019 constructed a novel algorithm for drug repurposing based on a multisimilarity fusion approach known as BiRWDDA [281]. Further, Fahimian et al. 2020 constructed a novel algorithm known as RepCOOL to identify promising repurposed drugs for breast cancer stage II. The results concluded that doxorubicin, paclitaxel, trastuzumab, and tamoxifen were potential therapeutic agents against breast cancer stage II [282]. Likewise, Li et al. 2020 constructed a computational framework of host-based drug repurposing for broad-spectrum antivirals against RNA virus. In this study, the authors investigated 2352 approved drugs and 1062 natural compounds against different viral pathogens and concluded that the repurposed drugs were effective against zika virus and coronavirus [283]. Further, Wu et al. 2020 applied ML models, namely structural profile prediction model and biological profile prediction model, to predict anti-fibrosis drug candidates. The results demonstrated that the area under the receiver operating characteristics curve were 0.879 and 0.972 in the training set, whereas 0.814 and 0.874 in the testing set. The results concluded that natural products possess anti-fibrosis characteristics and serve as potential anti-fibrosis drug targets [284]. Recently, COVID-19 emerged as a global pandemic and researchers around the globe started the hunt for promising therapeutic agents. In this regard AI-based drug repositioning plays a crucial role. For example, network-based drug repurposing identified 16 potential anti-HCoV repurposable drugs, whereas Hooshmand et al. 2020 identified 12 promising drug targets for COVID-19 based on the multimodal DL approach [285, 286]. In recent times, the development of neural networks, DL models, and pipelines for drug repositioning have increased to a great extent. For example, SNF-CVAE based on drug similarity network fusion identified promising therapeutic agents for Alzheimer’s disease (AD) and juvenile rheumatoid arthritis, whereas DTI-RCNN based on neural network algorithm and integrates long short-term memory predicts drug-target interactions [287, 288]. PhenoPredict and SDTNBI are two other ML-based algorithms used to identify disease phenome-wide drug repositioning for schizophrenia and prediction of drug-target interactions, respectively [289, 290]. Zang et al. 2019 developed a DL-based model known as deepDR (https://github.com/ChengF-Lab/deepDR) to predict in silico drug repositioning. In the study, the authors integrate 10 different types of biological networks such as drug-disease, drug-side effects, drug-target, and seven drug-drug networks. The results concluded that deepDR predicted approved drugs such as risperidone and aripiprazole for the treatment of Alzheimer's disease (AD), whereas methylphenidate and pergolide for treatment of Parkinson's disease (PD) [291]. Likewise, Chen et al. 2020 constructed an AI-based novel algorithm called as iDrug (https://github.com/Case-esaC/iDrug) for the integration of drug repositioning and drug-target prediction through cross-network embedding. The efficiency and effectiveness of iDrug allow users to understand novel clinical insights of drug-target-disease mechanisms [292]. Studies demonstrated that drug repurposing through an AI-based algorithm can be implemented in cancer. For example, Li et al. 2020 integrated transcriptomics data and chemical structure information using DL and identified that pimozide as a promising therapeutic candidate against non-small cell lung cancer [293]. Similarly, Kuenzi et al. 2020 predicted drug response and synergy using a DL model of human cancer cells. The results concluded that predicted combinations improve progression-free survival, and response predictions stratify ER-positive breast cancer patient clinical outcomes [294]. Another AI application in drug repurposing comes from the study performed by Wang et al. 2020, which used bipartite graph convolutional networks for in silico drug repurposing. The authors constructed a model known as BiFusion (https://github.com/zcwang0702/BiFusion) through DL and heterogeneous information fusion. The results demonstrated that BiFusion achieved improved performance than multiple baselines for drug repurposing [295]. The examples mentioned above concluded the potential role of AI-based algorithms in drug repurposing. Further, with the advancement in technology, chemical scientists, biological scientists, and computational scientists search the methods for improving the accuracy and precision of AI-based models. Moreover, both QSAR and drug repositioning methods of drug discovery are incomplete without the involvement of molecular docking, which is used to analyze the interaction between the target molecule and a ligand molecule. Initially, in the early 2000s molecular docking was developed as a standalone tool that is used to determine the interaction between two molecules that is a target molecule and a ligand molecule. However, with the advent of AI technology the applicability of molecular docking has changed. Now molecular docking is being used in conjugation with MD simulation and AI-based tools in different areas of drug discovery like VS, target identification, polypharmacology, and drug repurposing [296]. The implementation of MD simulation and AI-based algorithms can increase the efficiency and accuracy of molecular docking. In addition, over the years, limitations in the use of molecular docking have also been addressed. For instance, in drug designing, molecular docking can be used only for those biological targets whose crystal structures are available as there are many targets whose structures are not available. Thus, a technique like homology modeling has been developed to overcome this hindrance [297]. Further, crystal structure data in PDB are increasing exponentially, enhancing the applicability of molecular docking in drug discovery. Table 2 discusses the tools and algorithm that have been implemented in in silico QSAR and drug repositioning.
Prediction of physicochemical properties and bioactivity
It is a well-established fact that every chemical compound is associated with physicochemical properties such as solubility, partition coefficient, ionization degree, permeability coefficient, which may hinder the pharmacokinetic properties of the compound and drug-target binding efficiency. Thus, the physicochemical properties of compounds must be considered while designing a novel drug molecule [100, 298]. For this, different AI-based tools have been developed to predict the physicochemical properties of chemical compounds. The AI-based tools developed for predicting biophysical and biochemical properties of compounds include molecular fingerprinting, a SMILES format, Coulomb matrices, and potential energy measurements, which are used in the DNN training phase [299, 300]. Recently, Zhang et al. developed a QSAR model to predict the six different physiochemical properties of environmental agents extracted from environmental protection agency (EPA). Similarly, Lusci et al. 2013 constructed a neural network-based model to predict the molecular properties. In the study, molecules are described by undirected cyclic graphs, whereas the former approaches for predicting physicochemical properties use directed acyclic graphs [301]. Later on, six AI-based algorithms were constructed for the prediction of human intestinal absorption of compounds. The methods constructed are SVM, k-nearest neighbor, probabilistic neural network, ANN, PLS, and linear discriminate model. Among the above-said models, SVM has higher accuracy of 91.54% [302]. In 2016, Zang et al. developed an ML-based model for the prediction of physicochemical properties such as octanol–water partition coefficient, water solubility, boiling point, melting point, vapor pressure, and bioconcentration factors of environmental chemicals [303]. Moreover, different AI-based tools have been developed such as ALOGPS 2.1 (http://www.vcclab.org/lab/alogps/) [304], ASNN (http://www.vcclab.org/lab/asnn/) [305], E-BABEL (http://www.vcclab.org/lab/babel/) [304], PCLIENT (http://www.vcclab.org/lab/pclient/) [304], E-DRAGON (http://www.vcclab.org/lab/edragon/) [304], ChemSpider (http://www.chemspider.com/) [306], SPARC (http://sparc.chem.uga.edu/sparc/) [307], and OSIRIS property explorer (https://www.organic-chemistry.org/prog/peo/) [308]. In 2020, a study was conducted to design, synthesize, and ADMET prediction of bis-benzimidazole as anticancer agents. In the same study, the author calculated molecular properties of compounds through Lipinski’s rule of five and predicted the pre-ADMET properties of the synthetic compounds [309]. Further, Puratchikody et al. 2016 used ORISIS property explorer in their study to predict the quantitative structural toxicity of tyrosine derivates intended for safe, potent inflammation treatment. The results concluded that out of 55 potent molecules, only 19 molecules were considered as potent cyclooxygenase-2 inhibitors [310]. On similar lines, RF- and DNN-based models were constructed to predict human intestinal absorption of different chemical compounds. Thus, from the examples, it must be concluded that the AI-based approach has a significant role in drug discovery and development through the prediction of physicochemical properties.
Moreover, the therapeutic activity of drug molecules depends on their binding efficiency with the receptor or target, and thus, the chemical molecule, which are not able to show the binding affinity with the drug target, will not be considered as a therapeutic agent. For this reason, the prediction of the binding affinity of a chemical molecule with the therapeutic target is vital for drug discovery and development [311]. Recent advancements in AI algorithms enhance the process of binding affinity prediction, which uses similarity features of the drug and its associated target. Several web-based tools have been developed, such as ChemMapper and the similarity ensemble approach (SEA). Further, ML- and DL-based models for the identification of drug-target affinity have been constructed, such as KronRLS, SimBoost, DeepDTA, and PADME [312]. The KronRLS predicts the similarity between a drug and its target to calculate the drug-target binding affinity based on the ML algorithm. KronRLS considered both feature-based and similarity-based interaction while predicting drug-target binding affinity [313]. DL approaches such as DeepDTA (https://github.com/hkmztrk/DeepDTA) [314], and PADME [315] predict drug-target binding affinity, which depends on the 3-D structure of a protein. Beck et al. 2020 conducted a study to predict commercially available antiviral drugs as a potential therapeutic agent against novel coronavirus (SARS-CoV-2) through DeepDTA [316]. Similarly, Lee and Kim 2019 predicted the drug-target interactions by DNN based on large-scale drug-induced transcriptome data using PADME [317]. Another DL model that uses both RNN and CNN was constructed to predict drug-target binding affinity, which is called as DeepAffinity (https://github.com/Shen-Lab/DeepAffinity) [318]. Jiang et al. 2019, using DeepAffinity, proposed a novel protein descriptor for identifying drug-target interaction, whereas Born et al. 2020 with the help of Deep Affinity, identified antiviral candidates for SARS-CoV-2 [319, 320]. The above data validate the importance of ML and DL algorithms in physiochemical properties and bioactivity of drug molecules during drug designing. However, the validation and accuracy of such algorithms are still a significant drawback from a research perspective. Thus, extensive research should be done to maximize the accuracy and precision of AI-based algorithms through curated and extensive data input. In Table 2, we have summarized the tools and databases for physiochemical and bioactivity prediction based on AI algorithms, including DL, neural networks, SVM, and others.
Prediction of mode of action and toxicity of compounds
Drug toxicity refers to the chemical molecule's adverse effect on an organism or on any part of the organism due to the compound's mode of action or metabolism. The extended scope of AI has the potential to predict the off-target and on-target effects of drug molecules along with in vivo safety analysis of chemical compounds before their synthesis has fascinated the scientists associated with the drug development process. The involvement of AI has reduced drug development time, cost, attrition rates, and human resources. For this different web-based tools have been developed such as LimTox (http://limtox.bioinfo.cnio.es/) [321], pkCSM (http://biosig.unimelb.edu.au/pkcsm/) [322], admetSAR (http://lmmd.ecust.edu.cn/admetsar2/) [323], and Toxtree (http://toxtree.sourceforge.net/) [324]. Srivastava et al. 2020 used admetSAR to evaluate the toxicity of Withania somnifera as a therapeutic compound against COVID-19, whereas Uygun et al. 2021 incorporated pkCSM for the identification of the therapeutic effect and toxicological properties of pyrazolo[1,5-a]pyrazine-4(5H)-one derivative on lung adenocarcinoma cell line [325, 326]. Advancements in AI-based approaches led to the development of different toxicity prediction software and web-based tools such as Tox21 (https://ntp.niehs.nih.gov/whatwestudy/tox21/index.html) [327], SEA (http://sea.bkslab.org/) [328], eToxPred (https://www.brylinski.org/etoxpred-0) [329], and TargeTox (https://github.com/artem-lysenko/TargeTox) [330]. Tox21 evaluates the toxicity of 12,707 environmental compounds and drugs, whereas SEA forecasts the toxicity of 656 marketed drugs against 73 unintended targets. TargeTox predicts toxicity risk based on the target-drug biological network. In 2016, Huang et al. predicted the in vivo toxicity profile and mechanism characterization of more than 10,000 chemical compounds through modeling Tox21, whereas, in the same year, Zhou et al. predicted the cancer-relevant proteins using an improved molecular SEA [331, 332]. Further, Gupta and Rana. 2019 employed eToxPred to predict the toxicity of small molecules of androgen receptor. The authors incorporated 1444 characteristics features of small molecules on 10,273 drugs in which 461 are considered as active and 9812 are inactive [333].
DeepTox (http://bioinf.jku.at/research/DeepTox/tox21.html) [334] and PrOCTOR (https://github.com/kgayvert/PrOCTOR) [335], are used for prediction of toxicity of new compounds and prediction of the toxicity probability in clinical trials, respectively. For example, Robledo-Cadena et al. 2020 predicted the effect of non-steroidal anti-inflammatory drugs on cisplatin, paclitaxel, and doxorubicin efficacy against cervix cancer cells using PrOCTOR, whereas Gilvary et al. 2020 identified the novel indications for 2,576 small molecules incorporated with 16 different drug features for PD and Type 2 diabetes [336, 337]. Similarly, using DeepTox, Simm et al. 2018 analyzed and repurposed high-throughput imaging assay data to predict the biological activity of different chemical compounds that were targeting alternative biological pathways and processes [338]. Furthermore, DeepTox was used for the development of several ML and DL algorithms, which predicts the toxicity properties and chemical characteristics features of drug compounds such as SMILES2Vec (predicts chemical properties) [339], Chemception (DNN-based prediction of chemical properties) [245], DeepSynergy (prediction of anti-cancer drug synergy with DL) [340], and deepAOT (prediction of compound acute oral toxicity) [341]. However, the accuracy and precision of DeepTox and PrOCTOR could be increased by using large and refined data sets, which could be achieved with the pharmaceutical industry's involvement. Recently, other ML-based tools such as SPIDER [342] and read-across structure–activity relationships (RASAR) [343] were developed, which are capable of analyzing β-lapachone targets and linking molecular structures and toxic properties of an unknown compound, respectively.
Zhang et al. [344] developed different toxicity predictive models for drug-induced liver toxicity based on five ML algorithms combined with MACCS or FP4 fingerprinting. The results demonstrated that the best model yielded an accuracy rate of 75% against an external validation data set [344]. Similarly, several toxicity evaluation algorithms were constructed based on ML methods such as relevance vector machine (RVM), regularized-RF, C5.0 trees, eXtreme gradient boosting (XGBoost), AdaBoost, SVM boosting (SVMBoost), RVM Boosting (RVMBoost). The constructed models were used to evaluate rat oral acute toxicity, respiratory toxicity, and urinary tract toxicity [345,346,347,348]. In recent years, the execution of deep-learning algorithms has led to novel approaches for the molecular representation of chemical compounds, making DL methods suitable for predicting compound toxicity. Further, the potential for DL algorithms for toxicity prediction depends on the quality and quantity of data sets. In short, more research should be done to make AI-based algorithms reliable for toxicity prediction. However, the current ML-based predictors remain inappropriate to replace biological systems, but they are sufficient to extend the medicinal chemistry principles in the right direction, which reduces the number of synthesis cycles. Further, the detailed description of toxicity prediction AI-based algorithms and tools is discussed in Table 2.
Identification of molecular pathways and polypharmacology
One of the significant outcomes of AI and ML algorithms in drug discovery and development is the prediction and estimation of overall topology and dynamics of disease network or drug-drug interaction or drug-target relationships [349]. This methodology offers a vast avenue for the identification of novel molecular therapeutic targets for a particular disease. Text mining-driven databases like DisGeNET, STITCH, STRING are widely used to ascertain gene-disease associations, drug-target associations, and molecular pathways, respectively. For instance, Gu et al. 2020 used the similarity ensemble approach to identify targets for 197 most commonly used Chinese herbs. Later, the DisGeNET database was used to associate those drug targets with different diseases, thus linking herbs with diseases in which they can be used [350]. Further, chen et al. 2019 used the STITCH database to find targets of potential drugs shortlisted for esophageal carcinoma [351]. Likewise, Taha et al. 2020 used the STITCH database to find targets for active constituents of Nandina domestica, a plant used for treating various tumors. Later STRING database was used to construct compound-target pathways with the help of the cytoscape tool [352].
In medicinal chemistry, polypharmacology refers to designing a single drug molecule capable of interacting with multiple targets in a disease-related drug-target biological network. It is best suited for designing a promising therapeutic agent for more complex diseases such as cancer, neurodegenerative disease (NDDs), diabetes, heart failure, and many others [353,354,355]. ML-based methods have the potential to analyze guilt-by-association molecular networks due to strong mining capabilities and data analysis. Further, ML models assist in the rational design of multitarget ligand through the generation of chemical compounds with desired polypharmacological features as ML models generate a vast number of chemical structures with different chemical and topological features. Thus, the probability of discovering multi-target ligands increases. Furthermore, ML models help in the identification of multi-target ligands, where there are dissimilar binding pockets. Recent advancements in AI in drug discovery and development have led to the generation of web-based tools and stand-alone software packages for polypharmacology prediction such as polypharmacology browser (PPB) (http://www.gdb.unibe.ch/) [356], TarPred (http://www.dddc.ac.cn/tarpred/) [140], Self-Organizing Map Based Prediction of Drug Equivalence Relationship (SPiDER) (http://modlabcadd.ethz.ch/software/spider) [357], Targethunter (https://www.cbligand.org/TargetHunter3D/) [358], PharmMapper (http://lilab-ecust.cn/pharmmapper/) [359], ChemMapper (http://lilab.ecust.edu.cn/chemmapper/) [360], and Swiss Target Prediction (SwissTargetPrediction) (http://www.swisstargetprediction.ch/) [361]. Poirier et al. 2018 conducted an experiment using PPB for the identification of lysophosphatidic acid acyltransferase β as a therapeutic target of nanomolar angiogenesis, whereas Ozhathil et al. 2018 identified potent and selective small-molecule inhibitors of cation channel transient receptor potential cation channel subfamily M member 4 using PPB [362, 363]. Further, Vleet Van et al. 2018 implemented the TarPred tool for screening strategies and methods for improved off-target liability prediction, whereas, in the same year, Ratnawati et al. predicted the active compounds from SMILES codes using backpropagation algorithm [364, 365]. Among the above said web-based tools PharmMapper and ChemMapper were frequently used for current research. For example, synergistic mechanism of huangqi and huanglian for Diabetes Mellitus [366], investigation of blood enriching mechanism of danggui buxue decoction [367], and prediction of multiple mechanisms of Hedyotis diffusa Willd. On Colorectal Cancer [368], used PharmMapper. Similarly, identification of human copper trafficking blocker in cancer [369], identification of multi-target ligands through chemical-protein interaction in AD [370], prediction of the anticancer mechanism of Kushen Injection against Hepatocellular carcinoma [371], and discovery of Pteridin-7(8H)-one-Based as therapeutic compound against epidermal growth factor receptor kinase T790M/L858R mutant [372], were performed using ChemMapper. One major limitation of AI algorithms for polypharmacology prediction is inadequate data or reliability of the data set. Thus, quantum chemical calculations, which provide fine-tuned data set, should be done and, thus, which can increase the accuracy of a predictive model.
Moreover, AI in drug development opened the gates for identifying molecular pathways or molecular targets for the treatment of human disease through genomics information, biochemical features, and target specifications [373]. “OpenTargets” (https://www.opentargets.org/) [374], a freeware and ML-based tool, used for prioritizing potential therapeutic drug targets with over 71% accuracy. Recently, Nabirotchkin et al. identified the unfolded protein response and autophagy-related pathways of common approved drugs against COVID-19, whereas Lopez-Cortes et al. identified allele frequencies in colorectal cancer [375, 376]. Further, GWAS studies conducted by Isac-Lopez et al. [377] predicted the multiple risk loci and highlighted fibrotic and vasculopathy pathways. The results demonstrated that 27 independent genome-wide-associated signals and 13 novel risk loci were associated with systematic sclerosis. Martin et al. studied chromatin interactions to predict novel gene targets in rheumatic diseases. In the same study, the authors concluded that 454 high confidence genes were associated with rheumatic disease, in which 48 were drug targets, and 11 were existing targets. Finally, they demonstrated that 367 drugs were suitable for repositioning [378].
Implementation of artificial intelligence in de novo drug designing
The iterative process to design 3D structures of receptors to generate a novel molecule is termed as de novo drug designing, which is intended to produce new dynamics. However, de novo drug designing has not seen a boundless use in medication disclosure. Further, the field has seen some recovery recently because of advancements in the field of AI [421, 422]. VS has emerged as a massive tool in the drug improvement measure, as it conducts profitable in silico look in an enormous number of blends, further, extending yields of potential medicine leads. As a subset of AI, ML is a technique for coordinating VS for drug leads, which generally incorporates gathering a filtered set of compounds, containing known actives and inactive compounds to train a model [423, 424]. In the wake of setting up the model, it is tested and, if accurate enough, used on a previously unknown database, to identify novel drug. In this section, we discuss how AI has proved to be a boon for drug designing using the de novo technique.
In one study, the researchers utilized the indolent space portrayal to prepare a model dependent on the quantitative estimate of drug-likeness (QED) drug-similarity score and the manufactured availability score synthetic accessibility score (SAS) [425]. In another distribution, the presentation of such a variational autoencoder was contrasted with an antagonistic autoencoder [426]. The ill-disposed autoencoder comprises of a generative model delivering novel compound structures. A second discriminative antagonistic model is prepared to differentiate genuine particles from produced ones, while the generative model attempts to trick the discriminative one [427]. The antagonistic autoencoder created more substantial structures than the variational autoencoder in generation mode essentially. In mix with an in silico model, novel structures anticipated to be dynamic against the dopamine receptor type, 2 could be gotten. Researches utilized a generative ill-disposed organization (GAN) to propose mixes with putative anticancer properties [428].
RNN has likewise been effectively utilized for de novo drug design. Since SMILES strings encode substance structures in a grouping of letters, RNNs have been utilized to generate compound structures. It was observed that RNNs have the potential to utilize SMILES strings for drug designing [429]. A similar methodology was likewise effectively utilized for the development of novel peptide structures [430]. Neural network learning was effectively applied to inclination the created mixes toward wanted properties [431]. Similarly, transfer learning was utilized as another system to create novel synthetic structures with an ideal natural action. In the subsequent steps, the organization is prepared to get familiar with the SMILES syntax with a huge preparing set [432, 433]. In the subsequent advance, the preparation is proceeded with mixes having the ideal movement. Moreover, additional epochs of training were adequate to reach the stage of novel combinations into a compound space involved by dynamic atoms. Five atoms were combined in light of such a methodology, and the plan action could be affirmed for four particles against atomic, chemical receptors [434]. A few distinct designs have been proposed, which have created legitimate, important novel structures. The novel synthesis has been investigated by these strategies, with the property dissemination of the created molecules or atoms being similar to the extensive training set used. The primary application for this strategy was adequate, with 4 out of 5 atoms indicating the ideal action [435]. Optimization of AI and multi-objective has been a promising solution to bridge the chemical and biological phases. Novel pairs of multi-objectives based on RNN for the automated de novo design based on SMILES were developed to find the best possible match between physicochemical properties and their constrained biological targets. The results indicated that AI and multi-objective optimization allows capturing the latent links joining chemical and biological aspects, thus providing easy-to-use options for customizable design strategies, which proved especially effective for both lead generation and lead optimization [436].
ML models like SVM, RF, DNNs, and many others have been used for drug discovery for analyzing the pharmaceuticals applications from docking to VS [437]. Recently, drug repurposing has emerged as an innovative approach to minimize drug development duration that usually involves data mining and AI [438]. A group proposed a question–answer artificial system (QAAI) that had the capability to repurpose drugs that used Google semantic AI universal encoder to compute the sentence embedding in the red brain JSON database. The study validated prediction for the lipoxygenase inhibitor drug zileuton as a modulator of the NRF2 pathway in vitro, with potential applications to reduce macrophage M1 phenotype and reactive oxygen species production. This novel approach has been proved to effective for reposition in NDDs [439]. With the rapid development of systems-based pharmacology and polypharmacology, method development for the rational design of multi-target drugs has to become urgent. The first de novo multi-target drug configuration program known as LigBuilder V3 (http://www.pkumdl.cn/ligbuilder3/) has been devised to design ligands for different receptors, numerous coupling locales of one receptor, or different configurations of one receptor. LigBuilder V3 is again used for multi-target drug plans and enhancement, particularly for compact ligands for proteins with varying ligand binding sites [440]. De novo drug design actively seeks to use sets of chemical rules for the fast and efficient identification of structurally new chemotypes with the desired set of biological properties. Moreover, fragment-based de novo design tools have been successfully applied in the discovery of non-covalent inhibitors. Herein a new protocol, called Cov_FB3D, has been devised, which involves the in silico assembly of potential novel covalent inhibitors by identifying the active fragments in the covalently binding site of the target protein [441].
Artificial intelligence: possible role in pharmaceutical manufacturing and clinical trial design
The use of computational methods is quite well established in the pharmaceutical industries. However, the introduction of AI has given a broader scope to develop new approaches that can improve and optimize drug discovery [442]. This has not only encouraged the scientific community but has also resulted in the growing partnership between the pharmaceutical industry and AI companies [443]. A study stated that the overall success rate for 21,143 drugs was nearly 5.2% in 2013, which was less than 11.2% in 2005. Thus, the use of AI is mainly associated with a need to reduce attrition and costs [444]. It usually takes 12 years to bring a new drug to the market, which can cost up to 3 billion USD [445]. Further, it is a huge task to find a new drug when there are ~ 1060 existing drug-like molecules [446]. The current drug discovery challenges are related to the toxicity of the drug, its side effects, choosing the right target site, appropriate dosages, and even intellectual property [447]. The pharmaceutical industry mostly does not share pharmacokinetic and pharmacodynamic measurements of the drugs until they are approved. In addition to that, very less drug discovery data are available to train AI models [448]. There needs to be a community that can regulate and manage preclinical and clinical pharmacology data to accelerate the progress of AI in this field. Recent advances in AI have impacted clinical pharmacology in many ways like literature searching and processing, interactions with online predictive ML models, ML methods in framing policy to encourage healthcare in many countries and also to get predictive analysis for drug-related information [449, 450].
When a drug candidate successfully passes all preclinical tests, it is then administered to patients under clinical trials, which comprises of three phases: Phase 1, drug safety testing with a small number of people; Phase 2, drug efficacy testing with the small number of human subjects affected by a particular disease; Phase 3, efficacy studies with a large number of patients and after passing the clinical trials FDA reviews it for approval and commercialization [451, 452]. Further, the failure rate of clinical trials adds up to the drug development process's inefficiency, and each failed trial ruins the investment and impairs the costs of preclinical testing. The two main reasons behind high failure rates are improper patient selection and inefficient monitoring during trials. Furthermore, after the introduction of AI technology, the success rates of clinical trials have improved drastically [453]. A system for clinical trial matching has been developed by IBM Watson, which uses medical records of patients and an abundance of past clinical trial data to create detailed clinical findings profiles. It could also be used to keep a check on patients enrolled [454]. AI models can also reduce the cost of clinical trials by enhancing the success rate by analyzing toxicity, side effects, and other related parameters [455]. One such example, which predicted the outcome of phase I and phase II clinical trials, was based on DL and calculated the probability of possible side effects and pathway activation score, which was further used to train the model [456]. Similarly, another project named Visual Physiological Human was made to support in silico trials [457]. Further, development in AI technology will help in better management of clinical trial data, ultimately aiming to develop personalized medicines.
Involvement of artificial intelligence in drug development: a case of neurodegenerative diseases
NDDs are lethal, multifaceted, enervating disorders of the central nervous system and a major cause of death worldwide. AD, PD, Amyotrophic Lateral Sclerosis (ALS), and Huntington’s disease (HD) are some of the most commonly observed NDDs, which can ultimately lead to the death of the neurons in different areas of the central nervous system [458]. The aggregation of toxic, misfolded, cytoplasmic proteins in different brain regions is one of the primary reasons for the inception of these disorders [459]. Further, these disorders can exhibit varying symptoms like cognitive decline, slow movement, tremors, memory loss, depression, speaking problems, muscle stiffness [460, 461]. The major challenge posed by NDDs is in the area of drug discovery as to date, no drug has been discovered, which can arrest and revert the progression of this disorder. Hence, there is a dire need for new drug targets and drug compounds, which can alleviate the symptoms and mitigate the diseased conditions of the central nervous systems [462]. Nowadays, ML is extensively used to find novel targets and biomarkers associated with NDDs. For example, Martínez-Ballesteros et al. 2016 combined DT, quantitative association rules, and hierarchical clustering to determine potential risk genes with AD via gene expression profiling of patient and control samples. Further, [463] used a combination of protein–protein interaction networks, autoencoder, and SVM to predict novel target genes associated with PD. Likewise, [464] used ML models like RF, DT, generalized linear model, and rule induction to find out risk genes of HD through gene expression profiling. Moreover, [465] used a CNN trained on an extensive GWAS data set to find novel risk single nucleotide polymorphisms and genes associated with ALS.
Moreover, ML techniques are also being used to find suitable inhibitors of target proteins implicated in NDDs. For instance, [466] applied a combination of VS, ML, and molecular docking to find class 1 and class IIb histone deacetylase inhibitors, as HDAC enzymes have been reported to promote AD neurotoxicity. Here, ML was used for the classification of inhibitors and non-inhibitors post-VS. Further, [467] used descriptors derived from MD simulation trajectories of the caspase-8 protein–ligand complex to train ANN and RF models to find inhibitors of caspase 8 protease, a protease that has been implicated in AD pathogenesis. In another study, [468] used data from a traditional Chinese medicine database, followed by VS, molecular docking, and ML techniques, including DL, to find inhibitors of GSK3β, an enzyme implicated in AD. Further, MD simulation was used to assess the stability of GSK3β-ligand interactions. Additionally, Ponzoni et al. 2019 made a QSAR model for finding inhibitors of the BACE1 enzyme, which is responsible for β-amyloid (Aβ) aggregation in AD. Here, the QSAR model was built using an optimum set of molecular descriptors, which were sorted out using an amalgamation of ML algorithms, hybridization techniques, backward elimination strategy, and visual analysis [469]. Similarly, [470] used a cascade of Naïve Bayes networks to find potent and safe abelson tyrosine-protein kinase 1 (c-Abl) inhibitors, which promote neuroprotection in PD. Likewise, Shao et al. 2018 used integration of SVM algorithm and Tanimoto similarity-based clustering, followed by in vitro experiments, to find novel antagonists of both A2A adenosine receptor as well as Dopamine D2 receptor, as it has been observed that blocking these two receptors leads to neuroprotection in PD [471]. In addition, [472] implemented molecular docking, AI-QSAR, and MD simulations to find inhibitors of the NLR family pyrin domain containing 3 (NLRP3), an inflammasome involved in PD pathogenesis. Here, VS followed by docking was used to shortlist compounds from the traditional Chinese medicine database, whereas AI and QSAR models were used to ascertain bioactivity of the compounds, followed by assessing their binding stability via MD simulations [472]. Similarly, [473] used molecular docking, AI, and MD simulations to discover inhibitors of Galectin-3 a protein implicated for neuroinflammation in HD. Here, molecular docking was used for initial shortlisting, followed by evaluating the bioactivity of compounds through ML and assessing their binding stability through MD simulations. Further, different studies have used ML algorithms for drug repurposing in NDDs. Similarly, X. Zeng et al. 2019 developed a DL-based drug repurposing tool, called deepDR (https://github.com/ChengF-Lab/deepDR), which is used to find new repurposed drugs for AD and PD [291]. Furthermore, [474] proposed telmisartan as potential repurposed drug for AD by using a genetic network-driven classification model. In addition, [475] proposed a drug repurposing strategy for PD by scanning scientific literature through an integration of knowledge representation learning and ML algorithms .
Future challenges and possible solutions
At present, the major challenge for the pharmaceutical industry while developing a new drug is its increased costs and reduced efficiency. However, ML approaches and recent developments in DL come with great opportunities to reduce this cost, increase efficiency, and save time during the drug discovery and development process. Advances in AI algorithms, especially in DL approaches along with improving architectural hardware and easy accessibility of big data, are all indicating toward the third wave of AI. AI approaches in drug development have aroused great interest among researchers, such that many pharmaceutical companies have collaborated with AI companies. Moreover, the number of startups in this field has also escalated and reached 230 by June 2020 [476]. Further, DL approaches integrate data at multiple levels through nonlinear models, which is the shortcoming of the AI and ML approaches. However, integration of data at multiple levels makes DL algorithm advantageous as it provides great accuracy and precision. Moreover, in comparison with AI and ML algorithms, DL provides a much more flexible architecture to create a neural network for a specific problem [477,478,479,480]. Applications of AI like natural language processing, image, and voice recognition are easily doable these days, which has beaten humans in terms of performance [481]. So, it comes with no surprise that AI can very well be used in the drug discovery process. Today, AI is used in drug discovery for target identification, hit discovery, lead optimization, ADMET prediction, and structuring clinical trials. Despite great success, there are many remaining challenges like high-quality data acquisition under which there are two significant concerns. Firstly, labeling cannot be binary as the action of drugs in biological systems is complicated; secondly, the amount of data available in drug discovery is infinitesimal compared to the enormous amount of information available. Therefore, a community is required that not only provides quantity but the quality of data. In the pharmaceutical industry, open data sharing is not common, and Pistoia alliance has taken the initiative to start a movement that has encouraged many companies to share their data with others. They also intend to establish a uniform data format, which is technically challenging [161]. A possible solution to deal with this problem is to develop an algorithm that can handle sparse data; one such has been developed by Stanford University named “one-shot learning,” which predicts properties of a drug on the basis of heterogeneous data [482]. Moreover, the accuracy and uncertainty of the experimental data can be used for model building, that is instead of establishing new ML technologies, one can put efforts in training the existing one by tuning large number of hyperparameters and optimizing it for good results, although some studies indicated that some reasonable parameters can be used to start the optimization [435]. Molecular representation is also a challenge as it is one of the governing factors in model building. Few recently developed models learn task-related features from the raw data and refine the molecular representation to a standard. Earlier, drug repurposing used to rely only on clinical observations. However, the current large amount of data comprising of scientific literature, patents, and clinical trial results can collectively be used to improve the screening process. Additionally, DL-based VS can make full use of the data and reduce false-positive rates obtained due to imbalance in positive and negative data. Lead optimization is also a challenge in order to develop an efficient drug with good ADMET properties and target activities; however, these parameters are independent and at times mutually incompatible with each other. This problem can be solved by optimizing each parameter separately and further improving the model. Pharmaceutical companies’ faces trouble recruiting sufficient number of patients for clinical trials. AI approaches will help identify and recruit target patients and will also help in managing the collected data. Regarding drug discovery for neurodegenerative disorders, the major problem is their unknown pathophysiology which makes drug identification even more challenging. The “black box” nature of ML models is an additional challenge where even experts cannot explain that how the model arrives at a result and comprehend the biological mechanism behind it. Furthermore, the escalating numbers of ML models and their claim to be latest have left non-professional helpless as they cannot decide which model to choose to solve their problem. Thus, it will be better if users and developers agree upon standard objective evaluation and thereafter check the performance of the model. Further, it is important to note that most of the countries do not give patents to those inventions which are exclusively created by AI technology. Moreover, companies who use AI technology for drug discovery has to go through vigorous process to copyright their work so as to secure patent rights. Security is also a major concern, as AI-driven personalized medicine requires person’s genetic code for which personal information will be required. Finally, faster computation will be required for handling big data and it is said that in future the current supercomputers will be replaced by quantum computers or another technology which will do the job in minutes rather than taking hours. Although AI has given many novel targets and novel compounds for different diseases, still there has not been any success story where a compound generated through AI made it to the market for public use. Recently, for the first time ever, a novel target and its novel inhibitor has been proposed through AI-based tools. In silico medicine, a biotechnology company, proposed a novel target involved in idiopathic pulmonary fibrosis and made its novel inhibitor from scratch, through their AI-based tools. The identified small molecule inhibitor has showed good efficacy in human cells and animal models. In December 2020, in silico nominated their small molecule inhibitor for investigational new drug (IND) enabling studies and they are targeting clinical trials by early 2022. If the trials are successful, then it will be, for the first time ever, where a novel target and its inhibitor was proposed through AI-based tools and got approved. Though there are some unavoidable obstacles and tremendous amount of work has to be done to incorporate AI tools in drug discovery cycle, there is no doubt that in the near future AI will bring revolutionary changes in drug discovery and development process.
Abbreviations
- VS:
-
Virtual screening
- AI:
-
Artificial intelligence
- DL:
-
Deep learning
- ML:
-
Machine learning
- ANN:
-
Artificial neural network
- SVM:
-
Support vector machine
- LSTM:
-
Long short-term memory
- GPU:
-
Graphic processing unit
- GAN:
-
Generative adversarial networks
- HTS:
-
High-throughput sequencing
- GEO:
-
Gene Expression Omnibus
- TCGA:
-
The Cancer Genome Atlas
- GWAS:
-
Genome-wide association studies
- NCIGDC:
-
National Cancer Institute Genomic Data Commons
- LINCS:
-
Library of integrated network-based cellular signature
- PDB:
-
Protein data bank
- CADD:
-
Computer-aided drug design
- QSAR:
-
Quantitative structure–activity relationship
- PLS:
-
Partial least square
- HSVR:
-
Hierarchical SVM
- SARS:
-
Severe acute respiratory syndrome
- ADME:
-
Absorption, distribution, metabolism, and excretion
- MD:
-
Molecular dynamics
- FDA:
-
Food and drug administration
- PCA:
-
Principal component analysis
- LS-SVM:
-
Least-square SVM
- IACS:
-
Image-activated cell sorting
- SMILES:
-
Simplified molecular input line-entry system
- MMP:
-
Matched molecular pair
- RF:
-
Random forest
- GBM:
-
Gradient boosting machines
- AMPs:
-
Anti-microbial peptides
- GENTRL:
-
Generative tensorial reinforcement learning
- DEL:
-
DNA-encoded small molecule libraries
- PRS:
-
Parabolic response curve
- MIMIC II:
-
Multiparameter intelligent monitoring in intensive care II database
- KronRLS:
-
Kronecker-regularized least squares
- DTBA:
-
Drug target binding affinity
- PADME:
-
Protein and drug molecule interaction prediction
- PPIs:
-
Protein–protein interactions
- PPNIs:
-
Protein–protein non-interactions
- DT:
-
Decision tree
- EELM:
-
Ensemble extreme learning machine
- PBDs:
-
Proteins binding domains
- HSMM:
-
Hierarchical statistical mechanical modeling
- SBVS:
-
Structure-based VS
- LBVS:
-
Ligand-based VS
- Keap-Nrf2:
-
Kelch-like ECH-associated protein-nuclear factor erythroid 2-related factor 2
- GSK-3β:
-
Glycogen synthase kinase 3 beta
- MPTP:
-
Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MAO-B:
-
Monoamine oxidase B
- CNN:
-
Convolutional neural network
- AD:
-
Alzheimer’s disease
- EPA:
-
Environmental protection agency
- SEA:
-
Similarity ensemble approach
- RNN:
-
Recursive neural network
- PD:
-
Parkinson's disease
- RASAR:
-
Read-across structure–activity relationships
- RVM:
-
Relevance vector machine
- PPB:
-
Polypharmacology Browser
- SPiDER:
-
Self-Organizing Map-Based Prediction of Drug Equivalence Relationship
- QED:
-
Quantitative estimate of drug-likeness
- SAS:
-
Synthetic accessibility score
- GAN:
-
Generative ill-disposed organization
- QAAI:
-
Question–answer artificial system
- NDDs:
-
Neurodegenerative diseases
- ALS:
-
Amyotrophic Lateral Sclerosis
- HD:
-
Huntington’s disease
- BACE1:
-
Beta-secretase 1
- Aβ:
-
β-Amyloid
- NLRP3:
-
NLR family pyrin domain containing 3
- ADRs:
-
Adverse drug reactions
- HMM:
-
Hidden Markov models
- GO:
-
Gene ontology
References
Lipinski CF, Maltarollo VG, Oliveira PR et al (2019) Advances and perspectives in applying deep learning for drug design and discovery. Front Robot AI. https://doi.org/10.3389/frobt.2019.00108
Hamet P, Tremblay J (2017) Artificial intelligence in medicine. Metabolism. https://doi.org/10.1016/j.metabol.2017.01.011
Hassanzadeh P, Atyabi F, Dinarvand R (2019) The significance of artificial intelligence in drug delivery system design. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2019.05.001
Duch W, Swaminathan K, Meller J (2007) Artificial intelligence approaches for rational drug design and discovery. Curr Pharm Des. https://doi.org/10.2174/138161207780765954
Zhang L, Tan J, Han D, Zhu H (2017) From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov Today. https://doi.org/10.1016/j.drudis.2017.08.010
Jordan AM (2018) Artificial intelligence in drug design–the storm before the calm? ACS Med Chem Lett. https://doi.org/10.1021/acsmedchemlett.8b00500
Goel AK, Davies J (2019) Artificial intelligence. In: The Cambridge Handbook of Intelligence. Cambridge
Harrer S, Shah P, Antony B, Hu J (2019) Artificial Intelligence for Clinical Trial Design. Sci, Trends Pharmacol. https://doi.org/10.1016/j.tips.2019.05.005
Zhong F, Xing J, Li X et al (2018) Artificial intelligence in drug design. Sci China Life Sci. https://doi.org/10.1007/s11427-018-9342-2
Brown N, Ertl P, Lewis R et al (2020) Artificial intelligence in chemistry and drug design. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00317-x
Badillo S, Banfai B, Birzele F et al (2020) An introduction to machine learning. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.1796
Dutta Majumdar D (1985) Trends in pattern recognition and machine learning. Def Sci J. https://doi.org/10.14429/dsj.35.6027
Kubat M (2017) An Introduction to Machine Learning
Aggarwal M, Murty MN (2021) Deep Learning. In: SpringerBriefs in Applied Sciences and Technology. https://doi.org/10.1007/978-981-33-4022-0_3
Schmidhuber J (2015) Deep learning in neural networks: an overview. Neural Netw. https://doi.org/10.1016/j.neunet.2014.09.003
Hu YH, Hwang JN (2001) Introduction to neural networks for signal processing. In: Handbook of Neural Network Signal Processing. CRC Press, pp 12–41
Angermueller C, Pärnamaa T, Parts L, Stegle O (2016) Deep learning for computational biology. Mol Syst Biol. https://doi.org/10.15252/msb.20156651
McCulloch WS, Pitts W (1943) A logical calculus of the ideas immanent in nervous activity. Bull Math Biophys 5:115–133. https://doi.org/10.1007/BF02478259
Turing AM (2009) Computing machinery and intelligence. Parsing the Turing Test: Philosophical and Methodological Issues in the Quest for the Thinking Computer. Springer, Netherlands, pp 23–65
Samuel AL (1959) Some studies in machine learning using the game of checkers. IBM J Res Dev 3:210–229. https://doi.org/10.1147/rd.33.0210
Rosenblatt F (1957) The Perceptron: A Perceiving and Recognizing Automaton, Report 85–60–1
KELLEY HJ, (1960) Gradient theory of optimal flight paths. ARS J 30:947–954. https://doi.org/10.2514/8.5282
Dreyfus S (1962) The numerical solution of variational problems. J Math Anal Appl 5:30–45. https://doi.org/10.1016/0022-247X(62)90004-5
Fukushima K (1980) Neocognitron: a self-organizing neural network model for a mechanism of pattern recognition unaffected by shift in position. Biol Cybern 36:193–202. https://doi.org/10.1007/BF00344251
Fukushima K (1988) Neocognitron: a hierarchical neural network capable of visual pattern recognition. Neural Netw 1(2):119–130. https://doi.org/10.1016/0893-6080(88)90014-7
Rumelhart DE, Hinton GE, Williams RJ (1986) Learning representations by back-propagating errors. Nature 323:533–536. https://doi.org/10.1038/323533a0
LeCun Y, Boser B, Denker JS et al (1989) Backpropagation applied to handwritten zip code recognition. Neural Comput 1:541–551. https://doi.org/10.1162/neco.1989.1.4.541
Watkins CJCH, Dayan P (1992) Q-learning. Mach Learn 8:279–292. https://doi.org/10.1007/bf00992698
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20:273–297. https://doi.org/10.1023/A:1022627411411
Hochreiter S, Schmidhuber J (1997) Long short-term memory. Neural Comput 9:1735–1780. https://doi.org/10.1162/neco.1997.9.8.1735
Ilievski A, Zdraveski V, Gusev M (2018) How CUDA Powers the machine learning revolution. 2018 26th Telecommun Forum, TELFOR 2018 - Proc 420–425. https://doi.org/https://doi.org/10.1109/TELFOR.2018.8611982
Deng J, Dong W, Socher R et al (2010) ImageNet: a large-scale hierarchical image database. Inst Electric Electron Eng IEEE. https://doi.org/10.1109/CVPR.2009.5206848
Krizhevsky A, Sutskever I, Hinton GE (2012) ImageNet Classification with Deep Convolutional Neural Networks. In Proceedings of the 25th International Conference on Neural Information Processing Systems - Volume 1
Le Q V, Ranzato M’ A, Monga R, et al (2012) Building High-level Features Using Large Scale Unsupervised Learning. https://arxiv.org/abs/1112.6209v5
Jorda M, Valero-Lara P, Pena AJ (2019) Performance evaluation of cuDNN convolution algorithms on NVIDIA volta GPUs. IEEE Access 7:70461–70473. https://doi.org/10.1109/ACCESS.2019.2918851
Taigman Y, Yang M, Ranzato M, Wolf L (2014) DeepFace: Closing the gap to human-level performance in face verification. In: Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition. IEEE Computer Society, pp 1701–1708
Goodfellow I, Pouget-Abadie J, Mirza M et al (2020) Generative Adversarial Networks. Commun ACM. https://doi.org/10.1145/3422622
Gandomi A, Haider M (2015) Beyond the hype: Big data concepts, methods, and analytics. Int J Inf Manage 35:137–144. https://doi.org/10.1016/j.ijinfomgt.2014.10.007
Brazma A, Kapushesky M, Parkinson H et al (2006) [20] Data Storage and Analysis in ArrayExpress. Methods Enzymol 411:370–86. https://doi.org/10.1016/S0076-6879(06)11020-4
Lo Y-C, Ren G, Honda H, L. Davis K (2020) Artificial Intelligence-Based Drug Design and Discovery. In: Cheminformatics and its Applications: https://doi.org/10.5772/intechopen.89012
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. https://doi.org/10.1093/nar/30.1.207
Wang Z, Jensen MA, Zenklusen JC (2016) A practical guide to The Cancer Genome Atlas (TCGA). In: Methods in Molecular Biology 1418:111–41: https://doi.org/10.1007/978-1-4939-3578-9_6
Parkinson H, Kapushesky M, Shojatalab M et al (2007) ArrayExpress-a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. https://doi.org/10.1093/nar/gkl995
van IJzendoorn DGP, Szuhai K, Briaire-De Bruijn IH, et al (2019) Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Comput Biol 15:1–19. https://doi.org/10.1371/journal.pcbi.1006826
Lau A, So HC (2020) Turning genome-wide association study findings into opportunities for drug repositioning. Comput Struct Biotechnol J 18:1639–1650. https://doi.org/10.1016/j.csbj.2020.06.015
Beck T, Hastings RK, Gollapudi S et al (2014) GWAS Central: a comprehensive resource for the comparison and interrogation of genome-wide association studies. Eur J Hum Genet. https://doi.org/10.1038/ejhg.2013.274
Buniello A, Macarthur JAL, Cerezo M et al (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1120
Li J, Yuan X, March ME et al (2019) Identification of target genes at juvenile idiopathic arthritis GWAS loci in human neutrophils. Front Genet. https://doi.org/10.3389/fgene.2019.00181
Leinonen R, Sugawara H, Shumway M (2011) The sequence read archive. Nucleic Acids Res. https://doi.org/10.1093/nar/gkq1019
Jensen MA, Ferretti V, Grossman RL, Staudt LM (2017) The NCI genomic data commons as an engine for precision medicine. Blood 130(4):453–459. https://doi.org/10.1182/blood-2017-03-735654
Han Y, Yang J, Qian X et al (2019) DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz096
Guillaume JC (1998) PubMed. Ann Dermatol Venereol. https://doi.org/10.1002/9783527678679.dg10319
Canese K, Weis S (2013) PubMed: The bibliographic database. NCBI Handb
Kim S, Chen J, Cheng T et al (2021) PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa971
Kim S, Chen J, Cheng T et al (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1033
Mendez D, Gaulton A, Bento AP et al (2019) ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1075
Bento AP, Gaulton A, Hersey A et al (2014) The ChEMBL bioactivity database: an update. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1031
Wishart DS, Knox C, Guo AC et al (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm958
Wishart DS, Feunang YD, Guo AC et al (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1037
Keenan AB, Jenkins SL, Jagodnik KM et al (2018) The library of integrated network-based cellular signatures NIH program: system-level cataloging of human cells response to perturbations. Cell Syst 6(1):13–24. https://doi.org/10.1016/j.cels.2017.11.001
Duan Q, Reid SP, Clark NR et al (2016) L1000CDS2: LINCS L1000 characteristic direction signatures search engine. npj Syst Biol Appl 2:1–12. https://doi.org/10.1038/npjsba.2016.15
Rose PW, Prlić A, Altunkaya A et al (2017) The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw1000
Burley SK, Berman HM, Bhikadiya C et al (2019) RCSB Protein data bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1004
Xu Z, Yang L, Zhang X et al (2020) Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front Mol Biosci 7:1–8. https://doi.org/10.3389/fmolb.2020.556481
Fan Y, Zhang Y, Hua Y et al (2019) Investigation of machine intelligence in compound cell activity classification. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.9b00558
Chi CT, Lee MH, Weng CF, Leong MK (2019) In silico prediction of PAMPA effective permeability using a two-QSAR approach. Int J Mol Sci. https://doi.org/10.3390/ijms20133170
He S, Zhang X, Lu S et al (2019) A computational toxicology approach to screen the hepatotoxic ingredients in traditional chinese medicines: polygonum multiflorum thunb as a case study. Biomolecules. https://doi.org/10.3390/biom9100577
He S, Zhang C, Zhou P et al (2019) Herb-induced liver injury: Phylogenetic relationship, structure-toxicity relationship, and herb-ingredient network analysis. Int. J Mol Sci. 20(15):3633. https://doi.org/10.3390/ijms20153633
Zhang D, hai, Wu K lun, Zhang X, et al (2020) In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J Integr Med. https://doi.org/10.1016/j.joim.2020.02.005
Baldi A (2010) Computational approaches for drug design and discovery: an overview. Syst Rev Pharm 1(1):99. https://doi.org/10.4103/0975-8453.59519
Lavecchia A, Cerchia C (2016) In silico methods to address polypharmacology: current status, applications and future perspectives. Drug Discov Today 21(2):288–298. https://doi.org/10.1016/j.drudis.2015.12.007
Smith JS, Roitberg AE, Isayev O (2018) Transforming computational drug discovery with machine learning and AI. ACS Med Chem Lett 9(11):1065–1069. https://doi.org/10.1021/acsmedchemlett.8b00437
Jing Y, Bian Y, Hu Z et al (2018) Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. AAPS J 20(3):58. https://doi.org/10.1208/s12248-018-0210-0
Powles J, Hodson H (2017) Google deepmind and healthcare in an age of algorithms. Health Technol (Berl). https://doi.org/10.1007/s12553-017-0179-1
Senior AW, Evans R, Jumper J et al (2020) Improved protein structure prediction using potentials from deep learning. Nature 577:706–710. https://doi.org/10.1038/s41586-019-1923-7
AlQuraishi M (2019) End-to-End differentiable learning of protein structure. Cell Syst 8:292-301.e3. https://doi.org/10.1016/j.cels.2019.03.006
Kalaiarasi C, Manjula S, Kumaradhas P (2019) Combined quantum mechanics/molecular mechanics (QM/MM) methods to understand the charge density distribution of estrogens in the active site of estrogen receptors. RSC Adv. https://doi.org/10.1039/c9ra08607b
Schütt KT, Gastegger M, Tkatchenko A et al (2019) Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat Commun. https://doi.org/10.1038/s41467-019-12875-2
Gastegger M, McSloy A, Luya M et al (2020) A deep neural network for molecular wave functions in quasi-atomic minimal basis representation. J Chem Phys DOI. https://doi.org/10.1063/5.0012911
De Vivo M, Masetti M, Bottegoni G, Cavalli A (2016) Role of molecular dynamics and related methods in drug discovery. J Med Chem 59(9):4035–4061. https://doi.org/10.1021/acs.jmedchem.5b01684
Bennett WFD, He S, Bilodeau CL et al (2020) Predicting small molecule transfer free energies by combining molecular dynamics simulations and deep learning. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00318
Bai Q, Tan S, Xu T et al (2020) MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 00:1–12. https://doi.org/10.1093/bib/bbaa161
Sterling T, Irwin JJ (2015) ZINC 15-ligand discovery for everyone. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.5b00559
Popova M, Isayev O, Tropsha A (2018) Deep reinforcement learning for de novo drug design. Sci Adv 4:1–15. https://doi.org/10.1126/sciadv.aap7885
Grzybowski BA, Szymkuć S, Gajewska EP et al (2018) Chematica: a story of computer code that started to think like a chemist. Chem 4:390–398. https://doi.org/10.1016/j.chempr.2018.02.024
Genheden S, Thakkar A, Chadimová V et al (2020) AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J Cheminform 12:1–9. https://doi.org/10.1186/s13321-020-00472-1
Segler MHS, Preuss M, Waller MP (2018) Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555:604–610. https://doi.org/10.1038/nature25978
Bøgevig A, Federsel HJ, Huerta F et al (2015) Route design in the 21st century: the IC SYNTH software tool as an idea generator for synthesis prediction. Org Process Res Dev 19:357–368. https://doi.org/10.1021/op500373e
Jang G, Lee T, Hwang S et al (2018) PISTON: predicting drug indications and side effects using topic modeling and natural language processing. J Biomed Inform 87:96–107. https://doi.org/10.1016/j.jbi.2018.09.015
Piñero J, Bravo Á, Queralt-Rosinach N et al (2017) DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw943
Szklarczyk D, Gable AL, Lyon D et al (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1131
Szklarczyk D, Santos A, Von Mering C et al (2016) STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res 44:D380–D384. https://doi.org/10.1093/nar/gkv1277
Davenport TH, Ronanki R (2018) Artificial intelligence for the real world. Harv Bus Rev
Zhavoronkov A, Vanhaelen Q, Oprea TI (2020) Will Artificial Intelligence for Drug Discovery Impact Clinical Pharmacology? Clin Pharmacol Ther. https://doi.org/10.1002/cpt.1795
Watson O, Cortes-Ciriano I, Taylor A, Watson JA (2018) A decision theoretic approach to model evaluation in computational drug discovery. arXiv. https://arxiv.org/abs/1807.08926
Tripathy RK, Mahanta S, Paul S (2014) Artificial intelligence-based classification of breast cancer using cellular images. RSC Adv 4:9349–9355. https://doi.org/10.1039/c3ra47489e
Samui P, Kothari DP (2011) Utilization of a least square support vector machine (LSSVM) for slope stability analysis. Sci Iran 18:53–58. https://doi.org/10.1016/j.scient.2011.03.007
Chan HCS, Shan H, Dahoun T et al (2019) Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci 40:592–604. https://doi.org/10.1016/j.tips.2019.06.004
Ho CWL, Soon D, Caals K, Kapur J (2019) Governance of automated image analysis and artificial intelligence analytics in healthcare. Clin Radiol 74:329–337. https://doi.org/10.1016/j.crad.2019.02.005
Andrysek T (2003) Impact of physical properties of formulations on bioavailability of active substance: Current and novel drugs with cyclosporine. In: Molecular Immunology; 39(17–18):1061–5. https://doi.org/10.1016/s0161-5890(03)00077-4.
Elton DC, Boukouvalas Z, Butrico MS et al (2018) Applying machine learning techniques to predict the properties of energetic materials. Sci Rep 8:9059. https://doi.org/10.1038/s41598-018-27344-x
Tyrchan C, Evertsson E (2017) Matched molecular pair analysis in short: algorithms, applications and limitations. Comput Struct Biotechnol J 15:86–90. https://doi.org/10.1016/j.csbj.2016.12.003
Turk S, Merget B, Rippmann F, Fulle S (2017) Coupling matched molecular pairs with machine learning for virtual compound optimization. J Chem Inf Model 57:3079–3085. https://doi.org/10.1021/acs.jcim.7b00298
Carpenter KA, Huang X (2018) Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: a review. Curr Pharm Des 24:3347–3358. https://doi.org/10.2174/1381612824666180607124038
Schyman P, Liu R, Desai V, Wallqvist A (2017) vNN web server for ADMET predictions. Front Pharmacol 8:889. https://doi.org/10.3389/fphar.2017.00889
Álvarez-Machancoses Ó, Fernández-Martínez JL (2019) Using artificial intelligence methods to speed up drug discovery. Expert Opin Drug Discov 14(8):769–777. https://doi.org/10.1080/17460441.2019.1621284
Fleming N (2018) How artificial intelligence is changing drug discovery. Nature. https://doi.org/10.1038/d41586-018-05267-x
Segler MHS, Kogej T, Tyrchan C, Waller MP (2018) Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci. https://doi.org/10.1021/acscentsci.7b00512
Bruno BJ, Miller GD, Lim CS (2013) Basics and recent advances in peptide and protein drug delivery. Ther. Deliv 4(11):1443–67. https://doi.org/10.4155/tde.13.104
Yan J, Bhadra P, Li A et al (2020) Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning. Mol Ther-Nucleic Acids 20:882–894. https://doi.org/10.1016/j.omtn.2020.05.006
Plisson F, Ramírez-Sánchez O, Martínez-Hernández C (2020) Machine learning-guided discovery and design of non-hemolytic peptides. Sci Rep 10:1–19. https://doi.org/10.1038/s41598-020-73644-6
Kavousi K, Bagheri M, Behrouzi S et al (2020) IAMPE: NMR-assisted computational prediction of antimicrobial peptides. J Chem Inf Model 60:4691–4701. https://doi.org/10.1021/acs.jcim.0c00841
Yi HC, You ZH, Zhou X et al (2019) ACP-DL: a deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Mol Ther-Nucleic Acids 17:1–9. https://doi.org/10.1016/j.omtn.2019.04.025
Yu L, Jing R, Liu F et al (2020) DeepACP: a novel computational approach for accurate identification of anticancer peptides by deep learning algorithm. Mol Ther-Nucleic Acids 22:862–870. https://doi.org/10.1016/j.omtn.2020.10.005
Tyagi A, Kapoor P, Kumar R et al (2013) In silico models for designing and discovering novel anticancer peptides. Sci Rep 3:1–8. https://doi.org/10.1038/srep02984
Rao B, Zhang L, Zhang G (2020) ACP-GCN: the identification of anticancer peptides based on graph convolution networks. IEEE Access 8:176005–176011. https://doi.org/10.1109/access.2020.3023800
Wu C, Gao R, Zhang Y, De Marinis Y (2019) PTPD: predicting therapeutic peptides by deep learning and word2vec. BMC Bioinformatics 20:1–8. https://doi.org/10.1186/s12859-019-3006-z
Zhavoronkov A, Ivanenkov YA, Aliper A et al (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 37:1038–1040. https://doi.org/10.1038/s41587-019-0224-x
McCloskey K, Sigel EA, Kearnes S et al (2020) Machine learning on DNA-encoded libraries: a new paradigm for hit finding. J Med Chem 63:8857–8866. https://doi.org/10.1021/acs.jmedchem.0c00452
Xing G, Liang L, Deng C et al (2020) Activity prediction of small molecule inhibitors for antirheumatoid arthritis targets based on artificial intelligence. ACS Comb Sci. https://doi.org/10.1021/acscombsci.0c00169
Dimmitt S, Stampfer H, Martin JH (2017) When less is more–efficacy with less toxicity at the ED50. Br J Clin Pharmacol 83(7):1365–1368. https://doi.org/10.1111/bcp.13281
Shen Y, Liu T, Chen J et al (2020) Harnessing artificial intelligence to optimize long-term maintenance dosing for antiretroviral-naive adults with HIV-1 Infection. Adv Ther 3:1900114. https://doi.org/10.1002/adtp.201900114
Pantuck AJ, Lee D-K, Kee T et al (2018) Modulating BET bromodomain inhibitor ZEN-3694 and Enzalutamide combination dosing in a metastatic prostate cancer patient using CURATE.AI an artificial intelligence platform. Adv Ther. https://doi.org/10.1002/adtp.201800104
Julkunen H, Cichonska A, Gautam P et al (2020) Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nat Commun. https://doi.org/10.1038/s41467-020-19950-z
Sharabiani A, Bress A, Douzali E, Darabi H (2015) Revisiting warfarin dosing using machine learning techniques. Comput Math Methods Med. https://doi.org/10.1155/2015/560108
Nemati S, Ghassemi MM, Clifford GD (2016) Optimal medication dosing from suboptimal clinical examples: a deep reinforcement learning approach. Proc Annu Int Conf IEEE Eng Med Biol Soc EMBS. https://doi.org/10.1109/EMBC.2016.7591355
Tang J, Liu R, Zhang YL et al (2017) Application of machine-learning models to predict tacrolimus stable dose in renal transplant recipients. Sci Rep. https://doi.org/10.1038/srep42192
Hu YH, Tai CT, Tsai CF, Huang MW (2018) Improvement of adequate digoxin dosage: an application of machine learning approach. J Healthc Eng. https://doi.org/10.1155/2018/3948245
Imai S, Takekuma Y, Miyai T, Sugawara M (2020) A new algorithm optimized for initial dose settings of vancomycin using machine learning. Biol Pharm Bull 43:188–193. https://doi.org/10.1248/bpb.b19-00729
Rollinger JM, Stuppner H, Langer T (2008) Virtual screening for the discovery of bioactive natural products. Prog Drug Res 65:212–249. https://doi.org/10.1007/978-3-7643-8117-2_6
Schuster D, Maurer EM, Laggner C et al (2006) The discovery of new 11β-hydroxysteroid dehydrogenase type 1 inhibitors by common feature pharmacophore modeling and virtual screening. J Med Chem 49:3454–3466. https://doi.org/10.1021/jm0600794
Wu J, Zhang Q, Wu W et al (2018) WDL-RF: predicting bioactivities of ligand molecules acting with G protein-coupled receptors by combining weighted deep learning and random forest. Bioinformatics 34:2271–2282. https://doi.org/10.1093/bioinformatics/bty070
Cichonska A, Pahikkala T, Szedmak S et al (2018) Learning with multiple pairwise kernels for drug bioactivity prediction. Bioinformatics 34:i509–i518. https://doi.org/10.1093/bioinformatics/bty277
Babajide Mustapha I, Saeed F (2016) Bioactive molecule prediction using extreme gradient boosting. Molecules 21:1–11. https://doi.org/10.3390/molecules21080983
Merget B, Turk S, Eid S et al (2017) Profiling prediction of kinase inhibitors: toward the virtual assay. J Med Chem 60:474–485. https://doi.org/10.1021/acs.jmedchem.6b01611
Arshadi AK, Salem M, Collins J et al (2020) Deepmalaria: artificial intelligence driven discovery of potent antiplasmodials. Front Pharmacol. https://doi.org/10.3389/fphar.2019.01526
Sugaya N (2014) Ligand efficiency-based support vector regression models for predicting bioactivities of ligands to drug target proteins. J Chem Inf Model 54:2751–2763. https://doi.org/10.1021/ci5003262
Afolabi LT, Saeed F, Hashim H, Petinrin OO (2018) Ensemble learning method for the prediction of new bioactive molecules. PLoS ONE 13:1–14. https://doi.org/10.1371/journal.pone.0189538
Petinrin OO, Saeed F (2018) Bioactive molecule prediction using majority voting-based ensemble method. J Intell Fuzzy Syst 35:383–392. https://doi.org/10.3233/JIFS-169596
Liu X, Gao Y, Peng J et al (2015) TarPred: a web application for predicting therapeutic and side effect targets of chemical compounds. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv099
Liu M, Wu Y, Chen Y et al (2012) Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J Am Med Informatics Assoc 19:28–35. https://doi.org/10.1136/amiajnl-2011-000699
Jamal S, Goyal S, Shanker A, Grover A (2017) Predicting neurological adverse drug reactions based on biological, chemical and phenotypic properties of drugs using machine learning models. Sci Rep 7:1–12. https://doi.org/10.1038/s41598-017-00908-z
Xue R, Liao J, Shao X et al (2020) Prediction of adverse drug reactions by combining biomedical tripartite network and graph representation model. Chem Res Toxicol 33:202–210. https://doi.org/10.1021/acs.chemrestox.9b00238
Raja K, Patrick M, Elder JT, Tsoi LC (2017) Machine learning workflow to enhance predictions of adverse drug reactions (ADRs) through drug-gene interactions: application to drugs for cutaneous diseases. Sci Rep 7:1–11. https://doi.org/10.1038/s41598-017-03914-3
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. https://doi.org/10.1038/srep42717
Rost B, Liu J, Nair R et al (2003) Automatic prediction of protein function. Cell Mol Life Sci 60:2637–2650. https://doi.org/10.1007/s00018-003-3114-8
Browne F, Zheng H, Wang H, Azuaje F (2010) From experimental approaches to computational techniques: a review on the prediction of protein-protein interactions. Adv Artif Intell. https://doi.org/10.1155/2010/924529
Hale WH (1913) American association for the advancement of science. Sci Am 75:34–34. https://doi.org/10.1038/scientificamerican01181913-34supp
Troyanskaya OG, Dolinski K, Owen AB et al (2003) A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci U S A 100:8348–8353. https://doi.org/10.1073/pnas.0832373100
You ZH, Lei YK, Zhu L et al (2013) Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinformatics 14:1–11. https://doi.org/10.1186/1471-2105-14-S8-S10
Du X, Sun S, Hu C et al (2017) DeepPPI: boosting prediction of protein-protein interactions with deep neural networks. J Chem Inf Model 57:1499–1510. https://doi.org/10.1021/acs.jcim.7b00028
Cunningham JM, Koytiger G, Sorger PK, AlQuraishi M (2020) Biophysical prediction of protein–peptide interactions and signaling networks using machine learning. Nat Methods 17:175–183. https://doi.org/10.1038/s41592-019-0687-1
Chatterjee P, Basu S, Kundu M et al (2011) PPI_SVM: prediction of protein-protein interactions using machine learning, domain-domain affinities and frequency tables. Cell Mol Biol Lett 16:264–278. https://doi.org/10.2478/s11658-011-0008-x
Lu L, Lu H, Skolnick J (2002) Multiprospector: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins Struct Funct Genet 49:350–364. https://doi.org/10.1002/prot.10222
Singh R, Park D, Xu J et al (2010) Struct2Net: a web service to predict protein-protein interactions using a structure-based approach. Nucleic Acids Res 38:508–515. https://doi.org/10.1093/nar/gkq481
Dandekar T, Snel B, Huynen M, Bork P (1998) Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci 23:324–328. https://doi.org/10.1016/S0968-0004(98)01274-2
Keskin O, Tuncbag N, Gursoy A (2016) Predicting protein-protein interactions from the molecular to the proteome level. Chem Rev 116:4884–4909. https://doi.org/10.1021/acs.chemrev.5b00683
Lavecchia A, Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. https://doi.org/10.2174/09298673113209990001
Gonczarek A, Tomczak JM, Zaręba S et al (2018) Interaction prediction in structure-based virtual screening using deep learning. Comput Biol Med. https://doi.org/10.1016/j.compbiomed.2017.09.007
Goh GB, Hodas NO, Vishnu A (2017) Deep learning for computational chemistry. J Comput Chem 38(16):1291–1307. https://doi.org/10.1002/jcc.24764
Yang X, Wang Y, Byrne R et al (2019) Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev 119(18):10520–10594. https://doi.org/10.1021/acs.chemrev.8b00728
Arciniega M, Lange OF (2014) Improvement of virtual screening results by docking data feature analysis. J Chem Inf Model. https://doi.org/10.1021/ci500028u
Feinstein WP, Brylinski M (2015) Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J Cheminform. https://doi.org/10.1186/s13321-015-0067-5
Gazgalis D, Zaka M, Zaka M et al (2020) Protein binding pocket optimization for virtual high-throughput screening (vHTS) drug discovery. ACS Omega. https://doi.org/10.1021/acsomega.0c00522
Carpenter KA, Huang X (2018) Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: a review. Curr Pharm Des. https://doi.org/10.2174/1381612824666180607124038
Serafim MSM, Kronenberger T, Oliveira PR et al (2020) The application of machine learning techniques to innovative antibacterial discovery and development. Expert Opin Drug Discov. https://doi.org/10.1080/17460441.2020.1776696
Melville J, Burke E, Hirst J (2009) Machine learning in virtual screening. Comb Chem High Throughput Screen. https://doi.org/10.2174/138620709788167980
Wójcikowski M, Ballester PJ, Siedlecki P (2017) Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. https://doi.org/10.1038/srep46710
Carpenter KA, Cohen DS, Jarrell JT, Huang X (2018) Deep learning and virtual drug screening. Future Med Chem. 10(21):2557–2567. https://doi.org/10.4155/fmc-2018-0314
Labbé CM, Rey J, Lagorce D et al (2015) MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. https://doi.org/10.1093/nar/gkv306
Schellhammer I, Rarey M (2004) FlexX-Scan: Fast, structure-based virtual screening. Proteins Struct Funct Bioinforma 57:504–517. https://doi.org/10.1002/prot.20217
Perez-Castillo Y, Sotomayor-Burneo S, Jimenes-Vargas K et al (2019) CompScore: boosting structure-based virtual screening performance by incorporating docking scoring function components into consensus scoring. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00343
Skalic M, Martínez-Rosell G, Jiménez J, De Fabritiis G (2019) PlayMolecule bindscope: large scale CNN-based virtual screening on the web. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty758
Fang Y, Ding Y, Feinstein WP et al (2016) GeauxDock: accelerating structure-based virtual screening with heterogeneous computing. PLoS ONE. https://doi.org/10.1371/journal.pone.0158898
Pires DEV, Veloso WNP, Myung YC et al (2020) EasyVS: a user-friendly web-based tool for molecule library selection and structure-based virtual screening. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa480
Ibrahim TM, Bauer MR, Boeckler FM (2015) Applying DEKOIS 2.0 in structure-based virtual screening to probe the impact of preparation procedures and score normalization. J Cheminform. https://doi.org/10.1186/s13321-015-0074-6
Shin WH, Christoffer CW, Wang J, Kihara D (2016) PL-PatchSurfer2: improved local surface matching-based virtual screening method that is tolerant to target and ligand structure variation. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00163
Litfin T, Zhou Y, Yang Y (2017) SPOT-ligand 2: improving structure-based virtual screening by binding-homology search on an expanded structural template library. Bioinformatics. https://doi.org/10.1093/bioinformatics/btw829
Ropp PJ, Spiegel JO, Walker JL et al (2019) GypSUm-DL: An open-source program for preparing small-molecule libraries for structure-based virtual screening. J Cheminform. https://doi.org/10.1186/s13321-019-0358-3
Akbar R, Jusoh SA, Amaro RE, Helms V (2017) ENRI: a tool for selecting structure-based virtual screening target conformations. Chem Biol Drug Des. https://doi.org/10.1111/cbdd.12900
Kellenberger E, Springael JY, Parmentier M et al (2007) Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J Med Chem. https://doi.org/10.1021/jm061389p
De Graaf C, Rognan D (2008) Selective structure-based virtual screening for full and partial agonists of the β2 adrenergic receptor. J Med Chem. https://doi.org/10.1021/jm800710x
Vidler LR, Filippakopoulos P, Fedorov O et al (2013) Discovery of novel small-molecule inhibitors of BRD4 using structure-based virtual screening. J Med Chem. https://doi.org/10.1021/jm4011302
Liu LJ, Leung KH, Chan DSH et al (2014) Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. https://doi.org/10.1038/cddis.2014.250
Yang C, Wang W, Chen L et al (2016) Discovery of a VHL and HIF1α interaction inhibitor with: in vivo angiogenic activity via structure-based virtual screening. Chem Commun. https://doi.org/10.1039/c6cc04938a
Zhuang C, Narayanapillai S, Zhang W et al (2014) Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J Med Chem. https://doi.org/10.1021/jm4017174
Dou X, Jiang L, Wang Y et al (2018) Discovery of new GSK-3β inhibitors through structure-based virtual screening. Bioorganic Med Chem Lett. https://doi.org/10.1016/j.bmcl.2017.11.036
Liu Y, Ren Y, Cao Y et al (2017) Discovery of a low toxicity O-GlcNAc Transferase (OGT) inhibitor by structure-based virtual screening of natural products. Sci Rep. https://doi.org/10.1038/s41598-017-12522-0
Wang Y, Dou X, Jiang L et al (2019) Discovery of novel glycogen synthase kinase-3α inhibitors: Structure-based virtual screening, preliminary SAR and biological evaluation for treatment of acute myeloid leukemia. Eur J Med Chem. https://doi.org/10.1016/j.ejmech.2019.03.039
Wang Q, Xu J, Li Y et al (2018) Identification of a novel protein arginine methyltransferase 5 inhibitor in non-small cell lung cancer by structure-based virtual screening. Front Pharmacol. https://doi.org/10.3389/fphar.2018.00173
Sharma K, Patidar K, Ali MA et al (2018) Structure-based virtual screening for the identification of high affinity compounds as potent vegfr2 inhibitors for the treatment of renal cell carcinoma. Curr Top Med Chem. https://doi.org/10.2174/1568026619666181130142237
Yousuf Z, Iman K, Iftikhar N, Mirza MU (2017) Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer. Breast Cancer Targets Ther. https://doi.org/10.2147/BCTT.S132074
Leão M, Pereira C, Bisio A et al (2013) Discovery of a new small-molecule inhibitor of p53-MDM2 interaction using a yeast-based approach. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2013.01.032
Gahlawat A, Kumar N, Kumar R et al (2020) Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00546
Selvaraj C, Dinesh DC, Panwar U et al (2020) Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1778535
Cruz JV, Neto MFA, Silva LB et al (2018) Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules. https://doi.org/10.3390/molecules23020453
Kannan S, Melesina J, Hauser AT et al (2014) Discovery of inhibitors of schistosoma mansoni hdac8 by combining homology modeling, virtual screening, and in vitro validation. J Chem Inf Model. https://doi.org/10.1021/ci5004653
Zoete V, Daina A, Bovigny C, Michielin O (2016) SwissSimilarity: a web tool for low to ultra high throughput ligand-based virtual screening. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00174
Imbernón B, Cecilia JM, Pérez-Sánchez H, Giménez D (2018) METADOCK: a parallel metaheuristic schema for virtual screening methods. Int J High Perform Comput Appl. https://doi.org/10.1177/1094342017697471
Riniker S, Landrum GA (2013) Open-source platform to benchmark fingerprints for ligand-based virtual screening. J Cheminform. https://doi.org/10.1186/1758-2946-5-26
Li H, Leung KS, Wong MH, Ballester PJ (2016) USR-VS: a web server for large-scale prospective virtual screening using ultrafast shape recognition techniques. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw320
Suzuki SD, Ohue M, Akiyama Y (2018) PKRank: a novel learning-to-rank method for ligand-based virtual screening using pairwise kernel and RankSVM. Artif Life Robot. https://doi.org/10.1007/s10015-017-0416-8
Patel H, Brinkjost T, Koch O (2017) PyGOLD: a python based API for docking based virtual screening workflow generation. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx197
Banegas-Luna AJ, Cerón-Carrasco JP, Puertas-Martín S, Pérez-Sánchez H (2019) BRUSELAS: HPC generic and customizable software architecture for 3D ligand-based virtual screening of large molecular databases. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00279
Wang L, Pang X, Li Y et al (2017) RADER: a rapid decoy retriever to facilitate decoy based assessment of virtual screening. Bioinformatics. https://doi.org/10.1093/bioinformatics/btw783
Mochizuki M, Suzuki SD, Yanagisawa K et al (2019) QEX: target-specific druglikeness filter enhances ligand-based virtual screening. Mol Divers. https://doi.org/10.1007/s11030-018-9842-3
Zhang H, Liao L, Cai Y et al (2019) IVS2vec: a tool of inverse virtual screening based on word2vec and deep learning techniques. Methods. https://doi.org/10.1016/j.ymeth.2019.03.012
Arcon JP, Modenutti CP, Avendaño D et al (2019) AutoDock Bias: improving binding mode prediction and virtual screening using known protein-ligand interactions. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz152
Ebejer JP, Finn PW, Wong WK et al (2019) Ligity: a non-superpositional, knowledge-based approach to virtual screening. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.8b00779
Zhu Z, Wang X, Yang Y et al (2020) D3Similarity: a ligand-based approach for predicting drug targets and for virtual screening of active compounds against COVID-19. ChemRxiv. https://doi.org/10.26434/chemrxiv.11959323.v1
Bharti DR, Hemrom AJ, Lynn AM (2019) GCAC: Galaxy workflow system for predictive model building for virtual screening. BMC Bioinformatics. https://doi.org/10.1186/s12859-018-2492-8
Kong Y, Bender A, Yan A (2018) Identification of Novel Aurora Kinase A (AURKA) Inhibitors via Hierarchical Ligand-Based Virtual Screening. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.7b00300
Musumeci D, Amato J, Zizza P et al (2017) Tandem application of ligand-based virtual screening and G4-OAS assay to identify novel G-quadruplex-targeting chemotypes. Biochim Biophys Acta - Gen Subj. https://doi.org/10.1016/j.bbagen.2017.01.024
Yu M, Gu Q, Xu J (2018) Discovering new PI3Kα inhibitors with a strategy of combining ligand-based and structure-based virtual screening. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-017-0092-8
Halim SA, Khan S, Khan A et al (2017) Targeting dengue virus NS-3 Helicase by Ligand based Pharmacophore Modeling and structure based virtual screening. Front Chem. https://doi.org/10.3389/fchem.2017.00088
Debnath S, Debnath T, Bhaumik S et al (2019) Discovery of novel potential selective HDAC8 inhibitors by combine ligand-based, structure-based virtual screening and in-vitro biological evaluation. Sci Rep. https://doi.org/10.1038/s41598-019-53376-y
Fu Y, Sun YN, Yi KH et al (2017) 3D pharmacophore-based virtual screening and docking approaches toward the discovery of novel HPPD inhibitors. Molecules. https://doi.org/10.3390/molecules22060959
Krishna S, Shukla S, Lakra AD et al (2017) Identification of potent inhibitors of DNA methyltransferase 1 (DNMT1) through a pharmacophore-based virtual screening approach. J Mol Graph Model. https://doi.org/10.1016/j.jmgm.2017.05.014
Pérez-Nueno VI, Pettersson S, Ritchie DW et al (2009) Discovery of novel HIV entry inhibitors for the CXCR4 receptor by prospective virtual screening. J Chem Inf Model. https://doi.org/10.1021/ci800468q
Hofmarcher M, Mayr A, Rumetshofer E et al (2020) Large-scale ligand-based virtual screening for SARS-CoV-2 inhibitors using deep neural networks. SSRN Electron J. https://doi.org/10.2139/ssrn.3561442
Amin SA, Ghosh K, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: monte carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1780946
Ferraz WR, Gomes RA, Novaes ALS, Goulart Trossini GH (2020) Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med Chem. https://doi.org/10.4155/fmc-2020-0165
Choudhary S, Malik YS, Tomar S (2020) Identification of SARS-CoV-2 Cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front Immunol. https://doi.org/10.3389/fimmu.2020.01664
Xiao T, Qi X, Chen Y, Jiang Y (2018) Development of Ligand-based big data deep neural network models for virtual screening of large compound libraries. Mol Inform. https://doi.org/10.1002/minf.201800031
Hu J, Liu Z, Yu DJ, Zhang Y (2018) LS-align: An atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening. In: Bioinformatics 34(13): 2209–2218; https://doi.org/https://doi.org/10.1093/bioinformatics/bty081
Ha EJ, Lwin CT, Durrant JD (2020) LigGrep: a tool for filtering docked poses to improve virtual-screening hit rates. J Cheminform. https://doi.org/10.1186/s13321-020-00471-2
Spiegel JO, Durrant JD (2020) AutoGrow4: an open-source genetic algorithm for de novo drug design and lead optimization. J Cheminform. https://doi.org/10.1186/s13321-020-00429-4
Chen P, Ke Y, Lu Y et al (2019) Dligand2: an improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. J Cheminform. https://doi.org/10.1186/s13321-019-0373-4
Gattani S, Mishra A, Hoque MT (2019) StackCBPred: a stacking based prediction of protein-carbohydrate binding sites from sequence. Carbohydr Res. https://doi.org/10.1016/j.carres.2019.107857
Li X, Yan X, Yang Y et al (2019) LSA: a local-weighted structural alignment tool for pharmaceutical virtual screening. RSC Adv. https://doi.org/10.1039/c8ra08915a
Seifert MHJ (2005) ProPose: steered virtual screening by simultaneous protein-ligand docking and ligand-ligand alignment. J Chem Inf Model. https://doi.org/10.1021/ci0496393
Schellhammer I, Rarey M (2007) TrixX: Structure-based molecule indexing for large-scale virtual screening in sublinear time. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-007-9103-5
Lagarde N, Goldwaser E, Pencheva T et al (2019) A free web-based protocol to assist structure-based virtual screening experiments. Int J Mol Sci. https://doi.org/10.3390/ijms20184648
Rifaioglu AS, Nalbat E, Atalay V et al (2020) DEEPScreen: high performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem Sci. https://doi.org/10.1039/c9sc03414e
Obrezanova O, Segall MD (2010) Gaussian processes for classification: QSAR modeling of ADMET and target activity. J Chem Inf Model. https://doi.org/10.1021/ci900406x
Wu Z, Zhu M, Kang Y et al (2020) Do we need different machine learning algorithms for QSAR modeling? A comprehensive assessment of 16 machine learning algorithms on 14 QSAR data sets. Brief Bioinform. https://doi.org/10.1093/bib/bbaa321
Obrezanova O, Csányi G, Gola JMR, Segall MD (2007) Gaussian processes: a method for automatic QSAR modeling of ADME properties. J Chem Inf Model. https://doi.org/10.1021/ci7000633
Benfenati E, Manganaro A, Gini G (2013) VEGA-QSAR: AI inside a platform for predictive toxicology. In: CEUR Workshop Proceedings
Ambure P, Halder AK, González Díaz H, Cordeiro MNDS (2019) QSAR-Co: an open source software for developing robust multitasking or multitarget classification-based QSAR models. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00295
Chen S, Xue D, Chuai G et al (2020) FL-QSAR: a federated learning based QSAR prototype for collaborative drug discovery. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa1006
Olier I, Sadawi N, Bickerton GR et al (2018) Meta-QSAR: a large-scale application of meta-learning to drug design and discovery. Mach Learn. https://doi.org/10.1007/s10994-017-5685-x
Soufan O, Ba-Alawi W, Magana-Mora A et al (2018) DPubChem: a web tool for QSAR modeling and high-throughput virtual screening. Sci Rep. https://doi.org/10.1038/s41598-018-27495-x
Karpov P, Godin G, Tetko IV (2020) Transformer-CNN: swiss knife for QSAR modeling and interpretation. J Cheminform. https://doi.org/10.1186/s13321-020-00423-w
Wang Y-L, Wang F, Shi X-X et al (2020) Cloud 3D-QSAR: a web tool for the development of quantitative structure–activity relationship models in drug discovery. Brief Bioinform. https://doi.org/10.1093/bib/bbaa276
Goh GB, Siegel C, Vishnu A, et al (2017) Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv
Reis J, Cagide F, Chavarria D et al (2016) Discovery of new chemical entities for old targets: insights on the lead optimization of chromone-based monoamine oxidase B (MAO-B) inhibitors. J Med Chem. https://doi.org/10.1021/acs.jmedchem.6b00527
Hoelz L, Horta B, Araújo J et al (2010) Quantitative structure-activity relationships of antioxidant phenolic compounds. J Chem Pharm Res 2(5):291–306
Zhang Y, Han Z, Gao Q et al (2019) Prediction of K562 Cells Functional Inhibitors Based on Machine Learning Approaches. Curr Pharm Des. https://doi.org/10.2174/1381612825666191107092214
Halder AK, Giri AK, Dias Soeiro Cordeiro MN (2019) Multi-target chemometric modelling, fragment analysis and virtual screening with ERK inhibitors as potential anticancer agents. Molecules. https://doi.org/10.3390/molecules24213909
Halder AK, Cordeiro MNDS (2019) Development of multi-target chemometric models for the inhibition of class I PI3K enzyme isoforms: a case study using QSAR-Co tool. Int J Mol Sci. https://doi.org/10.3390/ijms20174191
Kim S, Cho KH (2019) PyQSAR: a fast QSAR modeling platform using machine learning and jupyter notebook. Bull Korean Chem Soc. https://doi.org/10.1002/bkcs.11638
Ben Geoffrey AS, Christian Prasana J, Muthu S (2020) Structure-activity relationship of Quercetin and its tumor necrosis factor alpha inhibition activity by computational and machine learning methods. Mater Today Proc. https://doi.org/10.1016/j.matpr.2020.07.464
Ben Geoffrey A S, Rafal Madaj, Akhil Sanker, Mario Sergio Valdés Tresanco, Host Antony Davidd, Gitanjali Roy, Rinnu Sarah Saji, Abdulbasit Haliru Yakubu BM Automated In Silico Identification of Drug Candidates for Coronavirus Through a Novel Programmatic Tool and Extensive Computational (MD, DFT) Studies of Select Drug Candidatesl; https://doi.org/https://doi.org/10.26434/chemrxiv.12423638.v3
Žuvela P, David J, Wong MW (2018) Interpretation of ANN-based QSAR models for prediction of antioxidant activity of flavonoids. J Comput Chem. https://doi.org/10.1002/jcc.25168
Ding Q, Hou S, Zu S et al (2020) VISAR: an interactive tool for dissecting chemical features learned by deep neural network QSAR models. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa187
Gadaleta D, Manganelli S, Roncaglioni A et al (2018) QSAR modeling of ToxCast assays relevant to the molecular initiating events of AOPs leading to hepatic steatosis. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.8b00297
Hermansyah O, Bustamam A, Yanuar A (2020) Virtual Screening of DPP-4 Inhibitors Using QSAR-Based Artificial Intelligence and Molecular Docking of Hit Compounds to DPP-8 and DPP-9 Enzymes. https://doi.org/10.21203/rs.2.22282/v1
Tian Y, Zhang S, Yin H, Yan A (2020) Quantitative structure-activity relationship (QSAR) models and their applicability domain analysis on HIV-1 protease inhibitors by machine learning methods. Chemom Intell Lab Syst. https://doi.org/10.1016/j.chemolab.2019.103888
Wei Y, Li W, Du T et al (2019) Targeting HIV/HCV coinfection using a machine learning-based multiple quantitative structure-Activity Relationships (Multiple QSAR) Method. Int J Mol Sci. https://doi.org/10.3390/ijms20143572
Michel Kana (2020) Handling Missing Data For Advanced Machine Learning
Kumar S (2020) 7 Ways to Handle Missing Values in Machine Learning | by Satyam Kumar | Towards Data Science
Gad SC (2014) QSAR. In: Third E (ed) Wexler PBT- Encyclopedia of Toxicology. Academic Press, Oxford, pp 1–9
Neves BJ, Braga RC, Melo-Filho CC et al (2018) QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front Pharmacol 9:1275. https://doi.org/10.3389/fphar.2018.01275
Roy K, Kar S, Das RN (2015) Chapter 9 - Newer QSAR Techniques. In: Roy K, Kar S, Das RN, Book Title- Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment (eds). Academic Press, Boston,
Kwon S, Bae H, Jo J, Yoon S (2019) Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinformatics 20:521. https://doi.org/10.1186/s12859-019-3135-4
Roy K, Kar S, Das RN (2015) Chapter 12 - Future Avenues. In: Roy K, Kar S, Das RN, Book Title- Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment (eds). Academic Press, Boston, pp 455–462. https://doi.org/https://doi.org/10.1016/B978-0-12-801505-6.00012-0
Paolini GV, Shapland RHB, Van Hoorn WP et al (2006) Global mapping of pharmacological space. Nat Biotechnol. https://doi.org/10.1038/nbt1228
Koch U, Hamacher M, Nussbaumer P (2014) Cheminformatics at the interface of medicinal chemistry and proteomics. Biochim Biophys Acta-Proteins Proteomics 1844(1):156–61; https://doi.org/10.1016/j.bbapap.2013.05.010
Makhouri FR, Ghasemi JB (2019) Combating diseases with computational strategies used for drug design and discovery. Curr Top Med Chem. https://doi.org/10.2174/1568026619666190121125106
Würth R, Thellung S, Bajetto A et al (2016) Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today 21(1):190–199. https://doi.org/10.1016/j.drudis.2015.09.017
Joachim Haupt V, Schroeder M (2011) Old friends in new guise: repositioning of known drugs with structural bioinformatics. Brief Bioinform. https://doi.org/10.1093/bib/bbr011
Butcher EC (2005) Can cell systems biology rescue drug discovery? Nat Rev Drug Discov. https://doi.org/10.1038/nrd1754
Iyengar R, Zhao S, Chung SW et al (2012) Merging systems biology with pharmacodynamics. Sci Transl Med 4(126):126ps7. https://doi.org/10.1126/scitranslmed.3003563
Martínez V, Navarro C, Cano C et al (2015) DrugNet: network-based drug-disease prioritization by integrating heterogeneous data. Artif Intell Med. https://doi.org/10.1016/j.artmed.2014.11.003
Zhang W, Xu H, Li X et al (2020) DRIMC: an improved drug repositioning approach using Bayesian inductive matrix completion. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa062
Luo H, Zhang P, Cao XH et al (2016) DPDR-CPI, a server that predicts drug positioning and drug repositioning via chemical-protein interactome. Sci Rep. https://doi.org/10.1038/srep35996
Zhu Q, Tao C, Shen F, Chute CG (2014) Exploring the pharmacogenomics knowledge base (pharmgkb) for repositioning breast cancer drugs by leveraging Web ontology language (owl) and cheminformatics approaches. In: Pacific Symposium on Biocomputing
Gallo K, Goede A, Eckert A et al (2020) PROMISCUOUS 2.0: a resource for drug-repositioning. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa1061
Luo H, Li M, Wang S et al (2018) Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty013
Yella JK, Jegga AG (2020) MGATRx: discovering drug repositioning candidates using multi-view graph attention. biorxiv. https://doi.org/10.1101/2020.06.29.171876
Yan CK, Wang WX, Zhang G et al (2019) BiRWDDA: a novel drug repositioning method based on multisimilarity fusion. J Comput Biol. https://doi.org/10.1089/cmb.2019.0063
Fahimian G, Zahiri J, Arab SS, Sajedi RH (2019) RepCOOL: computational drug repositioning via integrating heterogeneous biological networks. biorxiv. https://doi.org/10.1101/817882
Li Z, Yao Y, Cheng X, et al (2020) A Computational Framework of Host-Based Drug Repositioning for Broad-Spectrum Antivirals against RNA Viruses. https://doi.org/10.26434/chemrxiv.12927260.v1
Wu D, Gao W, Li X et al (2020) Dr AFC: drug repositioning through anti-fibrosis characteristic. Brief Bioinform. https://doi.org/10.1093/bib/bbaa115
Hooshmand SA, Zarei Ghobadi M, Hooshmand SE et al (2020) A multimodal deep learning-based drug repurposing approach for treatment of COVID-19. Mol Divers. https://doi.org/10.1007/s11030-020-10144-9
Zhou Y, Hou Y, Shen J et al (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. https://doi.org/10.1038/s41421-020-0153-3
Zheng X, He S, Song X, et al (2018) DTI-RCNN: New efficient hybrid neural network model to predict drug–target interactions. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics)
Jarada TN, Rokne JG, Alhajj R (2020) SNF–CVAE: computational method to predict drug–disease interactions using similarity network fusion and collective variational autoencoder. Knowledge-Based Syst. https://doi.org/10.1016/j.knosys.2020.106585
Xu R, Wang QQ (2015) PhenoPredict: a disease phenome-wide drug repositioning approach towards schizophrenia drug discovery. J Biomed Inform. https://doi.org/10.1016/j.jbi.2015.06.027
Wu Z, Cheng F, Li J et al (2017) SDTNBI: an integrated network and chemoinformatics tool for systematic prediction of drug-target interactions and drug repositioning. Brief Bioinform. https://doi.org/10.1093/bib/bbw012
Zeng X, Zhu S, Liu X et al (2019) DeepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz418
Chen H, Cheng F, Li J (2020) IDrug: Integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1008040
Li B, Dai C, Wang L et al (2020) A novel drug repurposing approach for non-small cell lung cancer using deep learning. PLoS ONE. https://doi.org/10.1371/journal.pone.0233112
Kuenzi BM, Park J, Fong SH et al (2020) Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell. https://doi.org/10.1016/j.ccell.2020.09.014
Wang Z, Zhou M, Arnold C (2020) Toward heterogeneous information fusion: bipartite graph convolutional networks for in silico drug repurposing. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa437
Pinzi L, Rastelli G (2019) Molecular docking: Shifting paradigms in drug discovery. Int J Mol Sci. https://doi.org/10.3390/ijms20184331
Muhammed MT, Aki-Yalcin E (2019) Homology modeling in drug discovery: overview, current applications, and future perspectives. Chem Biol Drug Des 93:12–20. https://doi.org/10.1111/cbdd.13388
Lynch SR, Bothwell T, Campbell L et al (2007) A comparison of physical properties, screening procedures and a human efficacy trial for predicting the bioavailability of commercial elemental iron powders used for food fortification. Int J Vitam Nutr Res. https://doi.org/10.1024/0300-9831.77.2.107
Schneider P, Walters WP, Plowright AT et al (2020) Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov 19:353–364. https://doi.org/10.1038/s41573-019-0050-3
Chen H, Engkvist O, Wang Y et al (2018) The rise of deep learning in drug discovery. Drug Discov Today 23(6):1241–1250. https://doi.org/10.1016/j.drudis.2018.01.039
Lusci A, Pollastri G, Baldi P (2013) Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model. https://doi.org/10.1021/ci400187y
Kumar R, Sharma A, Siddiqui MH, Tiwari RK (2017) Prediction of human intestinal absorption of compounds using artificial intelligence techniques. Curr Drug Discov Technol. https://doi.org/10.2174/1570163814666170404160911
Zang Q, Mansouri K, Williams AJ et al (2017) In silico prediction of physicochemical properties of environmental chemicals using molecular fingerprints and machine learning. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00625
Tetko IV, Gasteiger J, Todeschini R et al (2005) Virtual computational chemistry laboratory-design and description. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-005-8694-y
Radchenko E V, Palyulin VA, Zefirov NS (2002) Virtual computational chemistry laboratory. System
Royal Society of Chemistry (2015) ChemSpider. Search and Share Chemistry. R. Soc, Chem
Kucukdereli H, Allen NJ, Lee AT et al (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1104977108
Ayati A, Falahati M, Irannejad H, Emami S (2012) Synthesis, in vitro antifungal evaluation and in silico study of 3-azolyl-4-chromanone phenylhydrazones. DARU, J Pharm Sci. https://doi.org/10.1186/2008-2231-20-46
Rashid M (2020) Design, synthesis and ADMET prediction of bis-benzimidazole as anticancer agent. Bioorg Chem. https://doi.org/10.1016/j.bioorg.2020.103576
Puratchikody A, Sriram D, Umamaheswari A, Irfan N (2016) 3-D structural interactions and quantitative structural toxicity studies of tyrosine derivatives intended for safe potent inflammation treatment. Chem Cent J. https://doi.org/10.1186/s13065-016-0169-9
Nascimento ACA, Prudêncio RBC, Costa IG (2016) A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinformatics. https://doi.org/10.1186/s12859-016-0890-3
Öztürk H, Özgür A, Ozkirimli E (2018) A chemical language based approach for protein-Ligand interaction prediction. arXiv https://doi.org/10.1002/minf.202000212
Nascimento ACA, Prudêncio RBC, Costa IG (2019) A drug-target network-based supervised machine learning repurposing method allowing the use of multiple heterogeneous information sources. Methods Mol Biol 1903:281–289. https://doi.org/10.1007/978-1-4939-8955-3_17
Öztürk H, Özgür A, Ozkirimli E (2018) DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 34(17):i821–i829. https://doi.org/10.1093/bioinformatics/bty593
Feng Q, Dueva E, Cherkasov A, Ester M (2018) PADME: A deep learning-based framework for drug-target interaction prediction. arXiv https://arxiv.org/abs/1807.09741v4
Beck BR, Shin B, Choi Y et al (2020) Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. https://doi.org/10.1016/j.csbj.2020.03.025
Lee H, Kim W (2019) Comparison of target features for predicting drug-target interactions by deep neural network based on large-scale drug-induced transcriptome data. Pharmaceutics. https://doi.org/10.3390/pharmaceutics11080377
Karimi M, Wu D, Wang Z, Shen Y (2019) DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz111
Born J, Manica M, Cadow J, et al (2020) PaccMannRL on SARS-CoV-2: Designing antiviral candidates with conditional generative models. arXiv https://arxiv.org/abs/2005.13285v3
Jiang M, Li Z, Bian Y, Wei Z (2019) A novel protein descriptor for the prediction of drug binding sites. BMC Bioinformatics. https://doi.org/10.1186/s12859-019-3058-0
Cañada A, Capella-Gutierrez S, Rabal O et al (2017) LimTox: a web tool for applied text mining of adverse event and toxicity associations of compounds, drugs and genes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx462
Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. https://doi.org/10.1021/acs.jmedchem.5b00104
Cheng F, Li W, Zhou Y et al (2012) AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. https://doi.org/10.1021/ci300367a
Patlewicz G, Jeliazkova N, Safford RJ et al (2008) An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ Res. https://doi.org/10.1080/10629360802083871
Uygun MT, Amudi K, Turaçlı İD, Menges N (2021) A new synthetic approach for pyrazolo[1,5-a]pyrazine-4(5H)-one derivatives and their antiproliferative effects on lung adenocarcinoma cell line. Mol Divers. https://doi.org/10.1007/s11030-020-10161-8
Srivastava A, Siddiqui S, Ahmad R et al (2020) Exploring nature’s bounty: identification of Withania somnifera as a promising source of therapeutic agents against COVID-19 by virtual screening and in silico evaluation. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1835725
Attene-Ramos MS, Miller N, Huang R et al (2013) The Tox21 robotic platform for the assessment of environmental chemicals-From vision to reality. Drug Discov. Today 18(15–16):716–23. https://doi.org/10.1016/j.drudis.2013.05.015
Wang Z, Liang L, Yin Z, Lin J (2016) Improving chemical similarity ensemble approach in target prediction. J Cheminform. https://doi.org/10.1186/s13321-016-0130-x
Pu L, Naderi M, Liu T et al (2019) eToxPred: a machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacol Toxicol. https://doi.org/10.1186/s40360-018-0282-6
Lysenko A, Sharma A, Boroevich KA, Tsunoda T (2018) An integrative machine learning approach for prediction of toxicity-related drug safety. Life Sci Alliance. https://doi.org/10.26508/lsa.201800098
Zhou B, Sun Q, Kong DX (2016) Predicting cancer-relevant proteins using an improved molecular similarity ensemble approach. Oncotarget. https://doi.org/10.18632/oncotarget.8716
Huang R, Xia M, Sakamuru S et al (2016) Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun. https://doi.org/10.1038/ncomms10425
Gupta VK, Rana PS (2019) Toxicity prediction of small drug molecules of androgen receptor using multilevel ensemble model. J Bioinform Comput Biol. https://doi.org/10.1142/S0219720019500331
Mayr A, Klambauer G, Unterthiner T, Hochreiter S (2016) DeepTox: toxicity prediction using deep learning. Front Environ Sci. https://doi.org/10.3389/fenvs.2015.00080
Gayvert KM, Madhukar NS, Elemento O (2016) A data-driven approach to predicting successes and failures of clinical trials. Cell Chem Biol. https://doi.org/10.1016/j.chembiol.2016.07.023
Gilvary C, Elkhader J, Madhukar N et al (2020) A machine learning and network framework to discover new indications for small molecules. PLoS Comput Biol. https://doi.org/10.1371/JOURNAL.PCBI.1008098
Robledo-Cadena DX, Gallardo-Pérez JC, Dávila-Borja V et al (2020) Non-steroidal anti-inflammatory drugs increase cisplatin, paclitaxel, and doxorubicin efficacy against human cervix cancer cells. Pharmaceuticals (Basel). https://doi.org/10.3390/ph13120463
Simm J, Klambauer G, Arany A et al (2018) Repurposing high-throughput image assays enables biological activity prediction for drug discovery. Cell Chem Biol. https://doi.org/10.1016/j.chembiol.2018.01.015
Goh GB, Siegel C, Hodas N, Vishnu A (2017) SMILES2vec: An interpretable general-purpose deep neural network for predicting chemical properties. arXiv https://arxiv.org/abs/1712.02034v2
Preuer K, Lewis RPI, Hochreiter S et al (2018) Deepsynergy: predicting anti-cancer drug synergy with deep learning. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx806
Xu Y, Pei J, Lai L (2017) Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.7b00244
Rodrigues T, Werner M, Roth J et al (2018) Machine intelligence decrypts β-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem Sci. https://doi.org/10.1039/c8sc02634c
Luechtefeld T, Marsh D, Rowlands C, Hartung T (2018) Machine learning of toxicological big data enables read-across structure activity relationships (RASAR) outperforming animal test reproducibility. Toxicol Sci. https://doi.org/10.1093/toxsci/kfy152
Zhang C, Cheng F, Li W et al (2016) In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol Inform. https://doi.org/10.1002/minf.201500055
Lei T, Li Y, Song Y et al (2016) ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J Cheminform. https://doi.org/10.1186/s13321-016-0117-7
Lei T, Chen F, Liu H et al (2017) ADMET evaluation in drug discovery. Part 17: development of quantitative and qualitative prediction models for chemical-induced respiratory toxicity. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.7b00317
Lei T, Sun H, Kang Y et al (2017) ADMET evaluation in drug discovery. 18 reliable prediction of chemical-induced urinary tract toxicity by boosting machine learning approaches. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.7b00631
Pandya R, Pandya J (2015) C5.0 algorithm to improved decision tree with feature selection and reduced error pruning. Int J Comput Appl. https://doi.org/10.5120/20639-3318
Jenwitheesuk E, Horst JA, Rivas KL et al (2008) Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol Sci. https://doi.org/10.1016/j.tips.2007.11.007
Gu S, Lai L, hua, (2020) Associating 197 Chinese herbal medicine with drug targets and diseases using the similarity ensemble approach. Acta Pharmacol Sin 41:432–438. https://doi.org/10.1038/s41401-019-0306-9
Chen YT, Xie JY, Sun Q, Mo WJ (2019) Novel drug candidates for treating esophageal carcinoma: a study on differentially expressed genes, using connectivity mapping and molecular docking. Int J Oncol 54:152–166. https://doi.org/10.3892/ijo.2018.4618
Taha KF, Khalil M, Abubakr MS, Shawky E (2020) Identifying cancerrelated molecular targets of Nandina domestica Thunb. by network pharmacologybased analysis in combination with chemical profiling and molecular docking studies. J Ethnopharmacol 249:112413. https://doi.org/10.1016/j.jep.2019.112413
Anighoro A, Bajorath J, Rastelli G (2014) Polypharmacology: Challenges and opportunities in drug discovery. J Med Chem 57(19):7874–87. https://doi.org/10.1021/jm5006463
Zhang W, Pei J, Lai L (2017) Computational multitarget drug design. J Chem Inf Model 57(3):403–412. https://doi.org/10.1021/acs.jcim.6b00491
Proschak E, Stark H, Merk D (2019) Polypharmacology by design: a medicinal chemist’s perspective on multitargeting compounds. J Med Chem 62(2):420–444. https://doi.org/10.1021/acs.jmedchem.8b00760
Awale M, Reymond JL (2017) The polypharmacology browser: a web-based multi-fingerprint target prediction tool using ChEMBL bioactivity data. J Cheminform. https://doi.org/10.1186/s13321-017-0199-x
Reker D, Rodrigues T, Schneider P, Schneider G (2014) Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1320001111
Wang L, Ma C, Wipf P et al (2013) Targethunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. https://doi.org/10.1208/s12248-012-9449-z
Xia W, Chenxu P, Honglin L (2016) PharmMapper. In: Enhancing Enrich. Pharmacophore-Based Target Predict. Polypharmacological Profiles Drugs 56(6):1175–83. https://doi.org/10.1021/acs.jcim.5b00690
Gong J, Cai C, Liu X et al (2013) ChemMapper: a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics. https://doi.org/10.1093/bioinformatics/btt270
Gfeller D, Grosdidier A, Wirth M et al (2014) SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. https://doi.org/10.1093/nar/gku293
Poirier M, Awale M, Roelli MA et al (2019) Identifying lysophosphatidic acid acyltransferase β (LPAAT-β) as the target of a nanomolar angiogenesis inhibitor from a phenotypic screen using the polypharmacology browser PPB2. ChemMedChem. https://doi.org/10.1002/cmdc.201800554
Ozhathil LC, Delalande C, Bianchi B et al (2018) Identification of potent and selective small molecule inhibitors of the cation channel TRPM4. Br J Pharmacol. https://doi.org/10.1111/bph.14220
Ratnawati DE, Marjono M, Anam S (2018) Prediction of active compounds from SMILES codes using backpropagation algorithm. In: AIP Conference Proceedings
Van Vleet TR, Liguori MJ, Lynch JJ et al (2019) Screening strategies and methods for better offtarget liability prediction and identification of small-molecule pharmaceuticals. SLAS Discov 24(1):1–24. https://doi.org/10.1177/2472555218799713
Yue SJ, Liu J, Feng WW et al (2017) System pharmacology-based dissection of the synergistic mechanism of huangqi and huanglian for diabetes mellitus. Front Pharmacol. https://doi.org/10.3389/fphar.2017.00694
Shi XQ, Yue SJ, Tang YP et al (2019) A network pharmacology approach to investigate the blood enriching mechanism of Danggui buxue Decoction. J Ethnopharmacol. https://doi.org/10.1016/j.jep.2019.01.027
Liu X, Wu J, Zhang D et al (2018) A network pharmacology approach to uncover the multiple mechanisms of hedyotis diffusa willd on colorectal cancer. EvidenceBased Complem Altern Med. https://doi.org/10.1155/2018/6517034
Wang J, Luo C, Shan C et al (2015) Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat Chem. https://doi.org/10.1038/nchem.2381
Fang J, Li Y, Liu R et al (2015) Discovery of multitarget-directed ligands against Alzheimer’s disease through systematic prediction of chemical-protein interactions. J Chem Inf Model. https://doi.org/10.1021/ci500574n
Gao L, Wang KX, Zhou YZ et al (2018) Uncovering the anticancer mechanism of compound Kushen Injection against HCC by integrating quantitative analysis, network analysis and experimental validation. Sci Rep. https://doi.org/10.1038/s41598-017-18325-7
Zhou W, Liu X, Tu Z et al (2013) Discovery of pteridin-7(8H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) kinase T790M/L858R Mutant. J Med Chem 56:7821–7837. https://doi.org/10.1021/jm401045n
Wang Q, Feng YH, Huang JC et al (2017) A novel framework for the identification of drug target proteins: combining stacked auto-encoders with a biased support vector machine. PLoS ONE. https://doi.org/10.1371/journal.pone.0176486
Carvalho-Silva D, Pierleoni A, Pignatelli M et al (2019) Open targets platform: new developments and updates two years on. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1133
López-Cortés A, Paz-y-Miño C, Guerrero S et al (2020) Pharmacogenomics, biomarker network, and allele frequencies in colorectal cancer. Pharmacogenomics J 20(1):136–158. https://doi.org/10.1038/s41397-019-0102-4
Nabirotchkin S, Peluffo AE, Bouaziz J, Cohen D (2020) Focusing on the unfolded protein response and autophagy related pathways to reposition common approved drugs against COVID-19. Preprints
López-Isac E, Acosta-Herrera M, Kerick M et al (2019) GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat Commun. https://doi.org/10.1038/s41467-019-12760-y
Martin P, Ding J, Duffus K et al (2019) Chromatin interactions reveal novel gene targets for drug repositioning in rheumatic diseases. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2018-214649
Dong J, Cao DS, Miao HY et al (2015) ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation. J Cheminform. https://doi.org/10.1186/s13321-015-0109-z
Angelo RM, Io AK, Almeida MP, et al (2020) OntoQSAR: An ontology for interpreting chemical and biological data in quantitative structure-activity relationship studies. In: Proceedings-14th IEEE International Conference on Semantic Computing, ICSC 2020
Oldenhof M, Arany A, Moreau Y, Simm J (2020) Chemgrapher: optical graph recognition of chemical compounds by deep learning. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00459
Dong J, Yao ZJ, Zhu MF et al (2017) ChemSAR: An online pipelining platform for molecular SAR modeling. J Cheminform. https://doi.org/10.1186/s13321-017-0215-1
Buyukbingol E, Sisman A, Akyildiz M et al (2007) Adaptive neuro-fuzzy inference system (ANFIS): a new approach to predictive modeling in QSAR applications: a study of neuro-fuzzy modeling of PCP-based NMDA receptor antagonists. Bioorg Med Chem 15:4265–4282. https://doi.org/10.1016/j.bmc.2007.03.065
Jiang HJ, Huang YA, You ZH (2019) Predicting drug-disease associations via using Gaussian interaction profile and kernel-based autoencoder. Biomed Res Int. https://doi.org/10.1155/2019/2426958
Wang YY, Cui C, Qi L et al (2019) DrPOCS: drug repositioning based on projection onto convex sets. IEEE/ACM Trans Comput Biol Bioinforma. https://doi.org/10.1109/TCBB.2018.2830384
Xuan P, Cui H, Shen T et al (2019) HeteroDualNet: a dual convolutional neural network with heterogeneous layers for drug-disease association prediction via chou’s five-step rule. Front Pharmacol. https://doi.org/10.3389/fphar.2019.01301
Sadeghi SS, Keyvanpour M (2019) RCDR: A Recommender Based Method for Computational Drug Repurposing. In: 2019 IEEE 5th Conference on Knowledge Based Engineering and Innovation, KBEI 2019
Zhu Q, Luo J, Ding P, Xiao Q (2018) GRTR: Drug-disease association prediction based on graph regularized transductive regression on heterogeneous network. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics)
Jiang HJ, Huang YA, You ZH (2020) SAEROF: an ensemble approach for large-scale drug-disease association prediction by incorporating rotation forest and sparse autoencoder deep neural network. Sci Rep. https://doi.org/10.1038/s41598-020-61616-9
Wang MN, You ZH, Li LP, et al (2020) WGMFDDA: A Novel Weighted-Based Graph Regularized Matrix Factorization for Predicting Drug-Disease Associations. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics),
Liu H, Zhang W, Song Y et al (2020) HNet-DNN: inferring new drug-disease associations with deep neural network based on heterogeneous network features. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b01008
Lee I, Keum J, Nam H (2019) DeepConv-DTI: prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1007129
Abdel-Basset M, Hawash H, Elhoseny M et al (2020) DeepH-DTA: deep learning for predicting drug-target interactions: a case study of COVID-19 drug repurposing. IEEE Access. https://doi.org/10.1109/access.2020.3024238
Yang J, He S, Zhang Z, Bo X (2020) NegStacking: drug-target interaction prediction based on ensemble learning and logistic regression. IEEE/ACM Trans Comput Biol Bioinforma. https://doi.org/10.1109/TCBB.2020.2968025
King MD, Long T, Pfalmer DL et al (2018) SPIDR: small-molecule peptide-influenced drug repurposing. BMC Bioinformatics. https://doi.org/10.1186/s12859-018-2153-y
Huang K, Fu T, Glass LM et al (2020) DeepPurpose: a deep learning library for drug–target interaction prediction. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa1005
Chu Y, Kaushik AC, Wang X et al (2019) DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features. Brief Bioinform. https://doi.org/10.1093/bib/bbz152
Shar PA, Tao W, Gao S et al (2016) Pred-binding: large-scale protein–ligand binding affinity prediction. J Enzyme Inhib Med Chem 31:1443–1450. https://doi.org/10.3109/14756366.2016.1144594
Capuzzi SJ, Kim ISJ, Lam WI et al (2017) Chembench: a publicly accessible, integrated cheminformatics portal. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00462
Pires DEV, Blundell TL, Ascher DB (2016) MCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep. https://doi.org/10.1038/srep29575
Pires DEV, Ascher DB (2016) CSM-lig: a web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw390
Pires DEV, Ascher DB (2016) mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw458
Kaminskas LM, Pires DEV, Ascher DB (2019) dendPoint: a web resource for dendrimer pharmacokinetics investigation and prediction. Sci Rep. https://doi.org/10.1038/s41598-019-51789-3
Patel RD, Prasanth Kumar S, Pandya HA, Solanki HA (2018) MDCKpred: a web-tool to calculate MDCK permeability coefficient of small molecule using membrane-interaction chemical features. Toxicol Mech Methods. https://doi.org/10.1080/15376516.2018.1499840
Montanari F, Knasmüller B, Kohlbacher S et al (2020) Vienna LiverTox workspace—a set of machine learning models for prediction of interactions profiles of small molecules with transporters relevant for regulatory agencies. Front Chem. https://doi.org/10.3389/fchem.2019.00899
Kochev N, Avramova S, Jeliazkova N (2018) Ambit-SMIRKS: a software module for reaction representation, reaction search and structure transformation. J Cheminform. https://doi.org/10.1186/s13321-018-0295-6
Hornig M, Klamt A (2005) COSMOfrag: a novel tool for high-throughput ADME property prediction and similarity screening based on quantum chemistry. J Chem Inf Model. https://doi.org/10.1021/ci0501948
Hassan-Harrirou H, Zhang C, Lemmin T (2020) RosENet: improving binding affinity prediction by leveraging molecular mechanics energies with an ensemble of 3D convolutional neural networks. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00075
Rifaioglu AS, Cetin Atalay R, Cansen Kahraman D et al (2020) MDeePred: novel multi-channel protein featurization for deep learning-based binding affinity prediction in drug discovery. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa858
Banerjee P, Eckert AO, Schrey AK, Preissner R (2018) ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. https://doi.org/10.1093/nar/gky318
Dong J, Wang NN, Yao ZJ et al (2018) Admetlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform. https://doi.org/10.1186/s13321-018-0283-x
Maunz A, Gütlein M, Rautenberg M et al (2013) Lazar: a modular predictive toxicology framework. Front Pharmacol. https://doi.org/10.3389/fphar.2013.00038
Yao ZJ, Dong J, Che YJ et al (2016) TargetNet: a web service for predicting potential drug–target interaction profiling via multi-target SAR models. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-016-9915-2
Meng C, Hu Y, Zhang Y, Guo F (2020) PSBP-SVM: a machine learning-based computational identifier for predicting polystyrene binding peptides. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2020.00245
Shen C, Luo J, Ouyang W et al (2020) IDDkin: network-based influence deep diffusion model for enhancing prediction of kinase inhibitors. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa1058
Jewison T, Su Y, Disfany FM et al (2014) SMPDB 2.0: big improvements to the small molecule pathway database. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1067
Dalabira E, Viennas E, Daki E et al (2014) DruGeVar: an online resource triangulating drugs with genes and genomic biomarkers for clinical pharmacogenomics. Public Health Genom. https://doi.org/10.1159/000365895
Verma J, Luo H, Hu J, Zhang P (2017) DrugPathSeeker: Interactive UI for exploring drug-ADR relation via pathways. In: IEEE Pacific Visualization Symposium
Jarada T, Rokne J, Alhajj R (2021) SNF-NN: Computational Method To Predict Drug-Disease Interactions Using Similarity Network Fusion and Neural Networks. Res Sq. https://doi.org/10.21203/rs.3.rs-56433/v1
Cao X, Fan R, Zeng W (2020) DeepDrug: a general graph-based deep learning framework for drug relation prediction. biorxiv. https://doi.org/10.1101/2020.11.09.375626
Hartenfeller M, Schneider G (2011) Enabling future drug discovery by de novo design. Wiley Interdiscip Rev Comput Mol Sci 1:742–759. https://doi.org/10.1002/wcms.49
Schneider P, Schneider G (2016) De Novo design at the edge of chaos. J Med Chem 59:4077–4086. https://doi.org/10.1021/acs.jmedchem.5b01849
Lavecchia A (2015) Machine-learning approaches in drug discovery: methods and applications. Drug Discov Today 20(3):318–331. https://doi.org/10.1016/j.drudis.2014.10.012
Vyas V, Jain A, Jain A, Gupta A (2008) Virtual screening: a fast tool for drug design. Sci Pharm 76(3):333–360. https://doi.org/10.3797/scipharm.0803-03
Ertl P, Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform 1:1–11. https://doi.org/10.1186/1758-2946-1-8
Blaschke T, Olivecrona M, Engkvist O et al (2018) Application of generative autoencoder in De novo molecular design. Mol Inform 37:1–11. https://doi.org/10.1002/minf.201700123
Jaakkola TS, Haussler D (1999) Exploiting generative models in discriminative classifiers. In: Advances in Neural Information Processing Systems
Kadurin A, Aliper A, Kazennov A et al (2017) The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8:10883–10890. https://doi.org/10.18632/oncotarget.14073
Müller AT, Hiss JA, Schneider G (2018) Recurrent neural network model for constructive peptide design. J Chem Inf Model 58:472–479. https://doi.org/10.1021/acs.jcim.7b00414
Olivecrona M, Blaschke T, Engkvist O, Chen H (2017) Molecular de-novo design through deep reinforcement learning. J Cheminform 9:1–14. https://doi.org/10.1186/s13321-017-0235-x
Merk D, Friedrich L, Grisoni F, Schneider G (2018) De novo design of bioactive small molecules by artificial intelligence. Mol Inform 37:3–6. https://doi.org/10.1002/minf.201700153
Sarkar D (2018) A comprehensive hands-on guide to transfer learning with real-world applications in deep learning. Medium
Li X, Fourches D (2020) Inductive transfer learning for molecular activity prediction: next-gen QSAR models with MolPMoFiT. J Cheminform. https://doi.org/10.1186/s13321-020-00430-x
Engkvist O, Norrby PO, Selmi N et al (2018) Computational prediction of chemical reactions: current status and outlook. Drug Discov Today 23:1203–1218. https://doi.org/10.1016/j.drudis.2018.02.014
Hessler G, Baringhaus KH (2018) Artificial intelligence in drug design. Molecules. https://doi.org/10.3390/molecules23102520
Domenico A, Nicola G, Daniela T et al (2020) De novo drug design of targeted chemical libraries based on artificial intelligence and pair-based multiobjective optimization. J Chem Inf Model 60:4582–4593. https://doi.org/10.1021/acs.jcim.0c00517
Ekins S, Puhl AC, Zorn KM et al (2019) Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 18:435–441. https://doi.org/10.1038/s41563-019-0338-z
Pushpakom S, Iorio F, Eyers PA et al (2018) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18(1):41–58. https://doi.org/10.1038/nrd.2018.168
Kubick N, Pajares M, Enache I et al (2020) Repurposing Zileuton as a depression drug using an AI and in vitro approach. Molecules. https://doi.org/10.3390/molecules25092155
Yuan Y, Pei J, Lai L (2020) LigBuilder V3: a multi-target de novo drug design approach. Front Chem 8:1–18. https://doi.org/10.3389/fchem.2020.00142
Wei L, Wen W, Rao L et al (2020) Cov_FB3D: a de novo covalent drug design protocol integrating the Ba-SAMP strategy and machine-learning-based synthetic tractability evaluation. J Chem Inf Model 60:4388–4402. https://doi.org/10.1021/acs.jcim.9b01197
Jiménez-Luna J, Grisoni F, Schneider G (2020) Drug discovery with explainable artificial intelligence. Nat Mach Intell 2:573–584. https://doi.org/10.1038/s42256-020-00236-4
Cavasotto CN, Di Filippo JI (2021) Artificial intelligence in the early stages of drug discovery. Arch Biochem Biophys 698:108730. https://doi.org/10.1016/j.abb.2020.108730
Wong CH, Siah KW, Lo AW (2019) Estimation of clinical trial success rates and related parameters. Biostatistics 20:273–286. https://doi.org/10.1093/biostatistics/kxx069
DiMasi JA, Grabowski HG, Hansen RW (2016) Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 47:20–33. https://doi.org/10.1016/j.jhealeco.2016.01.012
Ruddigkeit L, Van Deursen R, Blum LC, Reymond JL (2012) Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J Chem Inf Model 52:2864–2875. https://doi.org/10.1021/ci300415d
Mohs RC, Greig NH (2017) Drug discovery and development: role of basic biological research. Alzheimer’s Dement Transl Res Clin Interv 3(4):651–657. https://doi.org/10.1016/2Fj.trci.2017.10.005
Vamathevan J, Clark D, Czodrowski P et al (2019) Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18(6):463–477. https://doi.org/10.1038/s41573-019-0024-5
Niel O, Bastard P (2019) Artificial intelligence in nephrology: core concepts, clinical applications, and perspectives. Am J Kidney Dis. https://doi.org/10.1053/j.ajkd.2019.05.020
Ahuja AS (2019) The impact of artificial intelligence in medicine on the future role of the physician. PeerJ. https://doi.org/10.7717/peerj.7702
Rubin EH, Gilliland DG (2012) Drug development and clinical trials-the path to an approved cancer drug. Nat Rev Clin Oncol 9:215–222. https://doi.org/10.1038/nrclinonc.2012.22
Rautio J, Kumpulainen H, Heimbach T et al (2008) Prodrugs: design and clinical applications. Nat Rev Drug Discov 7(3):255–270. https://doi.org/10.1038/nrd2468
Harrer S, Shah P, Antony B, Hu J (2019) Artificial intelligence for clinical trial design. Trends Pharmacol Sci 40:577–591. https://doi.org/10.1016/j.tips.2019.05.005
Fogel DB (2018) Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contemp Clin Trials Commun 11:156–164. https://doi.org/10.1016/2Fj.conctc.2018.08.001
Toh TS, Dondelinger F, Wang D (2019) Looking beyond the hype: applied AI and machine learning in translational medicine. EBioMedicine 47:607–615. https://doi.org/10.1016/j.ebiom.2019.08.027
Qi Y (2019) Predicting phase 3 clinical trial results by modeling phase 2 clinical trial subject level data using deep learning. Proc Mach Learn Res 106:1–14
Viceconti M, Henney A, Morley-Fletcher E (2016) In silico clinical trials: how computer simulation will transform the biomedical industry. Int J Clin Trials. https://doi.org/10.18203/2349-3259.ijct20161408
Magalingam KB, Radhakrishnan A, Ping NS, Haleagrahara N (2018) Current concepts of neurodegenerative mechanisms in Alzheimer’s disease. Biomed Res Int. https://doi.org/10.1155/2018/3740461
Hussain R, Zubair H, Pursell S, Shahab M (2018) Neurodegenerative diseases: regenerative mechanisms and novel therapeutic approaches. Brain Sci. https://doi.org/10.3390/brainsci8090177
Levenson RW, Sturm VE, Haase CM (2014) Emotional and behavioral symptoms in neurodegenerative disease: a model for studying the neural bases of psychopathology. Annu Rev Clin Psychol 10:581–606. https://doi.org/10.1146/annurev-clinpsy-032813-153653
Gitler AD, Dhillon P, Shorter J (2017) Neurodegenerative disease: Models, mechanisms, and a new hope. DMM Dis Model Mech 10:499–502. https://doi.org/10.1242/dmm.030205
Mak KK, Pichika MR (2019) Artificial intelligence in drug development: present status and future prospects. Drug Discov Today 24(3):773–780. https://doi.org/10.1016/j.drudis.2018.11.014
Peng J, Guan J, Shang X (2019) Predicting Parkinson’s disease genes based on node2vec and autoencoder. Front Genet 10:1–6. https://doi.org/10.3389/fgene.2019.00226
Thomas SN, Funk KE, Wan Y et al (2012) Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. https://doi.org/10.1007/s00401-011-0893-0
Yousefian-Jazi A, Sung MK, Lee T et al (2020) Functional fine-mapping of noncoding risk variants in amyotrophic lateral sclerosis utilizing convolutional neural network. Sci Rep 10:1–12. https://doi.org/10.1038/s41598-020-69790-6
Gupta R, Ambasta RK, Kumar P (2020) Identification of novel class I and class IIb histone deacetylase inhibitor for Alzheimer’s disease therapeutics. Life Sci. https://doi.org/10.1016/j.lfs.2020.117912
Jamal S, Grover A, Grover S (2019) Machine learning from molecular dynamics trajectories to predict caspase-8 Inhibitors against Alzheimer’s disease. Front Pharmacol 10:1–13. https://doi.org/10.3389/fphar.2019.00780
Chen HY, Chen JQ, Li JY et al (2019) Deep learning and random forest approach for finding the optimal traditional Chinese medicine formula for treatment of Alzheimer’s Disease. J Chem Inf Model 59:1605–1623. https://doi.org/10.1021/acs.jcim.9b00041
Ponzoni I, Sebastián-Pérez V, Martínez MJ et al (2019) QSAR Classification models for predicting the activity of inhibitors of beta-secretase (BACE1) associated with Alzheimer’s disease. Sci Rep 9:1–13. https://doi.org/10.1038/s41598-019-45522-3
Kaiser TM, Dentmon ZW, Dalloul CE et al (2020) Accelerated discovery of novel Ponatinib Analogs with improved properties for the treatment of Parkinson’s disease. ACS Med Chem Lett 11:491–496. https://doi.org/10.1021/acsmedchemlett.9b00612
Shao YM, Ma X, Paira P et al (2018) Discovery of indolylpiperazinylpyrimidines with dual-target profiles at adenosine A2A and dopamine D2 receptors for Parkinson’s disease treatment. PLoS ONE 13:1–27. https://doi.org/10.1371/journal.pone.0188212
Chen ZD, Zhao L, Chen HY et al (2020) A novel artificial intelligence protocol to investigate potential leads for Parkinson’s disease. RSC Adv 10:22939–22958. https://doi.org/10.1039/d0ra04028b
Deng L, Zhong W, Zhao L et al (2020) Artificial intelligence-based application to explore inhibitors of neurodegenerative diseases. Front Neurorobot. https://doi.org/10.3389/fnbot.2020.617327
Oh M, Ahn J, Yoon Y (2014) A network-based classification model for deriving novel drug-disease associations and assessing their molecular actions. PLoS ONE 9:1–12. https://doi.org/10.1371/journal.pone.0111668
Zhu Y, Jung W, Wang F, Che C (2020) Drug repurposing against Parkinson’s disease by text mining the scientific literature. Libr Hi Tech 38:741–750. https://doi.org/10.1108/LHT-08-2019-0170
Vatansever S, Schlessinger A, Wacker D, et al (2020) Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: state-of-the-arts and future directions. Med Res Rev Online ahead of print
Stokes JM, Yang K, Swanson K et al (2020) A deep learning approach to antibiotic discovery. Cell. https://doi.org/10.1016/j.cell.2020.01.021
Mamoshina P, Vieira A, Putin E, Zhavoronkov A (2016) Applications of deep learning in biomedicine. Mol Pharm 13(5):1445–1454. https://doi.org/10.1021/acs.molpharmaceut.5b00982
Preuer K, Klambauer G, Rippmann F, et al (2019) Interpretable deep learning in drug discovery. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics)
Ramsundar B, Liu B, Wu Z et al (2017) Is multitask deep learning practical for pharma? J Chem Inf Model. https://doi.org/10.1021/acs.jcim.7b00146
Grace K, Salvatier J, Dafoe A, et al (2017) When Will AI Exceed Human Performance? evidence from AI experts. J Artif Intell Res 62:1–48 https://arxiv.org/abs/1705.08807
Altae-Tran H, Ramsundar B, Pappu AS, Pande V (2017) Low data drug discovery with one-shot learning. ACS Cent Sci 3:283–293. https://doi.org/10.1021/acscentsci.6b00367
Acknowledgements
We would like to thank the senior management of Delhi Technological University for their constant support and guidance.
Author information
Authors and Affiliations
Contributions
All authors have read the paper and agreed to submit. PK conceived the idea. RG, DS, MS, ST arranged the data. RG, DS, MS, and ST contributed equally to this work. RKA and PK given their critical comments and structured this paper. Art work is done by RG, RAK, and PK. Paper is written by PK.
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest declared by the authors.
Rights and permissions
About this article

Cite this article
Gupta, R., Srivastava, D., Sahu, M. et al. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol Divers 25, 1315–1360 (2021). https://doi.org/10.1007/s11030-021-10217-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10217-3